Mutation Rates and Intrinsic Fidelity of Retroviral Reverse Transcriptases

Abstract
1. Introduction
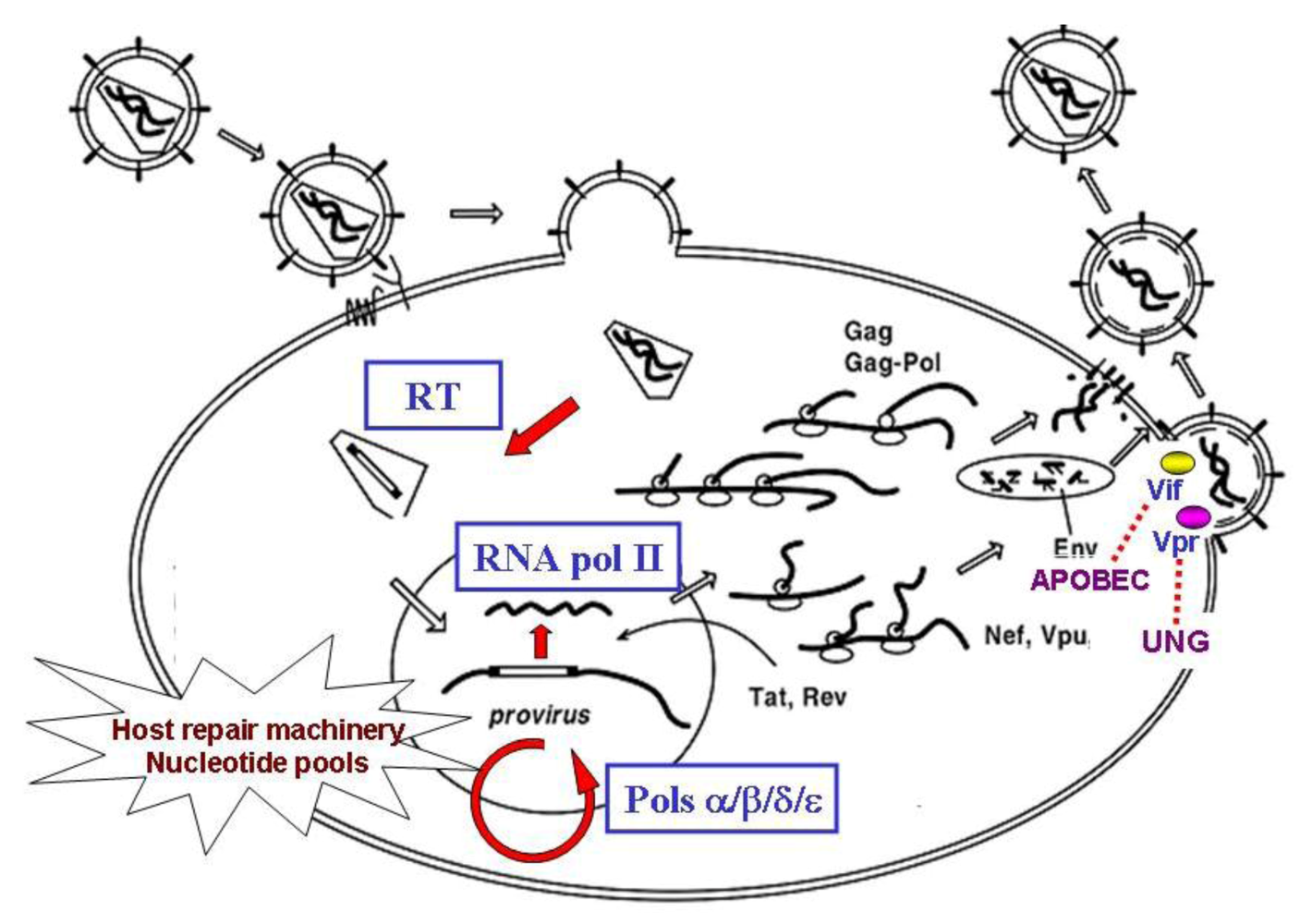
2. Viral and Host Factors Influencing Retroviral Mutation Rate

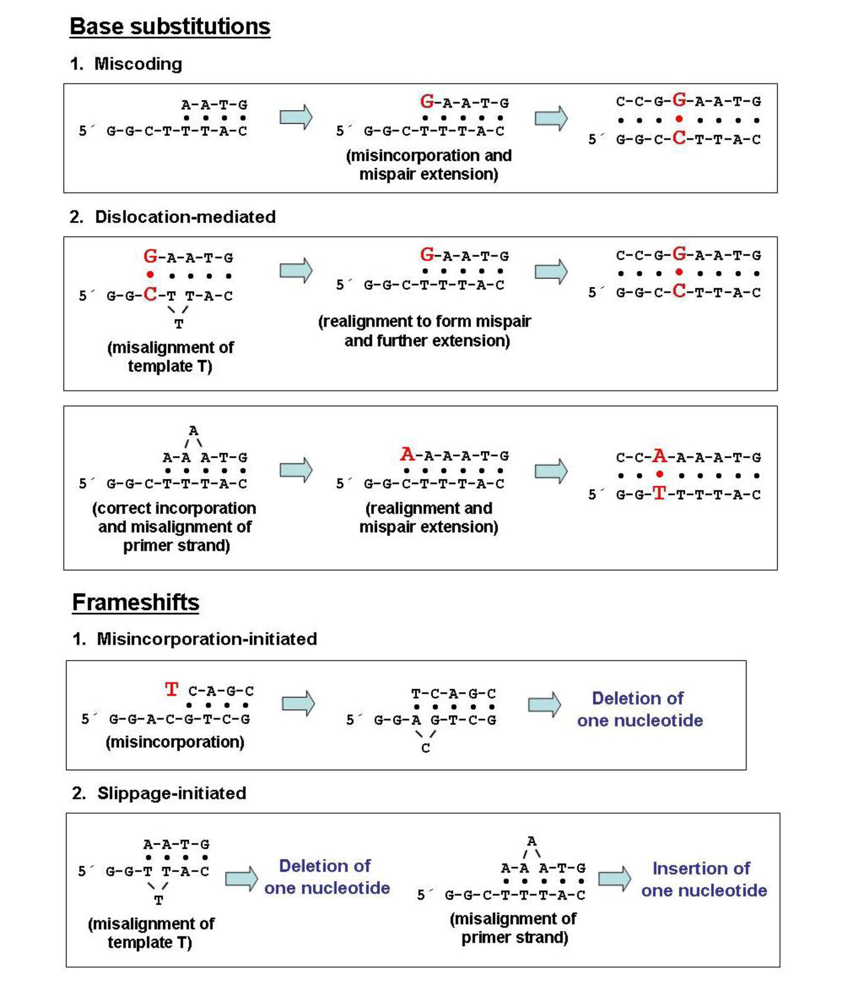
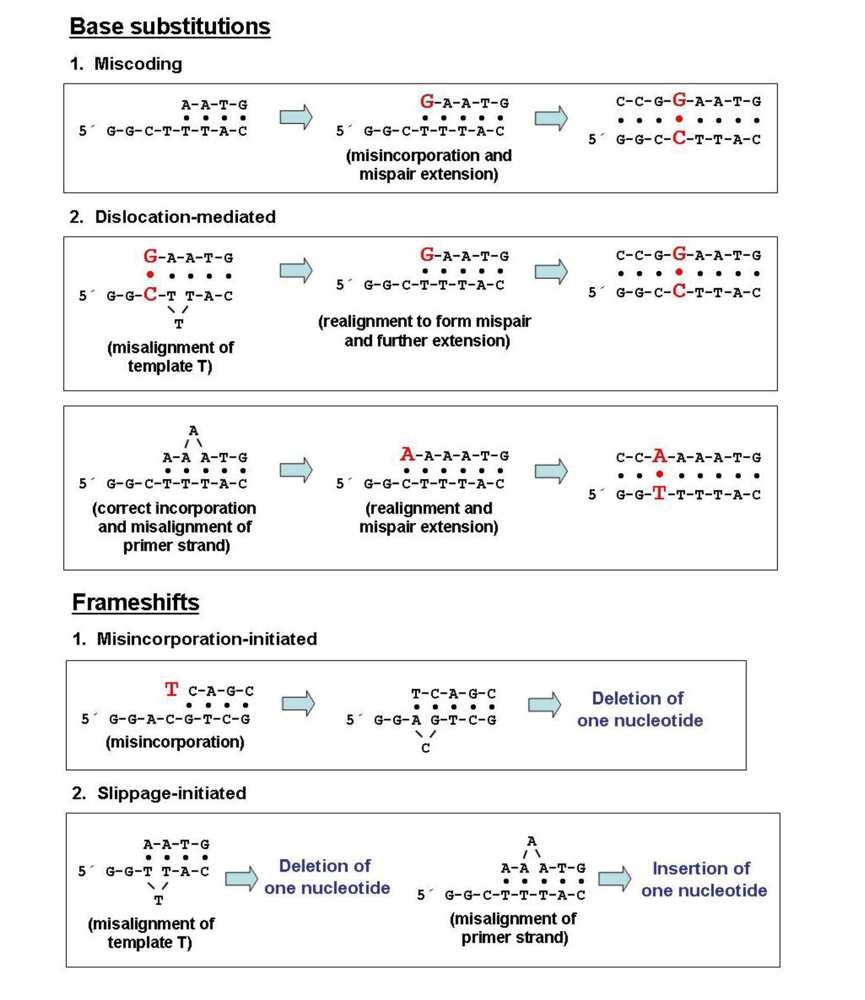
3. Intrinsic Fidelity of Retroviral RTs: M13mp2 lacZ Forward Mutation Assays

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Vector and reporter gene | Template | Error rate | References |
|---|---|---|---|---|
| HIV-1 RT (group M subtype B) | M13mp2, lac Zα | DNA | 0.6 – 6.7 x 10-4 | [69-79] |
| pBluescript, lac Z | DNA | 1.7 x 10-4 | [67] | |
| pBluescript, lac Z | RNA | 1.4 x 10-4 | [67] | |
| M13, env V1 | DNA | 1.9 x 10-4 | [66] | |
| M13, env V1 | RNA | 2.0 x 10-4 | [66] | |
| Litmus 29 (uracil-containing DNA), lac Z | DNA | 0.75 – 1.6 x 10-4 | [68,80] | |
| HIV-1 RT (group O) | M13mp2, lac Zα | DNA | 5.5 x 10-5 | [79] |
| SIVagm RT | M13mp2, lac Zα | DNA | 2.9 x 10-5 | [81] |
| SIVmne RT | M13mp2, lac Zα | DNA | 1.2 x 10-4 (CL8) a 1.6 x 10-5 (170) | [82] |
| PFV RT | Litmus 29, lac Z | DNA | 1.7 x 10-4 | [80] |
| FIV RT | M13mp2, lac Zα | DNA | 6.2 x 10-5 | [83] |
| AMV RT | M13mp2, lac Zα | DNA | 5.9 x 10-5 | [69,84] |
| Mo-MLV RT | M13mp2, lac Zα | DNA | 3.3 x 10-5 | [69,84] |
| pBluescript, lac Z | DNA | 3.4 x 10-5 | [67] | |
| pBluescript, lac Z | RNA | 2.7 x 10-5 | [67] | |
| FeLV RT | M13mp2, lac Zα | DNA | 5.8 x 10-6 | [83] |
4. Assessing Fidelity of DNA Synthesis Using Nucleotide Incorporation Kinetics
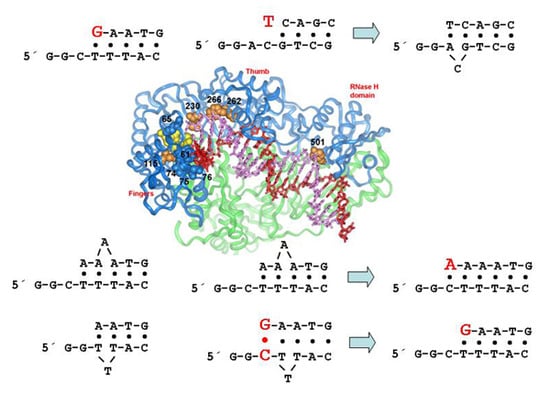
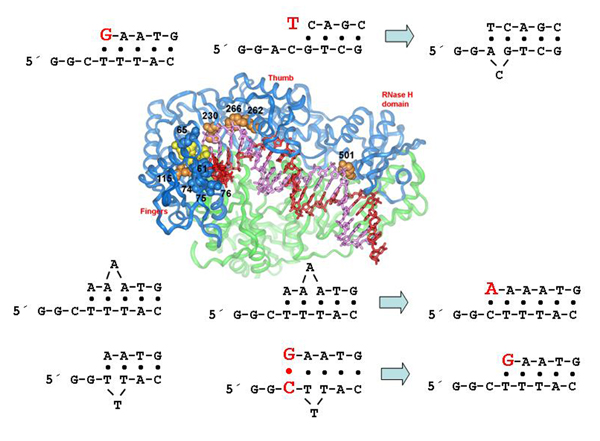
5. Structural Determinants of HIV-1 RT Fidelity
| Mutationa | RT subunit composition b | Subdomain location | Mutant frequency (x 10-4)c | Fold-Changed | References |
|---|---|---|---|---|---|
| M41L/T69SAG/AMs | p66/p51WT (HXB2) | fingers/palm | 6.3 (97) | ↓ 15.3 | [77] |
| M41L/T69SSG/AMs | p66/p51WT (HXB2) | fingers/palm | 5.9 (97) | ↓ 16.3 | [77] |
| F61A | p66/p51WT (HXB2) | fingers | 8.3 (97) | ↓ 11.7 | [75] |
| A62V/T69SAG/AMs | p66/p51WT (HXB2) | fingers/palm | 8.5 (97) | ↓ 11.4 | [77] |
| A62V/T69SSG/AMs | p66/p51WT (HXB2) | fingers/palm | 19 (97) | ↓ 5.0 | [77] |
| A62V/T69SSS/AMs | p66/p51WT (HXB2) | fingers/palm | 11 (97) | ↓ 8.8 | [77] |
| K65R | p66/p66 (BH10) | fingers | 10.6 (86) | ↓ 8.1 | [117] |
| T69SAG | p66/p51WT (HXB2) | fingers | 20 (97) | ↓ 4.8 | [77] |
| T69SSG | p66/p51WT (HXB2) | fingers | 12 (97) | ↓ 7.5 | [77] |
| T69SSS | p66/p51WT (HXB2) | fingers | 24 (97) | ↓ 4.0 | [77] |
| R72A | p66/p66 (HXB2) | fingers | 340 (210) | ↑ 1.6 | [118] |
| L74V | p66/p66 (BH10) | fingers | 50.5 (86) | ↓ 1.7 | [117] |
| p66/p51WT (BH10) | 55 (192) | ↓ 3.5 | [119] | ||
| V75A | p66/p51 (BH10) | fingers | 281 (206) | ↑ 1.4 | [78] |
| V75F | p66/p51 (BH10) | fingers | 112 (206) | ↓ 1.8 | [78] |
| V75I | p66/p51 (BH10) | fingers | 69.6 (206) | ↓ 3.0 | [78] |
| p66/p51 (ESP49) | 43.4 (83.1) | ↓ 1.9 | [79] | ||
| D76V | p66/p51 (BH10) | fingers | 26 (232) | ↓ 8.8 | [120] |
| R78A | p66/p51 (BH10) | fingers | 28 (250) | ↓ 8.9 | [121] |
| E89G | p66/p51WT (HXB2) | fingers | 62.6 (86) | ↓ 1.4 | [73] |
| p66/p51WT (BH10) | 96 (192) | ↓ 2.0 | [119] | ||
| E89G/M184V | p66/p51WT (HXB2) | fingers/palm | 123 (86) | ↑ 1.4 | [73] |
| E89K | p66/p51WT (HXB2) | fingers | 77 (86) | ↓ 1.2 | [122] |
| E89S | p66/p51WT (HXB2) | fingers | 53 (86) | ↓ 1.6 | [122] |
| E89V | p66/p51WT (HXB2) | fingers | 64 (86) | ↓ 1.3 | [122] |
| Y115A | p66/p51WT (BH10) | palm | 763 (192) | ↑ 4.0 | [119] |
| V148I | p66/p66 (HXB2) | palm | 30 (261) | ↓ 8.7 | [76] |
| p66/p66 (SIV-CL8) | 22 (178) | ↓ 8.1 | [82] | ||
| Q151M | p66/p66 (BH10) | palm | 55 (64) | ↓ 1.2 | [123] |
| Q151MCOMPLEX | p66/p66 (BH10) | fingers/palm | 31 (64) | ↓ 2.1 | [123] |
| Q151N | p66/p51 (BH10) | palm | 20 (261) | ↓ 13.1 | [124] |
| K154A | p66/p51 (BH10) | palm | 125 (261) | ↓ 2.1 | [124] |
| Y183F | p66/p51WT (BH10) | palm | 303 (192) | ↑ 1.6 | [119] |
| M184I | p66/p51 (HXB2) | palm | 24 (97) | ↓ 4.0 | [74] |
| M184V | p66/p51 (HXB2) | palm | 55.3 (86) | ↓ 1.6 | [73] |
| p66/p51WT (BH10) | 228 (192) | ↑ 1.2 | [119] | ||
| D256A | p66/p66 (HXB2) | thumb | 240 (200) | ↑ 1.2 | [125] |
| Q258A | p66/p66 (HXB2) | thumb | 390 (200) | ↑ 1.95 | [125] |
| K259A | p66/p66 (HXB2) | thumb | 300 (200) | ↑ 1.5 | [125] |
| L260A | p66/p66 (HXB2) | thumb | 230 (200) | ↑ 1.15 | [125] |
| G262A | p66/p66 (HXB2) | thumb | 860 (210) | ↑ 4.1 | [126] |
| K263A | p66/p66 (HXB2) | thumb | 290 (200) | ↑ 1.45 | [125] |
| W266A | p66/p66 (HXB2) | thumb | 630 (210) | ↑ 3.0 | [126] |
| Q269A | p66/p66 (HXB2) | thumb | 510 (200) | ↑ 2.55 | [125] |
| R277A | p66/p66 (HXB2) | thumb | 140 (160) | ↓ 1.1 | [127] |
| Q278A | p66/p66 (HXB2) | thumb | 190 (160) | ↑ 1.2 | [127] |
| L279A | p66/p66 (HXB2) | thumb | 150 (160) | ↓ 1.1 | [127] |
| C280A | p66/p66 (HXB2) | thumb | 300 (160) | ↑ 1.9 | [127] |
| K281A | p66/p66 (HXB2) | thumb | 140 (160) | ↓ 1.1 | [127] |
| L282A | p66/p66 (HXB2) | thumb | 120 (160) | ↓ 1.3 | [127] |
| R284A | p66/p66 (HXB2) | thumb | 170 (160) | ↑ 1.1 | [127] |
| G285A | p66/p66 (HXB2) | thumb | 160 (160) | = | [127] |
| K287A | p66/p66 (HXB2) | thumb | 120 (160) | ↓ 1.3 | [127] |
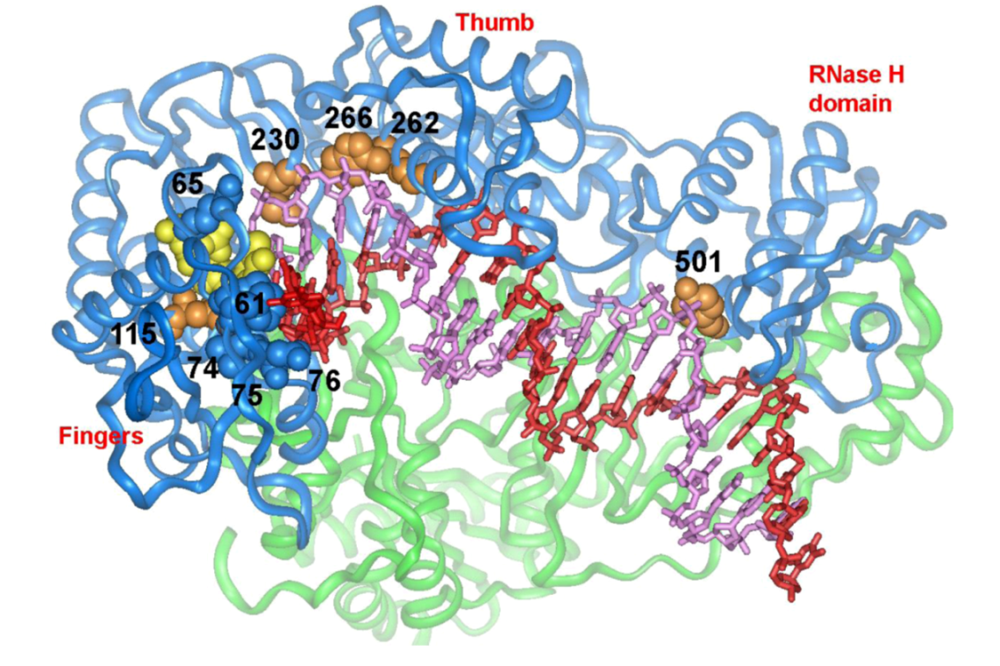
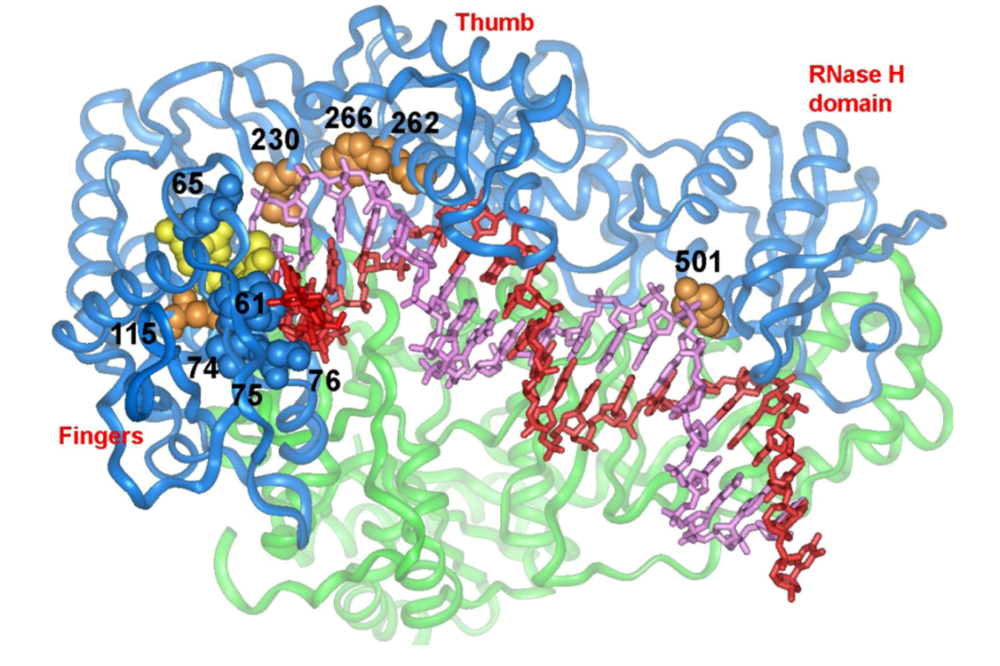
5.1. dNTP binding site residues
5.2. Residues that interact with the template strand
5.3. Residues that interact with the primer strand
5.4. Minor groove binding track residues
5.5. RNase H primer grip domain residues
6. Biological Consequences of Increasing or Decreasing Fidelity: Questions and Perspectives
Acknowledgments
References
- Wei, X.; Ghosh, S.K.; Taylor, M.E.; Johnson, V.A.; Emini, E.A.; Deutsch, P.; Lifson, J.D.; Bonhoeffer, S.; Nowak, M.A.; Hahn, B.H.; Saag, M.S.; Shaw, G.M. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 1995, 373, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.D.; Neumann, A.U.; Perelson, A.S.; Chen, W.; Leonard, J.M.; Markowitz, M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 1995, 373, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Coffin, J.M. HIV population dynamics in vivo: Implications for genetic variation, pathogenesis, and therapy. Science 1995, 267, 483–489. [Google Scholar] [PubMed]
- Svarovskaia, E.S.; Cheslock, S.R.; Zhang, W.-H.; Hu, W.-S.; Pathak, V.K. Retroviral mutation rates and reverse transcriptase fidelity. Front. Biosci. 2003, 8, D117–D134. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, J.P.; Temin, H.M. High mutation rate of a spleen necrosis virus-based retrovirus vector. Mol. Cell. Biol. 1986, 6, 4387–4395. [Google Scholar] [PubMed]
- Pathak, V.K.; Temin, H.M. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: Deletions and deletions with insertions. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 6024–6028. [Google Scholar] [CrossRef] [PubMed]
- Monk, R.J.; Malik, F.G.; Stokesberry, D.; Evans, L.H. Direct determination of the point mutation rate of a murine retrovirus. J. Virol. 1992, 66, 3683–3689. [Google Scholar] [PubMed]
- Varela-Echavarría, A.; Garvey, N.; Preston, B.D.; Dougherty, J.P. Comparison of Moloney murine leukemia virus mutation rate with the fidelity of its reverse transcriptase in vitro. J. Biol. Chem. 1992, 267, 24681–24688. [Google Scholar] [PubMed]
- Varela-Echavarría, A.; Prorock, C.M.; Ron, Y.; Dougherty, J.P. High rate of genetic rearrangement during replication of a Moloney murine leukemia virus-based vector. J. Virol. 1993, 67, 6357–6364. [Google Scholar] [PubMed]
- Mansky, L.M.; Temin, H.M. Lower mutation rate of bovine leukemia virus relative to that of spleen necrosis virus. J. Virol. 1994, 68, 494–499. [Google Scholar] [PubMed]
- Mansky, L.M.; Temin, H.M. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 1995, 69, 5087–5094. [Google Scholar] [PubMed]
- Parthasarathi, S.; Varela-Echevarría, A.; Ron, Y.; Preston, B.D.; Dougherty, J.P. Genetic rearrangements occurring during a single cycle of murine leukemia virus vector replication: characterization and implications. J. Virol. 1995, 69, 7991–8000. [Google Scholar] [PubMed]
- Mansky, L. Forward mutation rate of human immunodeficiency virus type 1 in a T lymphoid cell line. AIDS Res. Hum. Retroviruses 1996, 12, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M. In vivo analysis of human T-cell leukemia virus type 1 reverse transcription accuracy. J. Virol. 2000, 74, 9525–9531. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.J.; Wooley, D.P. A new cell-based assay for measuring the forward mutation rate of HIV-1. J. Virol. Methods 2005, 124, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Laakso, M.M.; Sutton, R.E. Replicative fidelity of lentiviral vectors produced by transient infection. Virology 2006, 348, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W. A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 7160–7164. [Google Scholar] [CrossRef] [PubMed]
- Sniegowski, P.D.; Gerrish, P.J.; Johnson, T.; Shaver, A. The evolution of mutation rates: separating causes from consequences. BioEssays 2000, 22, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Telesnitsky, A.; Goff, S.P. Reverse transcription and the generation of retroviral DNA. Coffin, J., Hughes, S.H., Varmus, H., Eds.; Cold Spring Harbor Laboratory Press: Plainview, NY, 1997. [Google Scholar]
- Basavapathruni, A.; Anderson, K. S. Reverse transcription of the HIV-1 pandemic. FASEB J., 2007, 21, 3795–3808. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Alexander, P.S. The base substitution fidelity of eucaryotic DNA polymerases. J. Biol. Chem. 1986, 261, 160–166. [Google Scholar] [PubMed]
- Matsuda, T.; Bebenek, K.; Masutani, C.; Hanaoka, F.; Kunkel, T.A. Low fidelity DNA synthesis by human DNA polymerase-η. Nature 2000, 404, 1011–1013. [Google Scholar] [CrossRef] [PubMed]
- de Mercoryol, L.; Corda, Y.; Job, C.; Job, D. Accuracy of wheat-germ RNA polymerase II: General enzymatic properties and effect of template conformational transition from right-handed B-DNA to left-handed Z-DNA. Eur. J. Biochem. 1992, 206, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Erie, D.A.; Hajiseyedjavadi, O.; Young, M.C.; von Hippel, P.H. Multiple RNA polymerase conformations and GreA: Control of the fidelity of transcription. Science 1993, 262, 867–873. [Google Scholar] [PubMed]
- Thomas, M.J.; Platas, A.A.; Hawley, D.K. Transcriptional fidelity and proofreading by RNA polymerase II. Cell 1998, 93, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Nesser, N.K.; Peterson, D.O.; Hawley, D.K. RNA polymerase II subunit Rpb9 is important for transcriptional fidelity in vivo. Proc. Natl. Acad. Sci. U.S.A., 2006, 103, 3268–3273. [Google Scholar] [CrossRef]
- Saxowsky, T.T.; Doetsch, P.W. RNA polymerase encounters with DNA damage: Transcription-coupled repair or transcriptional mutagenesis? Chem. Rev., 2006, 106, 474–488. [Google Scholar] [CrossRef]
- Sydow, J.F.; Brueckner, F.; Cheung, A.C.M.; Damsma, G.E.; Dengl, S.; Lehmann, E.; Vassylyev, D.; Cramer, P. Structural basis of transcription: Mismatch-specific fidelity mechanisms and paused RNA II polymerase II with frayed RNA. Mol. Cell, 2009, 34, 710–721. [Google Scholar] [CrossRef]
- Pugachev, K.V.; Guirakhoo, F.; Ocran, S.W.; Mitchell, F.; Parsons, M.; Penal, C.; Guirakhoo, S.; Pougatcheva, S.O.; Arroyo, J.; Trent, D.W.; Monath, T.P. High fidelity of yellow fever virus RNA polymerase. J. Virol., 2004, 78, 1032–1038. [Google Scholar] [CrossRef]
- O’Neil, P.K.; Sun, G.; Yu, H.; Ron, Y.; Dougherty, J.P.; Preston, B.D. Mutational analysis of HIV-1 long terminal repeats to explore the relative contribution of reverse transcriptase and RNA polymerase II to viral mutagenesis. J. Biol. Chem. 2002, 277, 38053–38061. [Google Scholar] [CrossRef] [PubMed]
- Elder, J.H.; Lerner, D.L.; Hasselkus-Light, C.S.; Fontenot, D.J.; Hunter, E.; Luciw, P.A.; Montelaro, R.C.; Phillips, T.R. Distinct subsets of retroviruses encode dUTPase. J. Virol. 1992, 66, 1791–1794. [Google Scholar] [PubMed]
- Köppe, B.; Menéndez-Arias, L.; Oroszlan, S. Expression and purification of the mouse mammary tumor virus gag-pro transframe protein p30 and characterization of its dUTPase activity. J. Virol. 1994, 68, 2313–2319. [Google Scholar] [PubMed]
- Lerner, D.L.; Wagaman, P.C.; Phillips, T.R.; Prospero-García, O.; Henriksen, S.J.; Fox, H.S.; Bloom, F.E.; Elder, J.H. Increased mutation frequency of feline immunodeficiency virus lacking a functional deoxyuridine-triphosphatase. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 7480–7484. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M. The mutation rate of human immunodeficiency virus type 1 is influenced by the vpr gene. Virology 1996, 222, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Morellet, N.; Bouaziz, S.; Petitjean, P.; Roques, B.P. NMR structure of the HIV-1 regulatory protein Vpr. J. Mol. Biol. 2003, 327, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Y.; Elder, R.T.; Bukrinsky, M. Interactions of HIV-1 viral protein R with host cell proteins. Adv. Pharmacol. 2007, 55, 233–260. [Google Scholar] [PubMed]
- Malim, M.H.; Emerman, M. HIV-1 accessory proteins – Ensuring viral survival in a hostile environment. Cell Host Microbe 2008, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Selig, L.; Benichou, S.; Rogel, M.E.; Wu, L.I.; Wodicka, M.A.; Sire, J.; Benarous, R.; Emerman, M. Uracil DNA glycosylase specifically interacts with Vpr of both human immunodeficiency virus type 1 and simian immunodeficiency virus of sooty mangabeys, but binding does not correlate with cell cycle arrest. J. Virol. 1997, 71, 4842–4846. [Google Scholar] [PubMed]
- Mansky, L.M.; Preveral, S.; Selig, L.; Benarous, R.; Benichou, S. The interaction of Vpr with uracil DNA glycosylase modulates the human immunodeficiency virus type 1 in vivo mutation rate. J. Virol. 2000, 74, 7039–7047. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M.; Le Rouzic, E.; Benichou, S.; Gajary, L.C. Influence of reverse transcriptase variants, drugs, and Vpr on human immunodeficiency virus type 1 mutant frequencies. J. Virol. 2003, 77, 2071–2080. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, H.; Otterlei, M.; Haug, T.; Solum, K.; Nagelhus, T.A.; Skorpen, F.; Krokan, H.E. Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res. 1997, 25, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Le Rouzic, E.; Kearney, J.A.; Mansky, L.M.; Benichou, S. Vpr-mediated incorporation of UNG2 into HIV-1 particles is required to modulate the virus mutation rate and for replication in macrophages. J. Biol. Chem. 2004, 279, 28419–38425. [Google Scholar] [CrossRef] [PubMed]
- Schröfelbauer, B.; Yu, Q.; Zeitlin, S.G.; Landau, N.R. Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J. Virol. 2005, 79, 10978–10987. [Google Scholar] [CrossRef] [PubMed]
- Langevin, C.; Maidou-Peindara, P.; Aas, P.A.; Jacquot, G.; Otterlei, M.; Slupphaug, G.; Benichou, S. Human immunodeficiency virus type 1 Vpr modulates cellular expresión of UNG2 via a negative transcriptional effect. J. Virol. 2009, 83, 10256–10263. [Google Scholar] [CrossRef] [PubMed]
- Willetts, K.E.; Rey, F.; Agostini, I.; Navarro, J.-M.; Baudat, Y.; Vigne, R.; Sire, J. DNA repair enzyme uracil DNA glycosylase is specifically incorporated into human immunodeficiency virus type 1 viral particles through a Vpr-independent mechanism. J. Virol. 1999, 73, 1682–1688. [Google Scholar] [PubMed]
- Priet, S.; Navarro, J.-M.; Gros, N.; Quérat, G.; Sire, J. Functional role of HIV-1 virion-associated uracil DNA glycosylase 2 in the correction of G:U mispairs to G:C pairs. J. Biol. Chem. 2003, 278, 4566–4571. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, R.S.; Peterlin, B.M. APOBEC3 proteins and reverse transcription. Virus Res. 2008, 134, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Santiago, M.L.; Greene, W.C. The role of the APOBEC3 family of cytidine deaminases in innate immunity, G-to-A hypermutation, and evolution of retroviruses, 2ndDomingo, E., Parrish, C.R., Holland, J.J., Eds.; Academic Press: London, UK, 2008. [Google Scholar]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113 2004, 116, 629 (erratum), 803–809. [Google Scholar] [CrossRef] [PubMed]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, B.; Pomerantz, R.J.; Zhang, C.; Arunachalam, S.C.; Gao, L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 2003, 424, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.N.; Holmes, R.K.; Malim, M.H. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 2006, 80, 8450–8458. [Google Scholar] [CrossRef] [PubMed]
- Holmes, R.K.; Koning, F.A.; Bishop, K.N.; Malim, M.H. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J. Biol. Chem. 2007, 282, 2587–2595. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.N.; Verma, M.; Kim, E.-Y.; Wolinsky, S.M.; Malim, M.H. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008, 4, e1000231. [Google Scholar] [CrossRef] [PubMed]
- Turelli, P.; Mangeat, B.; Jost, S.; Vianin, S.; Trono, D. Inhibition of hepatitis B virus replication by APOBEC3G. Science 2004, 303, 1829. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lilley, C.E.: Yu; Lee, D.V.; Chou, J.; Narvaiza, I.; Landau, N.R.; Weitzman, M.D. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr. Biol. 2006, 16, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Okeoma, C.M.; Lovsin, N.; Peterlin, B.M.; Ross, S.R. APOBEC3 inhibits mouse mammary tumour virus replication in vivo. Nature 2007, 445, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Holmes, R.K.; Malim, M.H.; Bishop, K.N. APOBEC-mediated viral restriction: not simply editing? Trends Biochem. Sci. 2007, 32, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.H.; Svarovskaia, E.S.; Barr, R.; Pathak, V.K. Y586F mutation in murine leukemia virus reverse transcriptase decreases fidelity of DNA synthesis in regions associated with adenine-thymine tracts. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 10090–10095. [Google Scholar] [CrossRef] [PubMed]
- Rascle, J.-B.; Ficheux, D.; Darlix, J.-L. Possible roles of nucleocapsid protein of MoMuLV in the specificity of proviral DNA synthesis and in the genetic variability of the virus. J. Mol. Biol. 1998, 280, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Post, K.; Kankia, B.; Gopalakrishnan, S.; Yang, V.; Cramer, E.; Saladores, P.; Gorelick, R.J.; Guo, J.; Musier-Forsyth, K.; Levin, J.G. Fidelity of plus-strand primer requires the nucleic acid chaperone activity of HIV-1 nucleocapsid protein. Nucleic Acids Res. 2009, 37, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Bakhanashvili, M. p53 enhances the fidelity of DNA synthesis by human immunodeficiency virus type 1 reverse transcriptase. Oncogene 2001, 20, 7635–7644. [Google Scholar] [CrossRef] [PubMed]
- Bakhanashvili, M.; Novitsky, E.; Lilling, G.; Rahav, G. p53 in cytoplasm may enhance the accuracy of DNA synthesis by human immunodeficiency virus type 1 reverse transcriptase. Oncogene 2004, 23, 6890–6899. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Arias, L. Molecular basis of fidelity of DNA synthesis and nucleotide specificity of retroviral reverse transcriptases. Prog. Nucleic Acid Res. Mol. Biol. 2002, 71, 91–147. [Google Scholar] [PubMed]
- Bebenek, K.; Kunkel, T.A. Analyzing fidelity of DNA polymerases. Methods Enzymol. 1995, 262, 217–232. [Google Scholar] [PubMed]
- Ji, J.; Loeb, L.A. Fidelity of HIV-1 reverse transcriptase copying a hypervariable region of the HIV-1 env gene. Virology 1994, 199, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Loeb, L.A. Fidelity of HIV-1 reverse transcriptase copying RNA in vitro. Biochemistry 1992, 31, 954–958. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.L.; Hughes, S.H. Effects of amino acid substitutions at position 115 on the fidelity of human immunodeficiency virus type 1 reverse transcriptase. J. Virol. 2000, 74, 6494–6500. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.D.; Bebenek, K.; Kunkel, T.A. The accuracy of reverse transcriptase from HIV-1. Science 1988, 242, 1171–1173. [Google Scholar] [PubMed]
- Bebenek, K.; Abbotts, J.; Roberts, J.D.; Wilson, S.H.; Kunkel, T.A. Specificity and mechanism of error-prone replication by human immunodeficiency virus-1 reverse transcriptase. J. Biol. Chem. 1989, 264, 16948–16956. [Google Scholar] [PubMed]
- Boyer, J.C.; Bebenek, K.; Kunkel, T.A. Unequal human immunodeficiency virus type 1 reverse transcriptase error rates with RNA and DNA templates. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 6919–6923. [Google Scholar] [CrossRef] [PubMed]
- Eckert, K.A.; Kunkel, T.A. Fidelity of DNA synthesis catalyzed by human DNA polymerase α and HIV-1 reverse transcriptase: effect of reaction pH. Nucleic Acids Res. 1993, 21, 5212–5220. [Google Scholar] [CrossRef] [PubMed]
- Drosopoulos, W.C.; Prasad, V.R. Increased misincorporation fidelity observed for nucleoside analog resistance mutations M184V and E89G in human immunodeficiency virus type 1 reverse transcriptase does not correlate with the overall error rate measured in vitro. J. Virol. 1998, 72, 4224–4230. [Google Scholar] [PubMed]
- Rezende, L.F.; Drosopoulos, W.C.; Prasad, V.R. The influence of 3TC resistance mutation M184I on the fidelity and error specificity of human immunodeficiency virus type 1 reverse transcriptase. Nucleic Acids Res. 1998, 26, 3066–3072. [Google Scholar] [CrossRef] [PubMed]
- Fisher, T.S.; Prasad, V.R. Substitutions of Phe61 located in the vicinity of template 5´-overhang influence polymerase fidelity and nucleoside analog sensitivity of HIV-1 reverse transcriptase. J. Biol. Chem. 2002, 277, 22345–22352. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.K.; Chen, R.; Skasko, M.; Reynolds, H.M.; Lee, K.; Bambara, R.A.; Mansky, L.M.; Kim, B. A role for dNTP binding of human immunodeficiency virus type 1 reverse transcriptase in viral mutagenesis. Biochemistry 2004, 43, 4490–4500. [Google Scholar] [CrossRef] [PubMed]
- Curr, K.; Tripathi, S.; Lennerstrand, J.; Larder, B.A.; Prasad, V.R. Influence of naturally occurring insertions in the fingers subdomain of human immunodeficiency virus type 1 reverse transcriptase on polymerase fidelity and mutation frequencies in vitro. J. Gen. Virol. 2006, 87, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Matamoros, T.; Kim, B.; Menéndez-Arias, L. Mechanistic insights into the role of Val75 of HIV-1 reverse transcriptase in misinsertion and mispair extension fidelity of DNA synthesis. J. Mol. Biol. 2008, 375, 1234–1248. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, M.M.; Matamoros, T.; Menéndez-Arias, L. Increased thermostability and fidelity of DNA synthesis of wild-type and mutant HIV-1 group O reverse transcriptases. J. Mol. Biol. 2009, 392, 872–874. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.L.; Stenbak, C.R.; Hoberman, D.; Linial, M.L.; Hughes, S.H. In vitro fidelity of the prototype primate foamy virus (PFV) RT compared to HIV-1 RT. Virology 2007, 367, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Stuke, A.W.; Ahmad-Omar, O.; Hoefer, K.; Hunsmann, G.; Jentsch, K.D. Mutations in the SIV env and the M13 lacZα gene generated in vitro by reverse transcriptases and DNA polymerases. Arch. Virol. 1997, 142, 1139–1154. [Google Scholar] [CrossRef] [PubMed]
- Diamond, T.L.; Souroullas, G.; Weiss, K.K.; Lee, K.Y.; Bambara, R.A.; Dewhurst, S.; Kim, B. Mechanistic understanding of an altered fidelity simian immunodeficiency virus reverse transcriptase mutation V148I, identified in a pig-tailed macaque. J. Biol. Chem. 2003, 278, 29913–29924. [Google Scholar] [CrossRef] [PubMed]
- Operario, D.J.; Reynolds, H.M.; Kim, B. Comparison of DNA polymerase activities between recombinant feline immunodeficiency and leukema virus reverse transcriptases. Virology 2005, 335, 106–121. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.D.; Preston, B.D.; Johnston, L.A.; Soni, A.; Loeb, L.A.; Kunkel, T.A. Fidelity of two retroviral reverse transcriptases during DNA-dependent DNA synthesis in vitro. Mol. Cell. Biol. 1989, 9, 469–476. [Google Scholar] [PubMed]
- Pathak, V.K.; Temin, H.M. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: substitutions, frameshifts, and hypermutations. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 6019–6023. [Google Scholar] [CrossRef] [PubMed]
- Bebenek, K.; Abbotts, J.; Wilson, S.H.; Kunkel, T.A. Error-prone polymerization by HIV-1 reverse transcriptase – Contribution of template-primer misalignment, miscoding, and termination probability to mutational hot spots. J. Biol. Chem. 1993, 268, 10324–10334. [Google Scholar] [PubMed]
- Quiñones-Mateu, M.E.; Soriano, V.; Domingo, E.; Menéndez-Arias, L. Characterization of the reverse transcriptase of a human immunodeficiency virus type 1 group O isolate. Virology 1997, 236, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Arias, L.; Abraha, A.; Quiñones-Mateu, M.E.; Mas, A.; Camarasa, M.-J.; Arts, E.J. Functional characterization of chimeric reverse transcriptases with polypeptide subunits of highly divergent HIV-1 group M and O strains. J. Biol. Chem. 2001, 276, 27470–27479. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, K.; Wiktorowicz, T.; Park, J.; Mergia, A.; Rethwilm, A.; Scheller, C. Accuracy estimation of foamy virus genome copying. Retrovirology 2009, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Hübner, A.; Kruhoffer, M.; Grosse, F.; Krauss, G. Fidelity of human immunodeficiency virus type I reverse transcriptase in copying natural RNA. J. Mol. Biol. 1992, 223, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Mudry Jr., R.A.; Rexrode 2nd, C.A. ; Pathak, V.K. Retroviral mutation rates and A-to-G hypermutations during different stages of retroviral replication. J. Virol. 1996, 70, 7594–7602. [Google Scholar] [PubMed]
- Echols, H.; Goodman, M.F. Fidelity mechanisms in DNA replication. Annu. Rev. Biochem. 1991, 60, 477–511. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A. Conformational coupling in DNA polymerase fidelity. Annu. Rev. Biochem. 1993, 62, 685–713. [Google Scholar] [PubMed]
- Kati, W.M.; Johnson, K.A.; Jerva, L.F.; Anderson, K.S. J. Biol. Chem. 1992, 267, 25988–25997. [PubMed]
- Zinnen, S.; Hsieh, J.-C.; Modrich, P. Misincorporation and mispaired primer extension by human immunodeficiency virus reverse transcriptase. J. Biol. Chem. 1994, 269, 24195–24202. [Google Scholar] [PubMed]
- Kerr, S.G.; Anderson, K.S. RNA dependent DNA replication fidelity of HIV-1 reverse transcriptase: Evidence of discrimination between DNA and RNA substrates. Biochemistry 1997, 36, 14056–14063. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.K.; Bambara, R.A.; Kim, B. Mechanistic role of residue Gln151 in error prone DNA synthesis by human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) – Pre-steady state kinetic study of the Q151N HIV-1 RT mutant with increased fidelity. J. Biol. Chem. 2002, 277, 22662–22669. [Google Scholar] [CrossRef] [PubMed]
- Skasko, M.; Weiss, K.K.; Reynolds, H.M.; Jamburuthugoda, V.; Lee, K.; Kim, B. Mechanistic differences in RNA-dependent DNA polymerization and fidelity between murine leukemia virus and HIV-1 reverse transcriptases. J. Biol. Chem. 2005, 280, 12190–12200. [Google Scholar] [CrossRef] [PubMed]
- Perrino, F.W.; Preston, B.D.; Sandell, L.L.; Loeb, L.A. Extension of mismatched 3' termini of DNA is a major determinant of the infidelity of human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl. Acad. Sci. U.S.A. 1989, 86, 8343–8347. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Goodman, M.F. Comparison of HIV-1 and avian myeloblastosis virus reverse transcriptase fidelity on RNA and DNA templates. J. Biol. Chem. 1992, 267, 10888–10896. [Google Scholar] [PubMed]
- Bakhanashvili, M.; Hizi, A. Fidelity of the reverse transcriptase of human immunodeficiency virus type 2. FEBS Lett. 1992, 306, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Bakhanashvili, M.; Hizi, A. Fidelity of the RNA-dependent DNA synthesis exhibited by the reverse transcriptases of human immunodeficiency virus types 1 and 2 and of murine leukemia virus: Mispair extension frequencies. Biochemistry 1992, 31, 9393–9398. [Google Scholar] [CrossRef] [PubMed]
- Bakhanashvili, M.; Hizi, A. Fidelity of DNA synthesis exhibited in vitro by the reverse transcriptase of the lentivirus equine infectious anemia virus. Biochemistry 1993, 32, 7559–7567. [Google Scholar] [CrossRef] [PubMed]
- Avidan, O.; Bochner, R.; Hizi, A. The catalytic properties of the recombinant reverse transcriptase of bovine immunodeficiency virus. Virology 2006, 351, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Avidan, O.; Meer, M.E.; Oz, I.; Hizi, A. The processivity and fidelity of DNA synthesis exhibited by the reverse transcriptase of bovine leukemia virus. Eur. J. Biochem. 2002, 269, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Taube, R.; Avidan, O.; Bakhanashvili, M.; Hizi, A. DNA synthesis exhibited by the reverse transcriptase of mouse mammary tumor virus: Processivity and fidelity of misinsertion and mispair extension. Eur. J. Biochem. 1998, 258, 1032–1039. [Google Scholar] [PubMed]
- Avidan, O.; Loya, S.; Tönjes, R.R.; Sevilya, Z.; Hizi, A. Expression and characterization of a recombinant novel reverse transcriptase of a porcine endogenous retrovirus. Virology 2003, 307, 341–357. [Google Scholar] [CrossRef] [PubMed]
- Boutabout, M.; Wilhelm, M.; Wilhelm, F.-X. DNA synthesis fidelity by the reverse transcriptase of the yeast retrotransposon Ty1. Nucleic Acids Res. 2001, 29, 2217–2222. [Google Scholar] [CrossRef] [PubMed]
- Conlan, L.H.; Stanger, M.J.; Ichiyanagi, K.; Belfort, M. Localization, mobility and fidelity of retrotransposed group II introns in rRNA genes. Nucleic Acids Res. 2005, 33, 5262–5270. [Google Scholar] [CrossRef] [PubMed]
- Kirshenboim, N.; Hayouka, Z.; Friedler, A.; Hizi, A. Expression and characterization of a novel reverse transcriptase of the LTR retrotransposon Tf1. Virology 2007, 366, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Arias, L. Mechanisms of resistance to nucleoside analogue inhibitors of HIV-1 reverse transcriptase. Virus Res. 2008, 134, 124–146. [Google Scholar] [CrossRef] [PubMed]
- Sarafianos, S.G.; Marchand, B.; Das, K.; Himmel, D.M.; Parniak, M.A.; Hughes, S.H.; Arnold, E. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009, 385, 693–713. [Google Scholar] [CrossRef] [PubMed]
- Kohlstaedt, L.A.; Wang, J.; Friedman, J.M.; Rice, P.A.; Steitz, T.A. Crystal structure at 3.5 Ã resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 1992, 256, 1783–1790. [Google Scholar] [PubMed]
- Huang, H.; Chopra, R.; Verdine, G.L.; Harrison, S.C. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: Implications for drug resistance. Science 1998, 282, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Mendieta, J.; Cases-González, C.E.; Matamoros, T.; Ramírez, G.; Menéndez-Arias, L. A Mg2+-induced conformational switch rendering a competent DNA polymerase catalytic complex. Proteins 2008, 71, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Steitz, T.A.; Smerdon, S.J.; Jäger, J.; Joyce, C.M. A unified polymerase mechanism for nonhomologous DNA and RNA polymerases. Science 1994, 266, 2022–2025. [Google Scholar] [PubMed]
- Shah, F.S.; Curr, K.A.; Hamburgh, M.E.; Parniak, M.; Mitsuya, H.; Arnez, J.G.; Prasad, V.R. Differential influence of nucleoside analog-resistance mutations K65R and L74V on the overall mutation rate and error specificity of human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem. 2000, 275, 27037–27044. [Google Scholar] [PubMed]
- Lewis, D.A.; Bebenek, K.; Beard, W.A.; Wilson, S.H.; Kunkel, T.A. Uniquely altered DNA replication fidelity conferred by an amino acid change in the nucleotide binding pocket of human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem. 1999, 274, 32924–32930. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, H.; De Clercq, E.; Anné, J. Fidelity analysis of HIV-1 reverse transcriptase mutants with an altered amino-acid sequence at residues Leu74, Glu89, Tyr115, Tyr183 and Met184. Eur. J. Biochem. 2000, 267, 2658–2665. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Hathaway, T.R.; Loeb, L.A. Fidelity of mutant HIV-1 reverse transcriptases: Interaction with the single-stranded template influences the accuracy of DNA synthesis. Biochemistry 1998, 37, 5831–5839. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Ayran, J.C.; Sagar, S.G.; Adman, E.T.; Fuller, S.M.; Tran, N.H.; Horrigan, J. New human immunodeficiency virus, type 1 reverse transcriptase (HIV-1 RT) mutants with increased fidelity of DNA synthesis – Accuracy, template binding, and processivity. J. Biol. Chem. 1999, 274, 27666–27673. [Google Scholar] [CrossRef] [PubMed]
- Hamburgh, M.E.; Curr, K.A.; Monaghan, M.; Rao, V.R.; Tripathi, S.; Preston, B.D.; Sarafianos, S.; Arnold, E.; Darden, T.; Prasad, V.R. Structural determinants of slippage-mediated mutations by human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem. 2006, 281, 7421–7428. [Google Scholar] [CrossRef] [PubMed]
- Rezende, L.F.; Curr, K.; Ueno, T.; Mitsuya, H.; Prasad, V.R. The impact of multidideoxynucleoside resistance-conferring mutations in human immunodeficiency virus type 1 reverse transcriptase on polymerase fidelity and error specificity. J. Virol. 1998, 72, 2890–2895. [Google Scholar] [PubMed]
- Weiss, K.K.; Isaacs, S.J.; Tran, N.H.; Adman, E.T.; Kim, B. Molecular architecture of the mutagenic active site of human immunodeficiency virus type 1 reverse transcriptase: roles of the β8-αE loop in fidelity, processivity, and substrate interactions. Biochemistry 2000, 39, 10684–10694. [Google Scholar] [CrossRef] [PubMed]
- Beard, W.A.; Stahl, S.J.; Kim, H.-R.; Bebenek, K.; Kumar, A.; Strub, M.-P.; Becerra, S.P.; Kunkel, T.A.; Wilson, S.H. Structure/function studies of human immunodeficiency virus type 1 reverse transcriptase – Alanine scanning mutagenesis of an α-helix in the thumb subdomain. J. Biol. Chem. 1994, 269, 28091–28097. [Google Scholar] [PubMed]
- Bebenek, K.; Beard, W.A.; Casas-Finet, J.R.; Kim, H.-R.; Darden, T.A.; Wilson, S.H.; Kunkel, T.A. Reduced frameshift fidelity and processivity of HIV-1 reverse transcriptase mutants containing alanine substitutions in helix H of the thumb subdomain. J. Biol. Chem. 1995, 270, 19516–19523. [Google Scholar] [CrossRef] [PubMed]
- Beard, W.A.; Minnick, D.T.; Wade, C.L.; Prasad, R.; Won, R.L.; Kumar, A.; Kunkel, T.A.; Wilson, S.H. Role of the “helix clamp” in HIV-1 reverse transcriptase catalytic cycling as revealed by alanine-scanning mutagenesis. J. Biol. Chem. 1996, 271, 12213–12220. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Orlova, M.; Georgiadis, M.M.; Hendrickson, W.A.; Goff, S.P. Conferring RNA polymerase activity to a DNA polymerase: A single residue in reverse transcriptase controls substrate selection. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Cases-González, C.E.; Gutiérrez-Rivas, M.; Menéndez-Arias, L. Coupling ribose selection to fidelity of DNA synthesis: The role of Tyr-115 of human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem. 2000, 275, 19759–19767. [Google Scholar] [CrossRef] [PubMed]
- Martín-Hernández, A.M.; Domingo, E.; Menéndez-Arias, L. Human immunodeficiency virus type 1 reverse transcriptase: role of Tyr115 in deoxynucleotide binding and misinsertion fidelity of DNA synthesis. EMBO J. 1996, 15, 4434–4442. [Google Scholar] [PubMed]
- Boyer, P.L.; Sarafianos, S.G.; Arnold, E.; Hughes, S.H. Analysis of mutations at positions 115 and 116 in the dNTP binding site of HIV-1 reverse transcriptase. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 3056–3061. [Google Scholar] [CrossRef] [PubMed]
- Martín-Hernández, A.M.; Gutiérrez-Rivas, M.; Domingo, E.; Menéndez-Arias, L. Mispair extension fidelity of human immunodeficiency virus type 1 reverse transcriptases with amino acid substitutions affecting Tyr115. Nucleic Acids Res. 1997, 25, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.L.; Hughes, S.H. Effects of amino acid substitutions at position 115 on the fidelity of human immunodeficiency virus type 1 reverse transcriptase. J. Virol. 2000, 74, 6494–6500. [Google Scholar] [CrossRef] [PubMed]
- Halvas, E.K.; Svarovskaia, E.S.; Pathak, V.K. Role of murine leukemia virus reverse transcriptase deoxyribonucleoside triphosphate-binding site in retroviral replication and in vivo fidelity. J. Virol. 2000, 74, 10349–10358. [Google Scholar] [CrossRef] [PubMed]
- Olivares, I.; Sánchez-Merino, V.; Martínez, M.A.; Domingo, E.; López-Galíndez, C.; Menéndez-Arias, L. Second-site reversion of a human immunodeficiency virus type 1 reverse transcriptase mutant that restores enzyme function and replication capacity. J. Virol. 1999, 73, 6293–6298. [Google Scholar] [PubMed]
- Gutiérrez-Rivas, M.; Ibáñez, A.; Martínez, M.A.; Domingo, E.; Menéndez-Arias, L. Mutational analysis of Phe-160 within the ‘palm’ subdomain of human immunodeficiency virus type 1 reverse transcriptase. J. Mol. Biol. 1999, 290, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Cases-González, C.E.; Menéndez-Arias, L. Nucleotide specificity of HIV-1 reverse transcriptases with amino acid substitutions affecting Ala-114. Biochem. J. 2005, 387, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, N.; Talele, T.T.; Pandey, P.K.; Harris, D.; Yadav, P.N.S.; Pandey, V.N. Role of glutamine 151 of human immunodeficiency virus type-1 reverse transcriptase in substrate selection as assessed by site-directed mutagenesis. Biochemistry 2000, 39, 2912–2920. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, M.A.; Drosopoulos, W.C.; Salomon, H.; Hsu, M.; Borkow, G.; Parniak, M.A.; Gu, Z.; Song, Q.; Manne, J.; Islam, S.; Castriota, G.; Prasad, V.R. Enhanced fidelity of 3TC-selected mutant HIV-1 reverse transcriptase. Science 1996, 271, 1282–1285. [Google Scholar] [PubMed]
- Hsu, M.; Inouye, P.; Rezende, L.; Richard, N.; Li, Z.; Prasad, V.R.; Wainberg, M.A. Higher fidelity of RNA-dependent DNA mispair extension by M184V drug-resistant than wild-type reverse transcriptase of human immunodeficiency virus type 1. Nucleic Acids Res. 1997, 25, 4532–4536. [Google Scholar] [CrossRef] [PubMed]
- Oude Essink, B.B.; Back, N.K.T.; Berkhout, B. Increased polymerase fidelity of the 3TC-resistant variants of HIV-1 reverse transcriptase. Nucleic Acids Res. 1997, 25, 3212–3217. [Google Scholar] [CrossRef] [PubMed]
- Hamburgh, M.E.; Drosopoulos, W.C.; Prasad, V.R. The influence of 3TC-resistance mutations E89G and M184V in the human immunodeficiency virus reverse transcriptase on mispair extension efficiency. Nucleic Acids Res. 1998, 26, 4389–4394. [Google Scholar] [CrossRef] [PubMed]
- Sarafianos, S.G.; Pandey, V.N.; Kaushik, N.; Modak, M.J. Site-directed mutagenesis of arginine 72 of HIV-1 reverse transcriptase – Catalytic role and inhibitor sensitivity. J. Biol. Chem. 1995, 270, 19729–19735. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, K.; Kaushik, N.; Pandey, V.N.; Modak, M.J. Elucidation of the role of Arg 110 of murine leukemia virus reverse transcriptase in the catalytic mechanism: Biochemical characterization of its mutant enzymes. Biochemistry 1996, 35, 16610–16620. [Google Scholar] [CrossRef] [PubMed]
- Agopian, A.; Depollier, J.; Lionne, C.; Divita, G. p66 Trp24 and Phe61 are essential for accurate association of HIV-1 reverse transcriptase with primer/template. J. Mol. Biol. 2007, 373, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Fisher, T.S.; Darden, T.; Prasad, V.R. Substitutions at Phe61 in the β3-β4 hairpin of HIV-1 reverse transcriptase reveal a role for the fingers subdomain in strand displacement DNA synthesis. J. Mol. Biol. 2003, 325, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Mandal, D.; Dash, C.; Le Grice, S.F.J.; Prasad, V.R. Analysis of HIV-1 replication block due to substitutions at F61 residue of reverse transcriptase reveals additional defects involving the RNase H function. Nucleic Acids Res. 2006, 34, 2853–2863. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, M.; Palaniappan, C.; Fu, Z.; Le Grice, S.F.J.; Fay, P.; Bambara, R.A. Mutations in the primer grip region of HIV reverse transcriptase can increase replication fidelity. J. Biol. Chem. 1999, 274, 28175–28184. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Rivas, M.; Menéndez-Arias, L. A mutation in the primer grip region of HIV-1 reverse transcriptase that confers reduced fidelity of DNA synthesis. Nucleic Acids Res. 2001, 29, 4963–4972. [Google Scholar] [CrossRef] [PubMed]
- Cases-González, C.E.; Menéndez-Arias, L. Increased GA transition frequencies displayed by primer grip mutants of human immunodeficiency virus type 1 reverse transcriptase. J. Virol. 2004, 78, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Das, K.; Hsiou, Y.; Sarafianos, S.G.; Clark Jr., A.D.; Jacobo-Molina, A.; Tantillo, C.; Hughes, S.H.; Arnold, E. Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 Ã resolution. J. Mol. Biol. 1998, 284, 1095–1111. [Google Scholar] [CrossRef] [PubMed]
- Sarafianos, S.G.; Das, K.; Tantillo, C.; Clark Jr., A.D.; Ding, J.; Whitcomb, J.M.; Boyer, P.L.; Hughes, S.H.; Arnold, E. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J. 2001, 20, 1449–1461. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, M.J.; Julias, J.G.; Sarafianos, S.G.; Alvord, W.G.; Arnold, E.; Hughes, S.H. Combining mutations in HIV-1 reverse transcriptase with mutations in the HIV-1 polypurine tract affects RNase H cleavages involved in PPT utilization. Virology 2006, 348, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Mbisa, J.L.; Nikolenko, G.N.; Pathak, V.K. Mutations in the RNase H primer grip domain of murine leukemia virus reverse transcriptase decrease efficiency and accuracy of plus-strand DNA transfer. J. Virol. 2005, 79, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, H.; Witvrouw, M.; De Clercq, E.; Anné, J. Lamivudine resistance of HIV type 1 does not delay development of resistance to nonnucleoside HIV type 1-specific reverse transcriptase inhibitors as compared with wild-type HIV type 1. AIDS Res. Hum. Retroviruses 1998, 14, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Keulen, W.; van Wijk, A.; Schuurman, R.; Berkhout, B.; Boucher, C.A.B. Increased polymerase fidelity of lamivudine-resistant HIV-1 variants does not limit their evolutionary potential. AIDS 1999, 13, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Diallo, K.; Brenner, B.; Oliveira, M.; Moisi, D.; Detorio, M.; Götte, M.; Wainberg, M.A. The M184V substitution in human immunodeficiency virus type 1 reverse transcriptase delays the development of resistance to amprenavir and efavirenz in subtype B and C clinical isolates. Antimicrob. Agents Chemother. 2003, 47, 2376–2379. [Google Scholar] [CrossRef] [PubMed]
- Preston, B.D. Reverse transcriptase fidelity and HIV-1 variation. Science 1997, 275, 228–229. [Google Scholar] [PubMed]
- Loeb, L.A.; Essigmann, J.M.; Kazazi, F.; Zhang, J.; Rose, K.D.; Mullins, J.I. Lethal mutagenesis of HIV with mutagenic nucleoside analogs. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Mullins, J.I. Lethal mutagenesis of HIV by mutagenic ribonucleoside analogs. AIDS Res. Hum. Retroviruses 2000, 16, 1–3. [Google Scholar] [PubMed]
- Domingo, E.; Escarmís, C.; Lázaro, E.; Manrubia, S.C. Quasispecies dynamics and RNA virus extinction. Virus Res. 2005, 107, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Pathak, V.K.; Temin, H.M. 5-Azacytidine and RNA secondary structure increase the retrovirus mutation rate. J. Virol. 1992, 66, 3093–3100. [Google Scholar] [PubMed]
- Crotty, S.; Cameron, C.E.; Andino, R. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 6895–6900. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.S.; Brabant, W.; Styrchak, S.; Gall, A.; Daifuku, R. KP-1212/1461, a nucleoside designed for the treatment of HIV by viral mutagenesis. Antiviral Res. 2005, 67, 1–9. [Google Scholar] [CrossRef]
- Graci, J.D.; Harki, D.A.; Korneeva, V.S.; Edathil, J.P.; Too, K.; Franco, D.; Smidansky, E.D.; Paul, A.V.; Peterson, B.R.; Brown, D.M.; Loakes, D.; Cameron, C.E. Lethal mutagenesis of poliovirus mediated by a mutagenic pyrimidine analogue. J. Virol. 2007, 81, 11256–11266. [Google Scholar] [CrossRef] [PubMed]
- LaCasse, R.A.; Remington, K.M.; North, T.W. The mutation frequency of feline immunodeficiency virus enhanced by 3'-azido-3'-deoxythymidine. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1996, 12, 26–32. [Google Scholar] [PubMed]
- Julias, J.G.; Kim, T.; Arnold, G.; Pathak, V.K. The antiretrovirus drug 3'-azido-3'-deoxythymidine increases the retrovirus mutation rate. J. Virol. 1997, 71, 4254–4263. [Google Scholar] [PubMed]
- Mansky, L.M.; Bernard, L.C. 3’-azido-3’-deoxythymidine (AZT) and AZT-resistant reverse transcriptase can increase the in vivo mutation rate of human immunodeficiency virus type 1. J. Virol. 2000, 74, 9532–9539. [Google Scholar] [CrossRef] [PubMed]
- Dapp, M.J.; Clouser, C.L.; Patterson, S.; Mansky, L.M. 5-Azacytidine can induce lethal muta-genesis in human immunodeficiency virus type 1. J. Virol. 2009, 83, 11950–11958. [Google Scholar] [CrossRef] [PubMed]
- Julias, J.G.; Pathak, V.K. Deoxyribonucleoside triphosphate pool imbalances in vivo are associated with an increased retroviral mutation rate. J. Virol. 1998, 72, 7941–7949. [Google Scholar] [PubMed]
- Balzarini, J.; Camarasa, M.-J.; Pérez-Pérez, M.-J.; San-Félix, A.; Velázquez, S.; Perno, C.-F.; De Clercq, E.; Anderson, J.N.; Karlsson, A. Exploitation of the low fidelity of human immunodeficiency virus type 1 (HIV-1) reverse transcriptase and the nucleotide composition bias in the HIV-1 genome to alter the drug resistance development of HIV. J. Virol. 2001, 75, 5772–5777. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Menéndez-Arias, L. Mutation Rates and Intrinsic Fidelity of Retroviral Reverse Transcriptases. Viruses 2009, 1, 1137-1165. https://doi.org/10.3390/v1031137
Menéndez-Arias L. Mutation Rates and Intrinsic Fidelity of Retroviral Reverse Transcriptases. Viruses. 2009; 1(3):1137-1165. https://doi.org/10.3390/v1031137
Chicago/Turabian StyleMenéndez-Arias, Luis. 2009. "Mutation Rates and Intrinsic Fidelity of Retroviral Reverse Transcriptases" Viruses 1, no. 3: 1137-1165. https://doi.org/10.3390/v1031137
APA StyleMenéndez-Arias, L. (2009). Mutation Rates and Intrinsic Fidelity of Retroviral Reverse Transcriptases. Viruses, 1(3), 1137-1165. https://doi.org/10.3390/v1031137