
Comprehensive Genomic Analysis of GRAS Transcription Factors Reveals Salt-Responsive Expression Profiles in Pecan (Carya illinoinensis)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Identification of GRAS Family Genes in Pecan
2.2. Phylogenetic and Structural Characterization of CiGRAS Genes
2.3. Promoter Cis-Element Analysis
2.4. Chromosomal Distribution and Duplication Analysis
2.5. Calculating Values of Ka, Ks and Ka/Ks
2.6. Expression Pattern Analysis in Tissues and Stress Conditions
2.7. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.8. Quantitative Real-Time PCR (qRT-PCR) Validation
3. Results
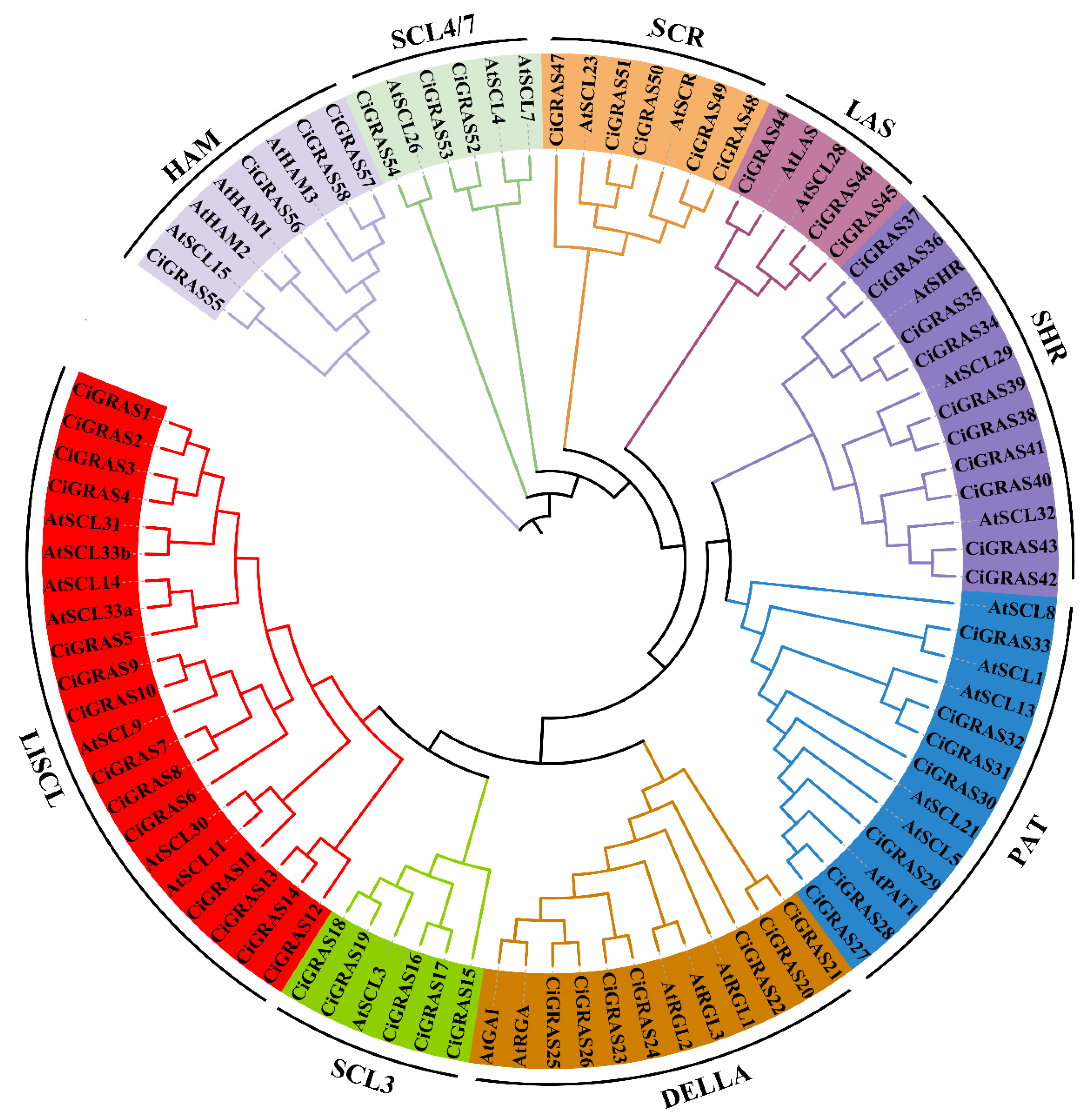
3.1. Identification and Phylogenetic Analysis of Carya illinoinensis GRAS Genes
3.2. Structural and Conserved Motif Analysis of CiGRAS Genes
3.3. Chromosomal Distribution of CiGRAS Genes
3.4. Promoter Cis-Element Analysis of CiGRAS Genes
3.5. Gene Duplication Events and Evolutionary Analysis of CiGRAS Genes
3.6. Tissue-Specific Expression Patterns of CiGRAS Genes
3.7. Expression Profiles of CiGRAS Genes Under Abiotic and Biotic Stresses
3.8. Co-Expression Network Analysis Reveals GRAS Gene-Enriched Functional Modules
3.9. qRT-PCR Validation of Selected Salt-Responsive CiGRAS Genes Under NaCl Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviation | Full Term |
| ABA | Abscisic Acid |
| bp | Base Pairs |
| CDS | Coding Sequence |
| CiGRAS | Carya illinoinensis GRAS Gene |
| DEG | Differentially Expressed Gene |
| DSD | Dispersed Duplication |
| GA | Gibberellin |
| GO | Gene Ontology |
| HMM | Hidden Markov Model |
| Ka/Ks | Nonsynonymous/Synonymous Substitution Ratio |
| MEME | Multiple EM for Motif Elicitation |
| NaCl | Sodium Chloride |
| NCBI | National Center for Biotechnology Information |
| PD | Proximal Duplication |
| qRT-PCR | Quantitative Real-Time Polymerase Chain Reaction |
| RPKM | Reads Per Kilobase per Million Mapped Reads |
| ROS | Reactive Oxygen Species |
| TF | Transcription Factor |
| TD | Tandem Duplication |
| TPM | Transcripts Per Million |
| TRD | Transposed Duplication |
| UTR | Untranslated Region |
| WGCNA | Weighted Gene Co-expression Network Analysis |
| WGD | Whole-Genome Duplication |
References
- Atanasov, A.G.; Sabharanjak, S.M.; Zengin, G.; Mollica, A.; Szostak, A.; Simirgiotis, M.; Huminiecki, Ł.; Horbanczuk, O.K.; Nabavi, S.M.; Mocan, A. Pecan nuts: A review of reported bioactivities and health effects. Trends Food Sci. Technol. 2018, 71, 246–257. [Google Scholar] [CrossRef]
- McWilliams, J. The Pecan: A History of America’s Native Nut; University of Texas Press: Austin, TX, USA, 2013. [Google Scholar]
- Hand, L.C.; Foshee III, W.G.; Monday, T.A.; Sibley, J.L. Long-term weed control for landscape pecan (Carya illinoinensis) trees. J. Environ. Hortic. 2018, 36, 82–84. [Google Scholar] [CrossRef]
- Wang, X.; Stein, L.; Black, M.; Kubenka, K.; Randall, J.; Ding, C. Phenotypic diversity and population structure of pecan (Carya illinoinensis) collections reveals geographic patterns. Sci. Rep. 2024, 14, 18592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Peng, F.; Li, Y. Pecan production in China. Sci. Hortic. 2015, 197, 719–727. [Google Scholar] [CrossRef]
- Jia, X.; Luo, H.; Xu, M.; Wang, G.; Xuan, J.; Guo, Z. Investigation of nut qualities of pecan cultivars grown in China. J. Plant Sci. 2019, 7, 117–124. [Google Scholar]
- Huang, C.; Wang, Z.; Ren, X.; Ma, X.; Zhou, M.; Ge, X.; Liu, H.; Fu, S. Evaluation of soil quality in a composite pecan orchard agroforestry system based on the smallest data set. Sustainability 2022, 14, 10665. [Google Scholar] [CrossRef]
- Wang, L.; Sun, X.; Li, S.; Zhang, T.; Zhang, W.; Zhai, P. Application of organic amendments to a coastal saline soil in North China: Effects on soil physical and chemical properties and tree growth. PLoS ONE 2014, 9, e89185. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.F.; Du, L.J.; Wang, X.F.; Han, L.W.; Xiong, Y.W.; Yin, Y.L. Effect of mixed salt stress on growth of common greening species in coastal area of Jiangsu Province and evaluation of their salt tolerance. Sci. Agric. Sin. 2015, 48, 2385–2395. [Google Scholar]
- Zhang, J.; Jiao, Y.; Sharma, A.; Shen, D.; Wei, B.; Hong, C.; Zheng, B.; Pan, C. Transcriptomic analysis reveals potential pathways associated with salt resistance in pecan (Carya illinoensis K. Koch). J. Biotechnol. 2021, 330, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Xu, Y.; Guan, S.L.; Zhang, J.; Jia, Z.; Hu, L.; Zhai, M.; Mo, Z.; Xuan, J. Comprehensive genomic analysis of CiPawPYL-PP2C-SnRK family genes in pecan (Carya illinoinensis) and functional characterization of CiPawSnRK2.1 under salt stress responses. Int. J. Biol. Macromol. 2024, 279, 135366. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, V.; Kakkar, M.; Kumari, P.; Zinta, G.; Gahlaut, V.; Kumar, S. Multifaceted roles of GRAS transcription factors in growth and stress responses in plants. iScience 2022, 25, 105026. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, S.; Oldroyd, G.E. GRAS-domain transcription factors that regulate plant development. Plant Signal. Behav. 2009, 4, 698–700. [Google Scholar] [CrossRef] [PubMed]
- Bolle, C. The role of GRAS proteins in plant signal transduction and development. Planta 2004, 218, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Khan, Y.; Xiong, Z.; Zhang, H.; Liu, S.; Yaseen, T.; Hui, T. Expression and roles of GRAS gene family in plant growth, signal transduction, biotic and abiotic stress resistance and symbiosis formation—A review. Plant Biol. 2022, 24, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Wan, P.; Sun, S.; Li, J.; Chen, M. Genome-wide analysis of the GRAS gene family in rice and Arabidopsis. Plant Mol. Biol. 2004, 54, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hu, X.; Hu, L.; Hou, X.; Xu, Z.; Yang, F.; Yuan, M.; Chen, F.; Wang, Y.; Tu, B. Wide Grain 3, a GRAS protein, interacts with DLT to regulate grain size and brassinosteroid signaling in rice. Rice 2022, 15, 55. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Agassin, R.H.; Ji, K. Transcriptome-wide identification of the GRAS transcription factor family in Pinus massoniana and its role in regulating development and stress response. Int. J. Mol. Sci. 2023, 24, 10690. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.; Li, M.; Chen, N.; Ren, G.; Qu, L.; Yue, H.; Wu, X.; Zhao, J. Genome-wide analysis and expression of the GRAS transcription factor family in Avena sativa. Genes 2023, 14, 164. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ding, X.; Gao, Y.; Yang, S. Genome-wide identification and characterization of GRAS genes in soybean (Glycine max). BMC Plant Biol. 2020, 20, 415. [Google Scholar] [CrossRef] [PubMed]
- Cenci, A.; Rouard, M. Evolutionary analyses of GRAS transcription factors in angiosperms. Front. Plant Sci. 2017, 8, 273. [Google Scholar] [CrossRef] [PubMed]
- Vera-Sirera, F.; Gomez, M.D.; Perez-Amador, M.A. DELLA proteins, a group of GRAS transcription regulators that mediate gibberellin signaling. In Plant Transcription Factors; Elsevier: Amsterdam, The Netherlands, 2016; pp. 313–328. [Google Scholar]
- Waseem, M.; Nkurikiyimfura, O.; Niyitanga, S.; Jakada, B.H.; Shaheen, I.; Aslam, M.M. GRAS transcription factors: Emerging regulators in plant growth, development, and multiple stresses. Mol. Biol. Rep. 2022, 49, 9673–9685. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Chen, S.; Li, T.; Ma, X.; Liang, X.; Ding, X.; Liu, H.; Luo, L. OsGRAS23, a rice GRAS transcription factor gene, is involved in drought stress response through regulating expression of stress-responsive genes. BMC Plant Biol. 2015, 15, 141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, X.; Wang, X.; Sun, M.; Song, R.; Mao, P.; Jia, S. Genome-wide identification of GRAS gene family and their responses to abiotic stress in Medicago sativa. Int. J. Mol. Sci. 2021, 22, 7729. [Google Scholar] [CrossRef] [PubMed]
- Fode, B.; Siemsen, T.; Thurow, C.; Weigel, R.; Gatz, C. The Arabidopsis GRAS protein SCL14 interacts with class II TGA transcription factors and is essential for the activation of stress-inducible promoters. Plant Cell 2008, 20, 3122–3135. [Google Scholar] [CrossRef] [PubMed]
- Lovell, J.T.; Bentley, N.B.; Bhattarai, G.; Jenkins, J.W.; Sreedasyam, A.; Alarcon, Y.; Bock, C.; Boston, L.B.; Carlson, J.; Cervantes, K.; et al. Four chromosome-scale genomes and a pan-genome annotation to accelerate pecan tree breeding. Nat. Commun. 2021, 12, 4125. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-H.; Jin, H.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Li, Q.; Yin, H.; Qi, K.; Li, L.; Wang, R.; Zhang, S.; Paterson, A.H. Gene duplication and evolution in recurring polyploidization–diploidization cycles in plants. Genome Biol. 2019, 20, 38. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Fan, P.; Liu, H.; Tan, P.; Ma, W.; Mo, Z.; Zhao, J.; Chu, G.; Peng, F. Insight into the CBL and CIPK gene families in pecan (Carya illinoinensis): Identification, evolution and expression patterns in drought response. BMC Plant Biol. 2022, 22, 221. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, K.; Zhang, C.; Han, X.; Zhang, X.; Ren, H.; Yao, X. Transcriptome analysis of resistant and susceptible pecan (Carya illinoinensis) reveals the mechanism of resistance to black spot disease (Colletotrichum fioriniae). J. Agric. Food Chem. 2023, 71, 5812–5822. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Hu, L.; Zhang, J.; Zhai, M.; Jia, Z.; Mo, Z.; Xuan, J. Comprehensive genomic analysis of the CDPK gene family in pecan (Carya illinoinensis) and their potential roles in salt stress response. Plants 2025, 14, 540. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Widmer, A. Genome-wide comparative analysis of the GRAS gene family in Populus, Arabidopsis and rice. Plant Mol. Biol. Rep. 2014, 32, 1129–1145. [Google Scholar] [CrossRef]
- Qiao, X.; Yin, H.; Li, L.; Wang, R.; Wu, J.; Wu, J.; Zhang, S. Different modes of gene duplication show divergent evolutionary patterns and contribute differently to the expansion of gene families involved in important fruit traits in pear (Pyrus bretschneideri). Front. Plant Sci. 2018, 9, 161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Mi, L.; Xu, L.; Yu, C.; Li, C.; Chen, C. Genome-wide identification, characterization, interaction network and expression profile of GRAS gene family in sweet orange (Citrus sinensis). Sci. Rep. 2019, 9, 2156. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Xian, Z.; Kang, X.; Tang, N.; Li, Z. Genome-wide identification, phylogeny and expression analysis of GRAS gene family in tomato. BMC Plant Biol. 2015, 15, 209. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, L.; Liu, Y.; Shen, X.; Guo, Y.; Ma, X.; Zhang, X.; Li, X.; Cheng, T.; Wen, H. RNA-Seq-based WGCNA and association analysis reveal the key regulatory module and genes responding to salt stress in wheat roots. Plants 2024, 13, 274. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, M.; Chen, J.; Qing, C.; He, S.; Zou, C.; Yuan, G.; Yang, C.; Peng, H.; Pan, G. GWAS and WGCNA uncover hub genes controlling salt tolerance in maize (Zea mays L.) seedlings. Theor. Appl. Genet. 2021, 134, 3305–3318. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Jiang, X.; Zhang, Y.; Zhai, M.; Hu, L.; Xuan, J. Weighted gene co-expression network analysis reveals key pathways and hub genes associated with successful grafting in pecan (Carya illinoinensis). Forests 2023, 14, 835. [Google Scholar] [CrossRef]
- Hou, M.; Li, Y.; Xuan, J.; Zhang, Y.; Wang, T.; Zhai, M.; Wang, G.; Hu, L.; Mo, Z. Weighted gene co-expression network analysis uncovers core drought responsive genes in pecan (Carya illinoinensis). Plants 2025, 14, 833. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, M.; Chen, Y.; Wang, G. Comprehensive Genomic Analysis of GRAS Transcription Factors Reveals Salt-Responsive Expression Profiles in Pecan (Carya illinoinensis). Forests 2025, 16, 1199. https://doi.org/10.3390/f16071199
Xu M, Chen Y, Wang G. Comprehensive Genomic Analysis of GRAS Transcription Factors Reveals Salt-Responsive Expression Profiles in Pecan (Carya illinoinensis). Forests. 2025; 16(7):1199. https://doi.org/10.3390/f16071199
Chicago/Turabian StyleXu, Ming, Yu Chen, and Guoming Wang. 2025. "Comprehensive Genomic Analysis of GRAS Transcription Factors Reveals Salt-Responsive Expression Profiles in Pecan (Carya illinoinensis)" Forests 16, no. 7: 1199. https://doi.org/10.3390/f16071199
APA StyleXu, M., Chen, Y., & Wang, G. (2025). Comprehensive Genomic Analysis of GRAS Transcription Factors Reveals Salt-Responsive Expression Profiles in Pecan (Carya illinoinensis). Forests, 16(7), 1199. https://doi.org/10.3390/f16071199