
Genetic Analyses of a Mixed Oak Stand at the Xeric Limit of Forest Climate and Its General Consequences for In Situ Conservation Management

 , , and
, , and 
Abstract
1. Introduction
- -
- What is the proportion of the drought-tolerant Q. virgiliana and/or putative hybrids of other taxa of the section Quercus based on morphological traits vs. genetic markers?
- -
- What is the proportion of trees within the population that are of vegetative (previous parent generation) and seed (progeny generation) origin, and what is the proportion of ‘identical genotypes’ (clones)?
- -
- Are there any differences in the genetic structure (gene pool?) between the previous generation (coppiced trees) and the offspring generation, e.g., allele number, heterozygosity values, possible loss of alleles, or the emergence of new alleles? Are there any genetic differences due to foreign origin (planted trees)?
- -
- Is the spatial distribution of trees, taxa, or putatively specific genotypes influenced by any microhabitat factors (topography, exposure, soil hydrology)?
- -
- Finally, is the gene pool of this population suitable for effective and long-term in situ gene conservation, and what aspects and factors should be considered? What silvicultural methods should or can be applied, particularly in a landscape dominated by arable lands and meadows?
2. Materials and Methods
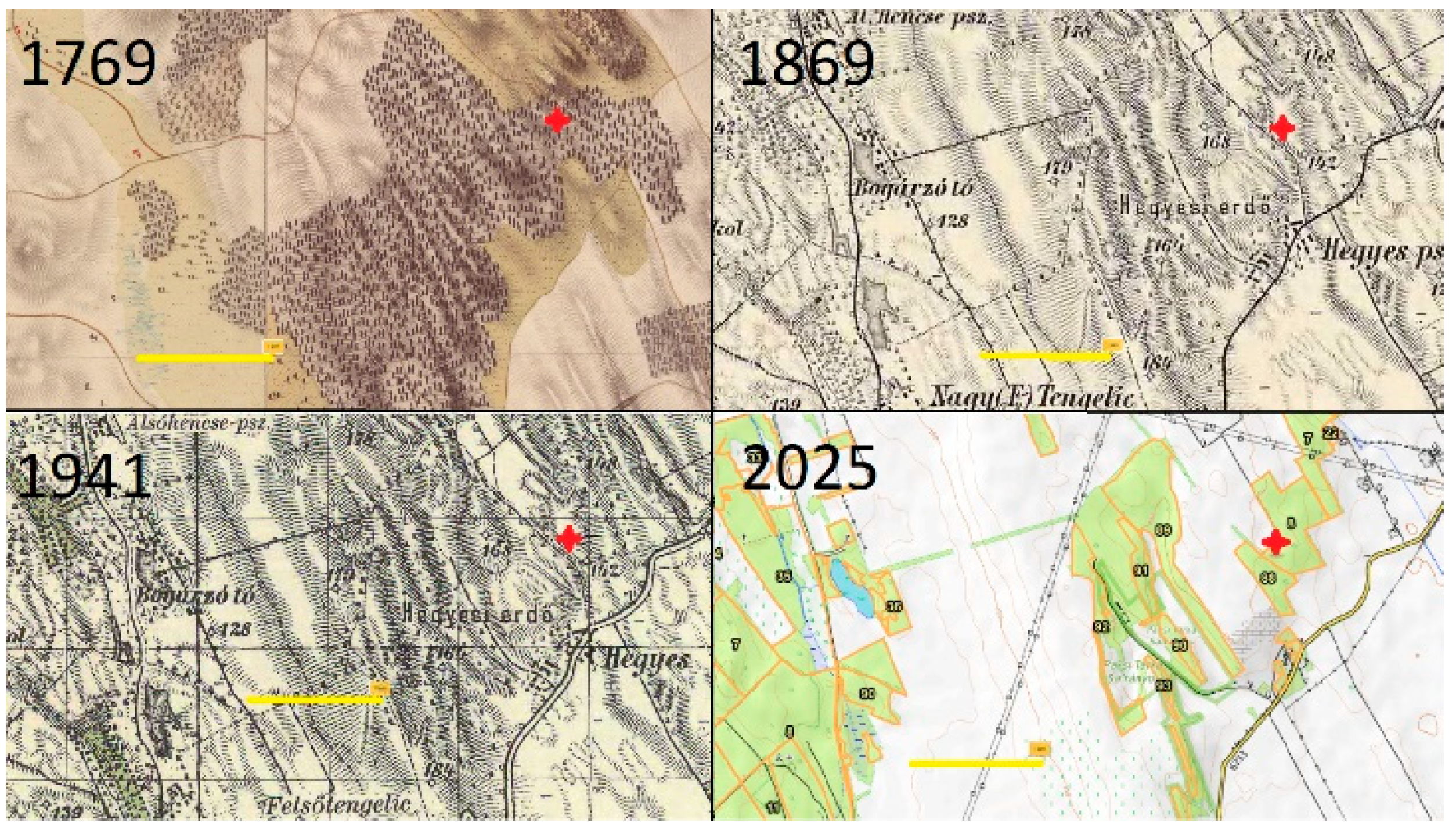
- First Military Land Survey of the Hungarian Kingdom, 1782–1785 [https://maps.arcanum.com/en/map/firstsurvey-hungary/?layers=147&bbox=2075541.607849373%2C6000935.425817063%2C2221001.2726760632%2C6054059.1604752615 (accessed on 29 March 2025)]
- Second Military Land Survey of the Hungarian Kingdom, 1819–1869 [https://maps.arcanum.com/hu/map/secondsurvey-hungary/?layers=5&bbox=2112256.0623183693%2C6020245.231900737%2C2130457.629678777%2C6027076.791303724 (accessed on 29 March 2025)]
- Military Land Survey of Hungary, 1941 [https://maps.arcanum.com/hu/map/hungary1941/?layers=29&bbox=2114166.9880255754%2C6020471.733854797%2C2132368.555385983%2C6027303.293257784 (accessed on 29 March 2025)]
- Forestry Map of Hungary, 2025 [https://erdoterkep.nebih.gov.hu/ (accessed on 28 March 2025)].
2.1. Sampling of Plant Material, Analyses of Morphological Traits
2.2. Statistical Evaluation of Taxonomic Descriptors
2.3. Microsatellite Analyses
2.4. Population Genetic Analyses
2.5. Test of Autochtonity
3. Results
3.1. Phenotype Classification Based on Morphological Traits
3.2. Genetic Analyses
3.2.1. Microsatellite Analyses
3.2.2. Analyses of Individual Genotypes
3.2.3. Genetic Structure of Coppiced vs. Seed-Originated Cohorts
3.3. Spatial Structure
3.4. Verification of Autochthony
4. Discussion
5. Conclusions
- The non-autochthonous (planted) trees should be removed whenever the next thinning is carried out to preserve the adapted local gene pool as much as possible.
- Preference should be given to trees with drought tolerance phenotypic traits of Q. virgiliana (e.g., hairiness, bark type, and oil-striped acorn) in silvicultural interventions (thinnings, natural regeneration).
- The seed vs. vegetative origin should not be a thinning consideration for trees. Traditional practice favors trees of seed origin over coppiced trees. In addition to the criteria described in the first two points, selection should be based on each tree’s general thinning aspects or health condition.
- The slight allelic decline in seed-originated trees (offspring generation) underlined the importance that the in situ gene conservation stands should not be maintained as an even-aged stand. Natural regeneration techniques typically carried out over a short period (3–10 years) and based on seedlings of a single crop year increase the potential for allelic loss, which may be exacerbated by the isolated and fragmented pattern of the studied gene conservation stand, which may also reduce the potential for external pollen flow.
- If natural regeneration by seed is not possible ₋ in whole or in part ₋, the use of local reproductive material must be preferred in the case of both reforestation and replacement (assisted regeneration, assisted gene flow). However, the first two priority points should also be considered.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Czúcz, B.; Gálhidy, L.; Mátyás, C. Present and forecasted xeric climatic limits of beech and sessile oak distribution at low altitudes in Central Europe. Ann. For. Sci. 2011, 68, 99–108. [Google Scholar] [CrossRef]
- Mátyás, C.; Sun, G. Forests in a water limited world under climate change. Environ. Res. Lett. 2014, 9, 085001. [Google Scholar] [CrossRef]
- Mátyás, C.; Berki, I.; Bidló, A.; Csóka, G.; Czimber, K.; Führer, E.; Gálos, B.; Gribovszki, Z.; Illés, G.; Hirka, A.; et al. Sustainability of Forest Cover under Climate Change on the Temperate-Continental Xeric Limits. Forests 2018, 9, 489. [Google Scholar] [CrossRef]
- Molnár, Z.; Biró, M.; Bartha, S.; Fekete, G. Past Trends, Present State and Future Prospects of Hungarian Forest-Steppes. In Eurasian Steppes. Ecological Problems and Livelihoods in a Changing World. Plant and Vegetation; Werger, M., van Staalduinen, M., Eds.; Springer: Dordrecht, The Netherlands, 2012; Volume 6. [Google Scholar] [CrossRef]
- Fodor, E.; Hâruta, O.; Dorog, S. Nurse plants and the regeneration niche of tree seedlings in wood-pastures from Western and North-Western Romania. Reforesta 2018, 41–59. [Google Scholar] [CrossRef]
- Lőrincz, Á.; Hábenczyus, A.A.; Kelemen, A.; Ratkai, B.; Tölgyesi, C.; Lőrinczi, G.; Frei, K.; Bátori, Z.; Maák, I.E. Wood-pastures promote environmental and ecological heterogeneity on a small spatial scale. Sci. Total Environ. 2024, 906, 167510. [Google Scholar] [CrossRef] [PubMed]
- Bordács, S.; Pintér, B.; Horváth, C.; Benedek, L.; Ladányi, M. Relic Vergilius Oak (Quercus virgiliana Ten.) Trees Could Preserve Microhabitats of Pannonian Forest–Steppe Vegetation. Diversity 2024, 16, 401. [Google Scholar] [CrossRef]
- Mátyás, C.; Vendramin, G.G.; Fady, B. Forests at the limit: Evolutionary–genetic consequencesof environmental changes at the receding (xeric) edge of distribution. Report from a research workshop. Ann. For. Sci. 2009, 66, 800. [Google Scholar] [CrossRef]
- Petit, R.J.; Bodénès, C.; Ducousso, A.; Roussel, G.; Kremer, A. Hybridization as a mechanism of invasion in oaks. New Phytol. 2003, 161, 151–164. [Google Scholar] [CrossRef]
- Petit, R.J.; Brewer, S.; Bordács, S.; Burg, K.; Cheddadi, R.; Coart, E.; Cottrell, J.; Csaikl, U.M.; Van Dam, B.; Deans, J.D.; et al. Identification of refugia and post-glacial colonisation routes of European white oaks based on chloroplast DNA and fossil pollen evidence. For. Ecol. Manag. 2002, 156, 49–74. [Google Scholar] [CrossRef]
- Petit, R.J.; Csaikl, U.M.; Bordács, S.; Burg, K.; Coart, E.; Cottrell, J.; van Dam, B.; Deans, J.D.; Dumolin-Lapègue, S.; Fineschi, S.; et al. Chloroplast DNA variation in European white oaks. Phylogeography and patterns of diversity based on data from over 2600 populations. For. Ecol. Manag. 2002, 156, 5–26. [Google Scholar] [CrossRef]
- Provan, J.; Bennett, K.D. Phylogeographic insights into cryptic glacial refugia. Trends Ecol. Evol. 2008, 23, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Bordács, S.; Popescu, F.; Slade, D.; Csaikl, U.M.; Lesur, I.; Borovics, A.; Kézdy, P.; König, A.O.; Gömöry, D.; Brewer, S.; et al. Chloroplast DNA variation of white oaks in northern Balkans and in the Carpathian Basin. For. Ecol. Manag. 2002, 156, 197–209. [Google Scholar] [CrossRef]
- Magyari, E.; Jakab, G.; Rudner, E.; Sümegi, P. Palynological and plant macrofossil data on Late Pleistocene short-term climatic oscillations in NE-Hungary. Acta Palaeobot. 1999, 2 (Suppl. 2), 252–264. [Google Scholar]
- Magyari, E.; Chapman, J.; Passmore, D.; Allen, J.; Huntley, J.; Huntley, B. Holocene persistence of wooded steppe in the Great Hungarian Plain. J. Biogeogr. 2010, 37, 915–935. [Google Scholar] [CrossRef]
- Denk, T.; Grimm, G.W.; Manos, P.S.; Deng, M.; Hipp, A.L. An Updated Infrageneric Classification of the Oaks: Review of Previous Taxonomic Schemes and Synthesis of Evolutionary Patterns. In Oaks Physiological Ecology. Exploring the Functional Diversity of Genus Quercus L. Tree Physiology; Gil-Pelegrín, E., Peguero-Pina, J., Sancho-Knapik, D., Eds.; Springer: Cham, Switzerland, 2017; Volume 7, pp. 13–38. [Google Scholar] [CrossRef]
- Bartha, D. Quercus virgiliana Ten. In (Hrsg.): Enzyklopädie der Holzgewächse. Handbuch und Atlas der Dendrologie; Schutt, P., Weisgerber, H., Schuck, H.J., Lang, U.M., Roloff, A., Eds.; Ecomed Verlagsgesellschaft: Landsberg, Germany, 2001; Band III/2/25; pp. 1–5. [Google Scholar]
- Bartha, D. Quercus dalechampii Ten. In (Hrsg.): Enzyklopädie der Holzgewächse. Handbuch und Atlas der Dendrologie; Schutt, P., Weisgerber, H., Schuck, H.J., Lang, U.M., Roloff, A., Eds.; Ecomed Verlagsgesellschaft: Landsberg, Germany, 2003; Band III/2/3; pp. 1–4. [Google Scholar]
- Matula, R. Comparison of general tree characteristics of less known oak species Quercus dalechampii Ten. and Quercus polycarpa Schur. J. For. Sci. 2008, 54, 333–339. [Google Scholar] [CrossRef]
- Di Pietro, R.; Conte, A.L.; Di Marzio, P.; Fortini, P.; Farris, E.; Gianguzzi, L.; Müller, M.; Rosati, L.; Spampinato, G.; Gailing, O. Does the genetic diversity among pubescent white oaks in southern Italy, Sicily and Sardinia islands support the current taxonomic classification? Eur. J. For. Res. 2021, 140, 355–371. [Google Scholar] [CrossRef]
- Kučera, P. New name for Central Europaean oak formerly labelled as Quercus dalechampii. Biologia 2018, 73, 313–317. [Google Scholar] [CrossRef]
- Kučera, P. Quercus banatus grows in Slovenia. THAISZIA J. Bot. 2019, 29, 61–69. [Google Scholar] [CrossRef]
- Bartha, D. A magyar tölgy (Quercus conferta Kit.) hibridjei. A nothotaxonok kritikai értékelése. Bot. Közlemények 2024, 111, 107–130. (In Hungarian) [Google Scholar] [CrossRef]
- Coldea, G.; Farcas, S.; Filipas, L.; Ursu, T.M.; Stoica, I.A. Syntaxonomic revision of Quercus virgiliana Ten. and Quercus pedunculiflora K. Koch forests from Romania. Studia UBB. Biol. 2010, LV.2, 39–50. [Google Scholar]
- Enescu, C.M.; Curtu, A.L.; Șofletea, N. Is Quercus virgiliana a distinct morphological and genetic entity among European white oaks? Turk. J. Agric. For. 2013, 37, 632–641. [Google Scholar] [CrossRef]
- Trinajstić, I. About the Problem of Differentiation Between the Oaks Quercus pubescens Willd. and Quercus virgiliana. (Ten.) Šumarski List 2007, 131, 57–60. [Google Scholar]
- Simonkai, L. Hazánk Tölgyfajai és Tölgyerdei = Quercus et Querceta Hungariae; Franklin-Társulat Nyomdája: Budapest, Hungary, 1890. 42 oldal. (In Hungarian) [Google Scholar]
- Soó, R. A Magyar Flóra és Vegetáció Rendszertani-Növényföldrajzi Kézikönyve I–VI; Akadémiai Kiadó: Budapest, Hungary, 1964–1980. (In Hungarian) [Google Scholar]
- Zólyomi, B.; Horváth, A.; Kevey, B.; Lendvai, G. Steppe woodlands with Tatarian Maple (Aceri tatarici-Quercetum pubescentis-roboris) on the great Hungarian plain and its neighbourhood. An unfinished synthesis with supplementary notes. Acta Bot. Hung. 2013, 55, 167–189. [Google Scholar] [CrossRef]
- Reif, A.; Schneider, E.; Oprea, A.; Laszlo Rakosy, L.; Luick, R. Romania’s natural forest types—A biogeographic and phytosociological overview in the context of politics and conservation. Tuexenia 2022, 42, 9–34. [Google Scholar] [CrossRef]
- Gafenco, I.M.; Pleșca, B.I.; Șofletea, N. The taxonomic structure of sessile oak (Quercus petraea (Matt.) Liebl.) in marginal populations from Eastern Romania—A bibliographic study. Rev. De Silvic. Şi Cinegetică 2023, 28, 39–45. [Google Scholar]
- Curtu, A.L.; Gailing, O.; Finkeldey, R. Evidence for hybridization and introgression within a species-rich oak (Quercus spp.) community. BMC Evol. Biol. 2007, 7, 218. [Google Scholar] [CrossRef]
- Antonecchia, G.; Fortini, P.; Lepais, O.; Gerber, S.; Legér, P.; Scippa, G.S.; Viscosi, V. Genetic structure of a natural oak community in central Italy: Evidence of gene flow between three sympatric white oak species (Quercus, Fagaceae). Ann. For. Res. 2015, 58, 205–216. [Google Scholar] [CrossRef]
- Pütz, J.; Jansen, S.; Reutimann, O.; Rellstab, C.; Bordács, S.; Neophytou, C. The influence of post-glacial migration and hybridization on the gene pool of marginal Quercus pubescens populations in Central Europe. Ann. Bot. 2025, 135, 867–884. [Google Scholar] [CrossRef]
- Leroy, T.; Roux, C.; Villate, L.; Bodénès, C.; Romiguier, J.; Paiva, J.A.P.; Dossat, C.; Aury, J.; Plomion, C.; Kremer, A. Extensive recent secondary contacts between four European white oak species. New Phytol. 2017, 214, 865–878. [Google Scholar] [CrossRef]
- Leroy, T.; Louvet, J.; Lalanne, C.; Le Provost, G.; Labadie, K.; Aury, J.; Delzon, S.; Plomion, C.; Kremer, A. Adaptive introgression as a driver of local adaptation to climate in European white oaks. New Phytol. 2020, 226, 1171–1182. [Google Scholar] [CrossRef]
- Dow, B.D.; Ashley, M.V. Microsatellite analysis of seed dispersal and parentage of saplings in bur oak, Quercus macrocarpa. Mol. Ecol. 1996, 5, 615–627. [Google Scholar] [CrossRef]
- Dow, B.D.; Ashley, M. High levels of gene flow in bur oak revealed by paternity analysis using microsatellites. J. Hered. 1998, 89, 62–70. [Google Scholar] [CrossRef]
- Gerber, S.; Chadœuf, J.; Gugerli, F.; Lascoux, M.; Buiteveld, J.; Cottrell, J.; Dounavi, A.; Fineschi, S.; Forrest, L.L.; Fogelqvist, J.; et al. High Rates of Gene Flow by Pollen and Seed in Oak Populations across Europe. PLoS ONE 2014, 9, e85130. [Google Scholar] [CrossRef]
- Craft, K.J.; Ashley, M.V. Pollen-mediated gene flow in isolated and continuous stand of bur oak, Quercus macrocarpa (Fagaceae). Am. J. Bot. 2010, 97, 1999–2006. [Google Scholar] [CrossRef] [PubMed]
- Cseke, K.; Bordács, S.; és Borovics, A. Egy elegyes tölgyes taxonómiai és genetikai szerkezetének elemzése. Erdészettudományi Közlemények 2011, 1, 95–105. (In Hungarian) [Google Scholar]
- Liesebach, H.; Eusemann, P.; Höltken, A.M.; Tröber, U.; Kuchma, O.; Karopka, M.; Becker, F.; Kätzel, R.; Fussi, B. Effective population size of adult and offspring cohorts as a genetic monitoring tool in two stand-forming and wind-pollinated tree species: Fagus sylvatica L. and Picea abies (L.) Karst. Conserv. Genet. 2024, 25, 739–753. [Google Scholar] [CrossRef]
- Leroy, T.; Rougemont, Q.; Dupouey, J.; Bodénès, C.; Lalanne, C.; Belser, C.; Labadie, K.; Le Provost, G.; Aury, J.; Kremer, A.; et al. Massive postglacial gene flow between European white oaks uncovered genes underlying species barriers. New Phytol. 2020, 226, 1183–1197. [Google Scholar] [CrossRef]
- Fady, B.; Aravanopoulos, F.A.; Alizoti, P.; Mátyás, C.; von Wühlisch, G.; Westergren, M.; Belletti, P.; Cvjetkovic, B.; Ducci, F.; Huber, G.; et al. Evolution-based approach needed for the conservation and silviculture of peripheral forest tree populations. For. Ecol. Manag. 2016, 375, 66–75. [Google Scholar] [CrossRef]
- Bakker, E.G.; Van Dam, B.C.; Van Eck, H.J.; Jacobsen, E. The Description of Clones of Quercus robur L. and Q. petraea (Matt.) Liebl. with Microsatellites and AFLP in an Ancient Woodland. Plant Biol. 2001, 3, 616–621. [Google Scholar] [CrossRef]
- Bordács, S.; Zhelev, P.; Schirone, B. EUFORGEN Technical Guidelines for genetic conservation and use of pubescent oak (Quercus pubescens). In European Forest Genetic Resources Programme (Euforgen); European Forest Institute: Joensuu, Finland, 2019; 6p, ISBN 978-952-5980-63-9. [Google Scholar]
- Bordács, S.; Gál, L. Erdészeti génmegőrzés a klímaváltozás tükrében. Erdészeti Lapok 2023, 158, 461–463. (In Hungarian) [Google Scholar]
- Koloszár, J. Site requirements and possibilities for planting of Slavonian oak in Hungary. Acta Fac. For. 1984, 1, 51–60. [Google Scholar]
- Soó, R.; Kárpáti, Z. Magyar Flóra, Harasztok—Virágos Növények; Tankönyvkiadó Budapest: Budapest, Hungary, 1968; pp. 649–652. (In Hungarian) [Google Scholar]
- Mátyás, V. A tölgyek dendrológiai ismertetése. In A tölgyek; Keresztesi, B., Ed.; Akadémiai Kiadó: Budapest, Hungary, 1967; pp. 51–90. (In Hungarian) [Google Scholar]
- Schwarz, O. Monographie der Eichen Europas und des Mittelmeergebietes. In Feddes Repertorium; Sonderbeiheft, D., Ed.; University and Research Library Catalog: Wageningen, The Netherlands, 1936. [Google Scholar]
- Borovics, A. A tölgyek hibridizációja, morfológiai és genetikai változatossága. Erdészeti Kut. 1998, 88, 89–108. (In Hungarian) [Google Scholar]
- Gencsi, I.; Vancsura, R. Dendrológia; Mezőgazda Kiadó: Budapest, Hungary, 1982; pp. 226–259. (In Hungarian) [Google Scholar]
- Kassambara, A.; Mundt, F. factoextra: Extract and Visualize the Results of Multivariate Data Analyses; R package Version 1.0.7. 2020. Available online: https://CRAN.R-project.org/package=factoextra (accessed on 20 May 2025).
- Galili, T. dendextend: An R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; Available online: https://ggplot2.tidyverse.org (accessed on 20 May 2025).
- Dumolin, S.; Demesure, B.; Petit, R.J. Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient PCR method. Theor. Appl. Genet. 1995, 91, 1253–1256. [Google Scholar] [CrossRef]
- Bruegmann, T.; Fladung, M.; Schroeder, H. Flexible DNA isolation procedure for different tree species as a convenient lab routine. Silvae Genet. 2022, 71, 20–30. [Google Scholar] [CrossRef]
- Steinkellner, H.; Fluch, S.; Turetschek, E.; Lexer, C.; Streiff, R.; Kremer, A.; Burg, K.; Glössl, J. Identification and characterization of (GA/CT)n- microsatellite loci from Quercus petraea. Plant Mol. Biol. 1997, 33, 1093–1096. [Google Scholar] [CrossRef] [PubMed]
- Kampfer, S.; Lexer, C.; Glössl, J.; Steinkellner, H. Characterization of (GA)n Microsatellite Loci from Quercus robur. Hereditas 1998, 129, 183–186. [Google Scholar] [CrossRef]
- Dow, B.D.; Ashley, M.V.; Howe, H.F. Characterization of highly variable (GA/CT) n microsatellites in the bur oak, Quercus macrocarpa. Theor. Appl. Genet. 1995, 91, 137–141. [Google Scholar] [CrossRef]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.O.D.; Smouse, P.E. genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Francis, R.M. pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2016, 17, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, F.; Carnevale, S.; Vendramin, G.G. A new set of mono- and dinucleotide chloroplast microsatellites in Fagaceae. Mol. Ecol. Notes 2004, 4, 259–261. [Google Scholar] [CrossRef]
- Weising, K.; Gardner, R.C. A set of conserved PCR primers for the analysis of simple sequence repeat polymorphisms in chloroplast genomes of dicotyledonous angiosperms. Genome 1999, 42, 9–19. [Google Scholar] [CrossRef]
- Deguilloux, M.; Dumolin-Lapègue, S.; Gielly, L.; Grivet, D.; Petit, R.J. A set of primers for the amplification of chloroplast microsatellites in Quercus. Mol. Ecol. Notes 2003, 3, 24–27. [Google Scholar] [CrossRef]
- Solymos, R. Improvement and silviculture of oaks in Hungary. Ann. For. Sci. 1993, 50, 607–614. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | N | Na | Ne | Np | I | Ho | He | uHe | F |
|---|---|---|---|---|---|---|---|---|---|
| DH | 7 | 7.563 | 5.765 | 0.313 | 1.823 | 0.768 | 0.800 | 0.862 | 0.047 |
| PETH | 13 | 10.000 | 6.491 | 0.625 | 1.980 | 0.750 | 0.807 | 0.839 | 0.088 |
| PETV | 5 | 5.438 | 4.254 | 0.188 | 1.497 | 0.700 | 0.728 | 0.808 | 0.064 |
| R | 4 | 5.188 | 4.272 | 0.500 | 1.493 | 0.781 | 0.729 | 0.833 | −0.079 |
| RPED | 14 | 10.313 | 6.636 | 1.688 | 1.956 | 0.763 | 0.787 | 0.816 | 0.032 |
| V | 8 | 7.875 | 5.742 | 0.375 | 1.820 | 0.680 | 0.789 | 0.841 | 0.160 |
| VH | 11 | 9.375 | 5.862 | 0.375 | 1.935 | 0.761 | 0.801 | 0.839 | 0.062 |
| VD | 10 | 8.563 | 6.032 | 0.250 | 1.910 | 0.813 | 0.813 | 0.856 | 0.007 |
| VDPET | 15 | 10.813 | 6.632 | 0.438 | 2.032 | 0.721 | 0.814 | 0.842 | 0.128 |
| VPET | 16 | 11.625 | 7.106 | 0.438 | 2.115 | 0.727 | 0.838 | 0.865 | 0.139 |
| VPETD | 14 | 10.688 | 6.652 | 0.625 | 2.032 | 0.781 | 0.817 | 0.848 | 0.040 |
| VR | 4 | 5.750 | 4.918 | 0.125 | 1.632 | 0.813 | 0.773 | 0.884 | −0.050 |
| Cluster | N | Na | Ne | Np | I | Ho | He | uHe | F |
|---|---|---|---|---|---|---|---|---|---|
| cluster 1 | 17 | 11.375 | 6.991 | 2.313 | 2.023 | 0.768 | 0.796 | 0.820 | 0.034 |
| cluster 2 | 9 | 6.438 | 4.160 | 0.438 | 1.507 | 0.736 | 0.695 | 0.736 | −0.064 |
| cluster 3 | 39 | 15.000 | 7.005 | 2.250 | 2.193 | 0.731 | 0.832 | 0.842 | 0.128 |
| cluster 4 | 8 | 4.438 | 3.239 | 0.313 | 1.236 | 0.758 | 0.650 | 0.693 | −0.157 |
| mixed | 48 | 16.938 | 8.009 | 2.438 | 2.297 | 0.766 | 0.844 | 0.853 | 0.098 |
| Cluster/Origin | N | Np | Ho | uHe | F | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Seed | Veg | Seed | Veg | Seed | Veg | Seed | Veg | Seed | Veg | |
| cluster 1 | 14 | 3 | 4.563 | 0.375 | 0.763 | 0.792 | 0.811 | 0.750 | 0.027 | −0.259 |
| cluster 2 | 2 | 7 | 0.375 | 0.313 | 0.688 | 0.750 | 0.750 | 0.727 | −0.191 | −0.110 |
| cluster 3 | 7 | 32 | 1.688 | 2.938 | 0.714 | 0.734 | 0.831 | 0.840 | 0.075 | 0.122 |
| cluster 4 | 2 | 6 | 0.438 | 0.375 | 0.719 | 0.771 | 0.667 | 0.715 | −0.431 | −0.164 |
| mixed | 7 | 41 | 2.313 | 3.250 | 0.795 | 0.761 | 0.864 | 0.849 | 0.010 | 0.099 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pintér, B.; Cseke, K.; Ladányi, M.; Lados, B.B.; Bordács, S. Genetic Analyses of a Mixed Oak Stand at the Xeric Limit of Forest Climate and Its General Consequences for In Situ Conservation Management. Forests 2025, 16, 939. https://doi.org/10.3390/f16060939
Pintér B, Cseke K, Ladányi M, Lados BB, Bordács S. Genetic Analyses of a Mixed Oak Stand at the Xeric Limit of Forest Climate and Its General Consequences for In Situ Conservation Management. Forests. 2025; 16(6):939. https://doi.org/10.3390/f16060939
Chicago/Turabian StylePintér, Beáta, Klára Cseke, Márta Ladányi, Botond Boldizsár Lados, and Sándor Bordács. 2025. "Genetic Analyses of a Mixed Oak Stand at the Xeric Limit of Forest Climate and Its General Consequences for In Situ Conservation Management" Forests 16, no. 6: 939. https://doi.org/10.3390/f16060939
APA StylePintér, B., Cseke, K., Ladányi, M., Lados, B. B., & Bordács, S. (2025). Genetic Analyses of a Mixed Oak Stand at the Xeric Limit of Forest Climate and Its General Consequences for In Situ Conservation Management. Forests, 16(6), 939. https://doi.org/10.3390/f16060939