Genetic Diversity of the Endangered Dalbergia odorifera Revealed by SSR Markers

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and DNA Extraction Materials and Methods
2.2. RNA Sequencing and Data Deposition
2.3. SSR Identification and Marker Development
2.4. Validation of SSR Marker by PCR and Capillary Electrophoresis
2.5. Statistical Analysis
3. Results
3.1. Distribution of SSR Loci in D. odorifera
3.2. Development of Polymorphic SSR Markers
3.3. Polymorphism of 19 SSR Loci
3.4. Genetic Diversity in D. odorifera
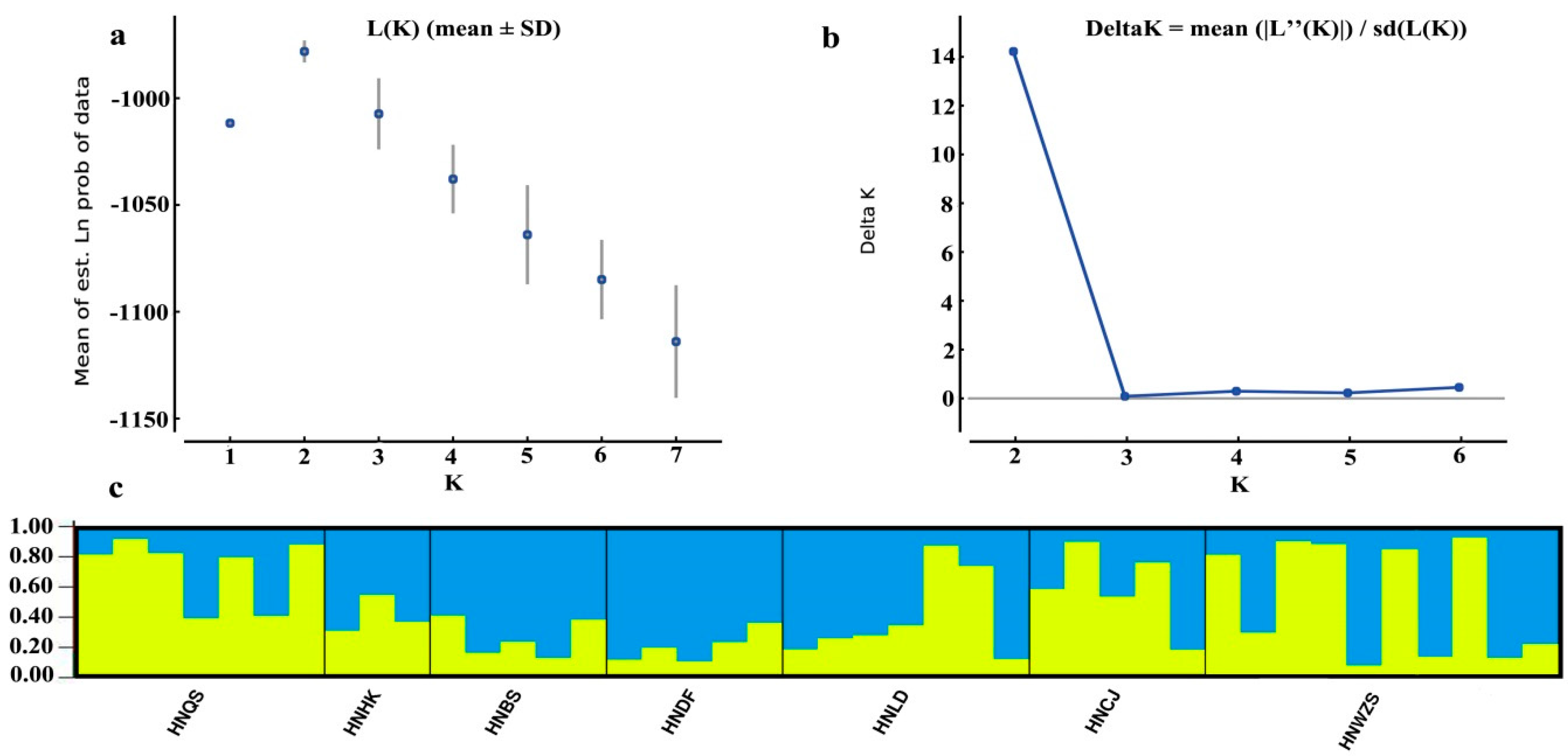
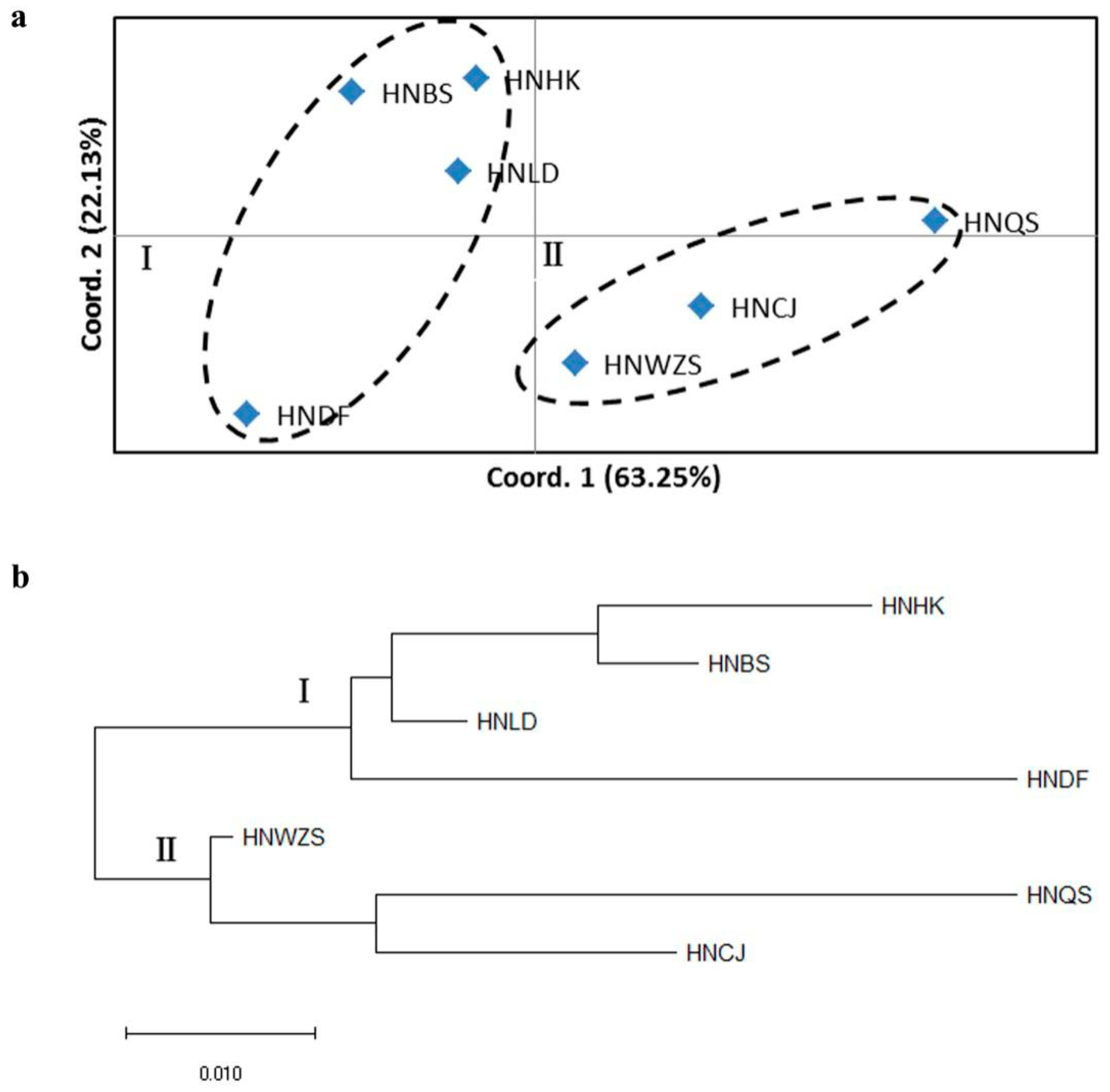
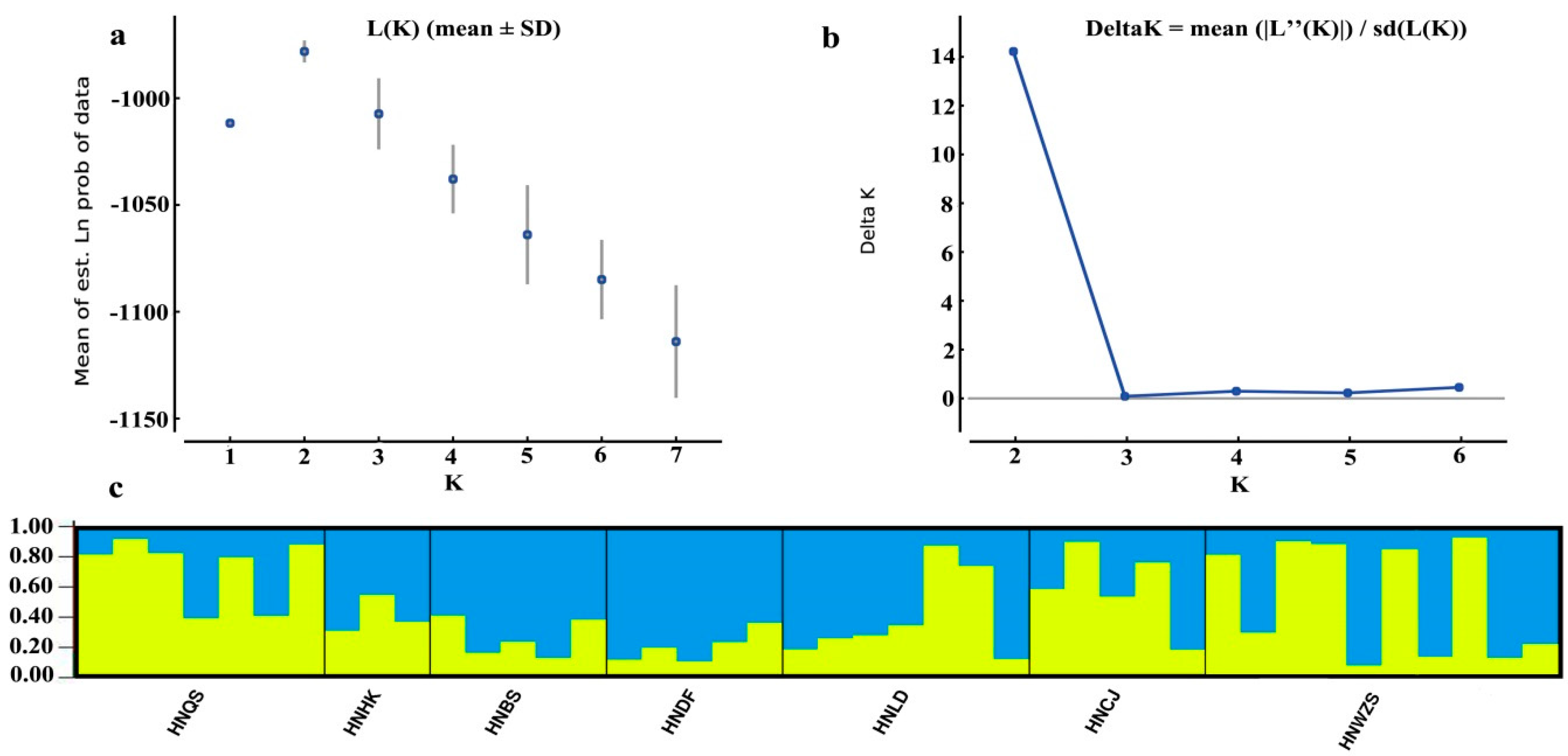
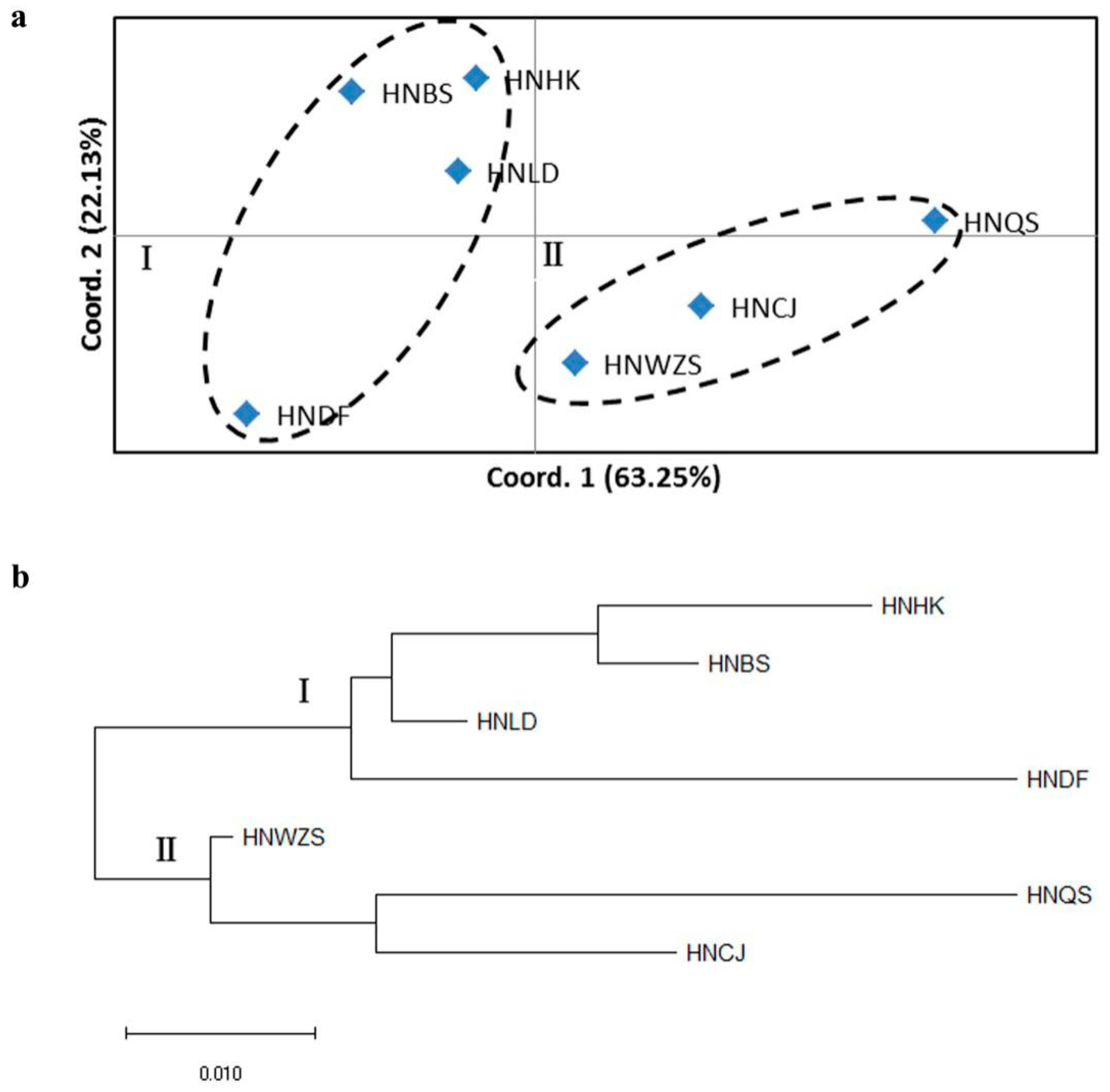
3.5. Population Structure of D. odorifera
4. Discussion
4.1. Development of SSR Marker for D. odorifera
4.2. Genetic Diversity of D. odorifera
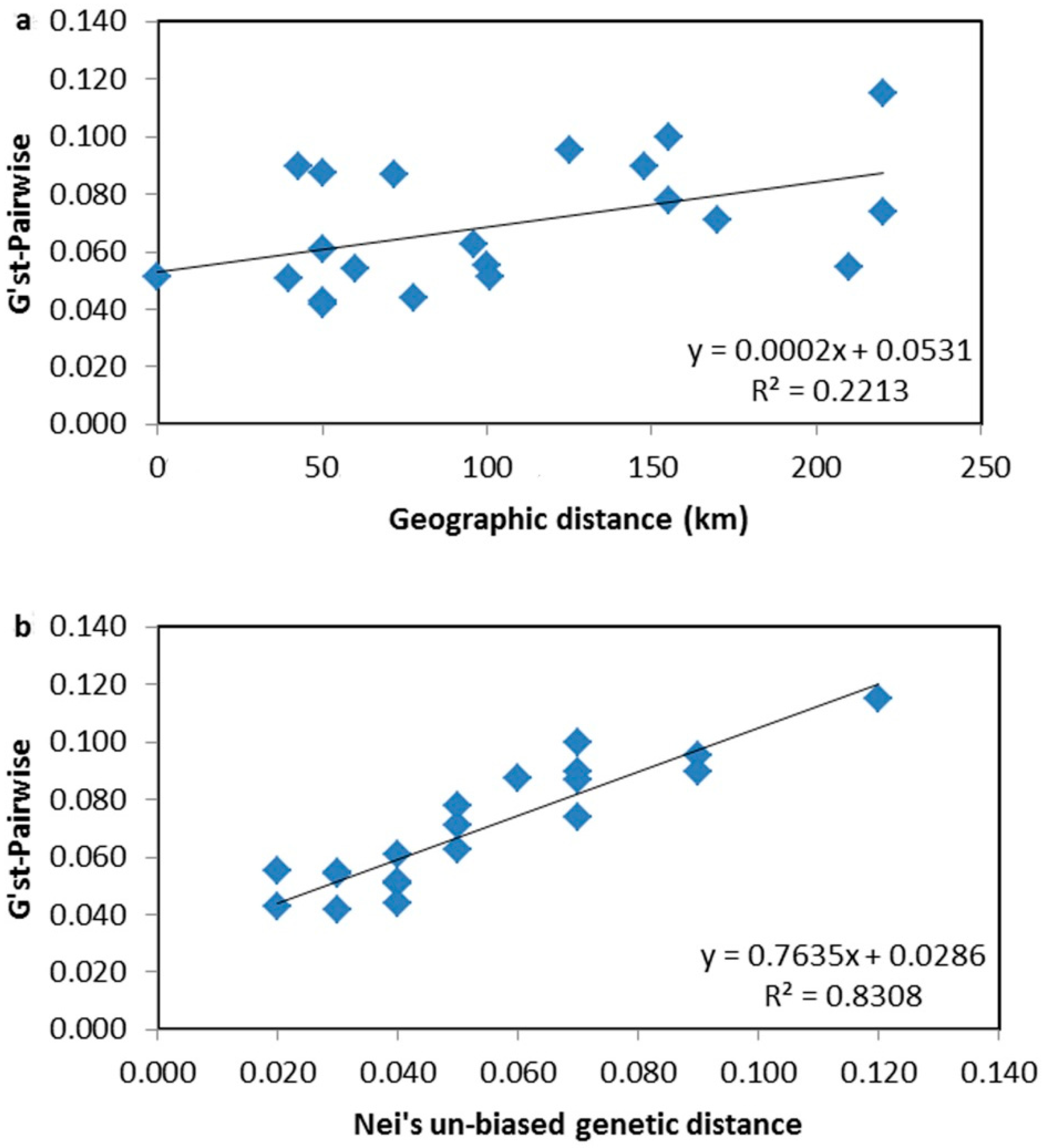
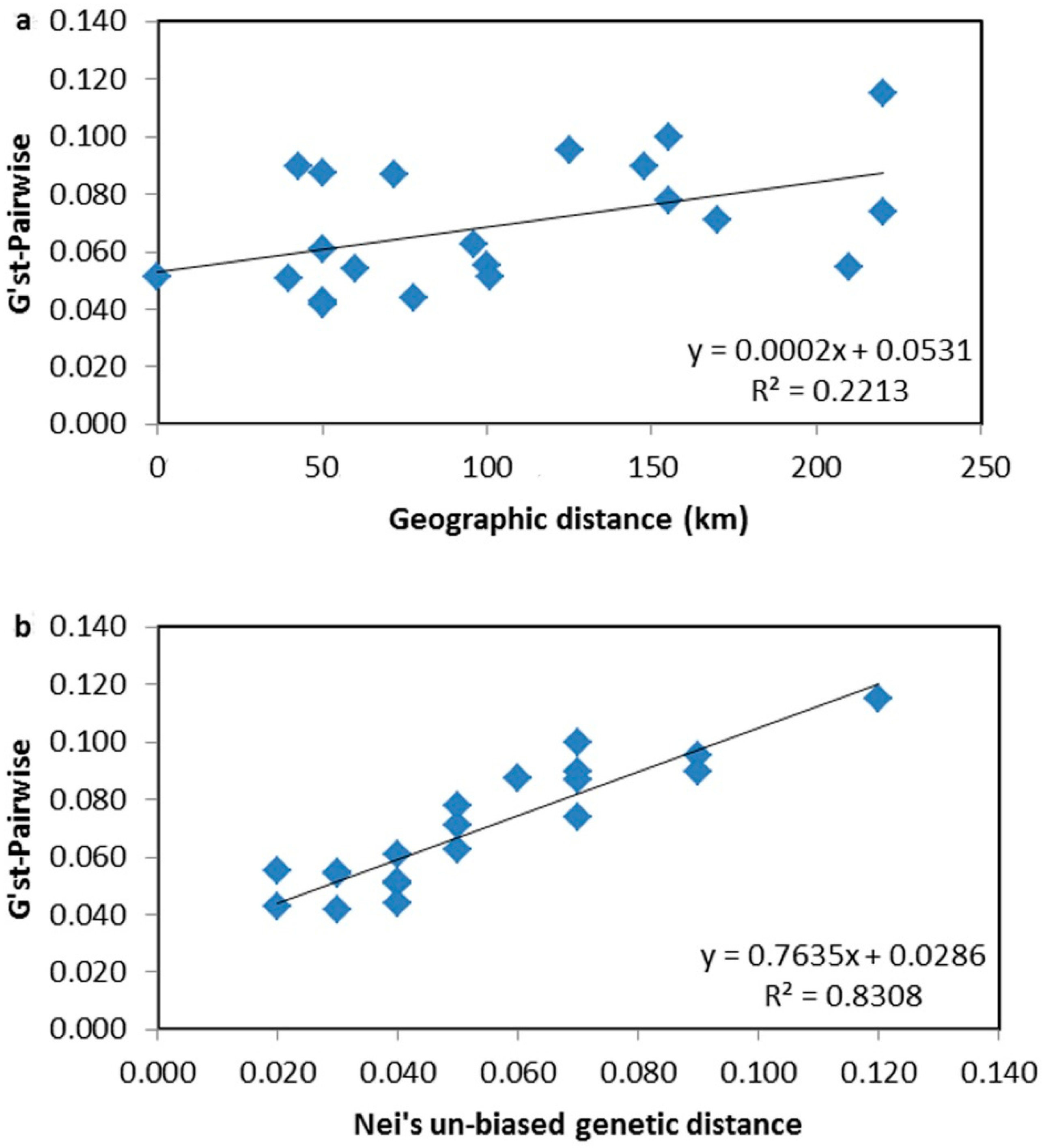
4.3. Genetic Differentiation and Population Structure
4.4. Conservation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, X.; Xu, D.; Yang, Z.; Zhang, N. Geographic variations in seed germination of Dalbergia odorifera T. Chen in response to temperature. Ind. Crop. Prod. 2017, 102, 45–50. [Google Scholar] [CrossRef]
- Lee, D.S.; Kim, K.S.; Ko, W.; Li, B.; Keo, S.; Jeong, G.S.; Oh, H.; Kim, Y.C. The neoflavonoid latifolin isolated from MeOH extract of Dalbergia odorifera attenuates inflammatory responses by inhibiting NF-kappaB activation via Nrf2-mediated heme oxygenase-1 expression. Phytother. Res. 2014, 28, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dong, W.H.; Zuo, W.J.; Wang, H.; Zhong, H.M.; Mei, W.L.; Dai, H.F. Three new phenolic compounds from Dalbergia odorifera. J. Asian Nat. Prod. Res. 2014, 16, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Chen, D.L.; Yang, Y.; Liu, Y.Y.; Wei, J.H. Sesquiterpenoids with cytotoxicity from heartwood of Dalbergia odorifera. J. Asian Nat. Prod. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Wang, Y. Bioactive sesquiterpenes isolated from the essential oil of Dalbergia odorifera T. Chen. Fitoterapia 2010, 81, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dong, W.H.; Zuo, W.J.; Liu, S.; Zhong, H.M.; Mei, W.L.; Dai, H.F. Five new sesquiterpenoids from Dalbergia odorifera. Fitoterapia 2014, 95, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.M.; Wang, D.Y.; Yang, J.M.; Lin, Z.X.; Lin, Y.X.; Yang, A.L.; Fan, H.; Cao, M.; Yuan, S.Y.; Liu, Z.J.; et al. Dalbergia odorifera extract promotes angiogenesis through upregulation of VEGFRs and PI3K/MAPK signaling pathways. J. Ethnopharmacol. 2017, 204, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.; Li, B.; Keo, S.; Kim, K.S.; Jeong, G.S.; Oh, H.; Kim, Y.C. Inhibitory effect of 9-hydroxy-6,7-dimethoxydalbergiquinol from Dalbergia odorifera on the NF-kappaB-related neuroinflammatory response in lipopolysaccharide-stimulated mouse BV2 microglial cells is mediated by heme oxygenase-1. Int. Immunopharmacol. 2013, 17, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Park, J.A.; Hwang, J.S.; Ham, S.A.; Yoo, T.; Lee, W.J.; Paek, K.S.; Shin, H.C.; Lee, C.H.; Seo, H.G. A Dalbergia odorifera extract improves the survival of endotoxemia model mice by inhibiting HMGB1 release. BMC Complement. Altern. Med. 2017, 17, 212. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Weng, X.C.; Cheng, D.L. Antioxidant activities of natural phenolic components from Dalbergia odorifera T. Chen. Food Chem. 2000, 71, 45–49. [Google Scholar] [CrossRef]
- Dalbergia Cochinchinensis. The IUCN Red List of Threatened Species. Available online: http://www.iucnredlist.org/details/32398/0 (accessed on 18 July 2018).
- Yang, Q.X.; Feng, J.D.; Wei, J.H.; Li, R.T.; He, M.J. Genetic diversity of China’s endangered medicinal plant Dalbergia odorifera. World Sci. Technol. Mod. Tradit. Chin. Med. Mater. Med. 2007, 9, 73–79. Available online: http://kns.cnki.net/KCMS/detail/detail.aspx?dbcode=CJFQ&dbname=CJFD2007&filename=SJKX200702016&v=MTgxNzJGeUhuVmJ2TE5pZkFkckc0SHRiTXJZOUVZb1I4ZVgxTHV4WVM3RGgxVDNxVHJXTTFGckNVUkxLZVplZHE= (accessed on 20 June 2018).
- Nybom, H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol. Ecol. 2004, 13, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Meng, H.; Wu, Y.; Chen, B.; Gan, B.C. Primer screening of SRAP molecular marker in Dalbergia odorifera. Acta Agric. Jiangxi. 2011, 23, 29–31. [Google Scholar]
- Fregene, M.A.; Suarez, M.; Mkumbira, J.; Kulembeka, H.; Ndedya, E.; Kulaya, A.; Mitchel, S.; Gullberg, U.; Rosling, H.; Dixon, A.G.O.; et al. Simple sequence repeat marker diversity in cassava landraces: Genetic diversity and differentiation in an asexually propagated crop. Theor. Appl. Genet. 2003, 107, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.; Morgante, M.; Andre, C.; Hanafey, M.; Vogel, J.; Tingey, S.; Rafalski, A. The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol. Breed. 1996, 2, 225–238. [Google Scholar] [CrossRef]
- Dai, F.; Tang, C.; Wang, Z.; Luo, G.; He, L.; Yao, L. De novo assembly, gene annotation, and marker development of mulberry (Morus atropurpurea) transcriptome. Tree. Genet. Genomes. 2015, 11, 26. [Google Scholar] [CrossRef]
- Huang, J.; Guo, X.; Hao, X.; Zhang, W.; Chen, S.; Huang, R.; Gresshoff, P.M.; Zheng, Y. De novo sequencing and characterization of seed transcriptome of the tree legume Millettia pinnata for gene discovery and SSR marker development. Mol. Breed. 2016, 36, 75. [Google Scholar] [CrossRef]
- Taheri, S.; Lee Abdullah, T.; Yusop, M.; Hanafi, M.; Sahebi, M.; Azizi, P.; Shamshiri, R. Mining and development of novel SSR markers using next generation sequencing (NGS) data in plants. Molecules 2018, 23, 399. Available online: https://www.mdpi.com/1420-3049/23/2/399 (accessed on 2 January 2019). [CrossRef] [PubMed]
- Dervishi, A.; Jakše, J.; Ismaili, H.; Javornik, B.; Štajner, N. Comparative assessment of genetic diversity in Albanian olive (Olea europaea L.) using SSRs from anonymous and transcribed genomic regions. Tree Genet. Genomes 2018, 14. [Google Scholar] [CrossRef]
- Dong, M.; Wang, Z.; He, Q.; Zhao, J.; Fan, Z.; Zhang, J. Development of EST-SSR markers in Larix principis-rupprechtii Mayr and evaluation of their polymorphism and cross-species amplification. Trees 2018. [Google Scholar] [CrossRef]
- Li, N.; Zheng, Y.; Ding, H.; Li, H.; Peng, H.; Jiang, B.; Li, H. Development and validation of SSR markers based on transcriptome sequencing of Casuarina equisetifolia. Trees-Struct. Funct. 2018, 32, 41–49. [Google Scholar] [CrossRef]
- Liu, F.-M.; Hong, Z.; Yang, Z.-J.; Zhang, N.-N.; Liu, X.-J.; Xu, D.-P. De novo transcriptome analysis of Dalbergia odorifera and transferability of SSR markers developed from the transcriptome. Forests 2019, 10, 98. Available online: https://www.mdpi.com/1999-4907/10/2/98 (accessed on 28 January 2019). [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3--new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. Micro-checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Popgene, Version 1.32; The User-Friendly Shareware for Population Genetic Analysis Molecular Biology and Biotechnology Center. University of Alberta Edmonton. Available online: http://www.ualberta.ca/~fyeh (accessed on 23 November 2017).
- Nagy, S.; Poczai, P.; Cernák, I.; Gorji, A.M.; Hegedűs, G.; Taller, J. PICcalc: An online program to calculate polymorphic information content for molecular genetic studies. Biochem. Genet. 2012, 50, 670–672. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2010, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Porras-Hurtado, L.; Ruiz, Y.; Santos, C.; Phillips, C.; Carracedo, A.; Lareu, M.V. An overview of STRUCTURE: Applications, parameter settings, and supporting software. Front. Genet. 2013, 4, 98. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; Vonholdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Jonathan, M.; Mattias, J.; Rosenberg, N.A.; Itay, M. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, M.; Hou, L.; Zhang, Z.; Pang, X.; Li, Y. De novo transcriptome assembly and population genetic analyses for an endangered Chinese endemic Acer miaotaiense (Aceraceae). Genes 2018, 9, 378. [Google Scholar] [CrossRef] [PubMed]
- Szmidt, A.E.; Wang, X.R.; Lu, M.Z. Empirical assessment of allozyme and RAPD variation in Pinus sylvestris (L.) using haploid tissue analysis. Heredity 1996, 76, 412–420. [Google Scholar] [CrossRef]
- Torales, S.L.; Rivarola, M.; Pomponio, M.F.; Gonzalez, S.; Acuña, C.V.; Fernández, P.; Lauenstein, D.L.; Verga, A.R.; Hopp, H.E.; Paniego, N.B.; et al. De novo assembly and characterization of leaf transcriptome for the development of functional molecular markers of the extremophile multipurpose tree species Prosopis alba. BMC. Genom. 2013, 14, 705. [Google Scholar] [CrossRef] [PubMed]
- Sathyanarayana, N.; Pittala, R.K.; Tripathi, P.K.; Chopra, R.; Singh, H.R.; Belamkar, V.; Bhardwaj, P.K.; Doyle, J.J.; Egan, A.N. Transcriptomic resources for the medicinal legume Mucuna pruriens: De novo transcriptome assembly, annotation, identification and validation of EST-SSR markers. BMC Genom. 2017, 18, 409. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, H.; Wu, A.; Hou, Y.; An, Y.; Wei, C. Construction of fingerprinting for tea plant (Camellia sinensis) accessions using new genomic SSR markers. Mol. Breed. 2017, 37, 93. [Google Scholar] [CrossRef]
- Chen, H.; Chen, X.; Tian, J.; Yang, Y.; Liu, Z.; Hao, X.; Wang, L.; Wang, S.; Liang, J.; Zhang, L.; et al. Development of gene-based SSR markers in rice bean (Vigna umbellata L.) based on transcriptome data. PLoS ONE 2016, 11, e0151040. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Wu, F.; Luo, K.; Zhao, Y.; Yan, Q.; Zhang, Y.; Wang, Y.; Zhang, J. Cross-species transferability of EST-SSR markers developed from the transcriptome of Melilotus and their application to population genetics research. Sci. Rep.-UK 2017, 7, 17959. [Google Scholar] [CrossRef] [PubMed]
- Vu Thi Thu, H. Genetic diversity among endangered rare Dalbergia cochinchinensis (Fabaceae) genotypes in Vietnam revealed by random amplified polymorphic DNA (RAPD) and inter simple sequence repeats (ISSR) markers. Afr. J. Biotechnol. 2012, 11. [Google Scholar] [CrossRef]
- Phong, D.T.; Hien, V.T.; Thanh, T.T.; Tang, D.V. Comparison of RAPD and ISSR markers for assessment of genetic diversity among endangered rare Dalbergia oliveri (Fabaceae) genotypes in Vietnam. Genet. Mol. Resh. 2011, 10, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; An, Y.; Li, F.; Li, S.; Liu, L.; Zhou, Q.; Zhao, S.; Wei, C. Genome-wide identification of simple sequence repeats and development of polymorphic SSR markers for genetic studies in tea plant (Camellia sinensis). Mol. Breed. 2018, 38, 59. [Google Scholar] [CrossRef]
- Väli, Ü.; Einarsson, A.; Waits, L.; Ellegren, H. To what extent do microsatellite markers reflect genome-wide genetic diversity in natural populations? Mol. Ecol. 2008, 17, 3808–3817. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.M.; Eguiarte, L.E.; Montana, C. Genetic structure and outcrossing rates in Flourensia cernua (Asteraceae) growing at different densities in the South-western Chihuahuan Desert. Ann. Bot. 2004, 94, 419–426. [Google Scholar] [CrossRef] [PubMed]
- White, T.L.; Adams, W.T.; Neale, D.B. Forest Genetics; CABI Publishing: Boston, MA, USA, 2007; pp. 149–186. [Google Scholar]
- Gadissa, F.; Tesfaye, K.; Dagne, K.; Geleta, M. Genetic diversity and population structure analyses of Plectranthus edulis (Vatke) Agnew collections from diverse agro-ecologies in Ethiopia using newly developed EST-SSRs marker system. BMC Genet. 2018, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Chung, M.G. Genetic variation and population structure in Korean endemic species: IV. Hemerocallis hakuunensis (Liliaceae). J. Plant. Res. 1997, 110, 209–217. [Google Scholar] [CrossRef]
- Zhong, T.; Zhao, G.; Lou, Y.; Lin, X.; Guo, X. Genetic diversity analysis of Sinojackia microcarpa, a rare tree species endemic in China, based on simple sequence repeat markers. J. For. Res. 2018. [Google Scholar] [CrossRef]
- Gichira, A.W.; Li, Z.-Z.; Saina, J.K.; Hu, G.-W.; Gituru, R.W.; Wang, Q.-F.; Chen, J.-M. Demographic history and population genetic structure of Hagenia abyssinica (Rosaceae), a tropical tree endemic to the Ethiopian highlands and eastern African mountains. Tree Genet. Genomes 2017, 13. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.J.W.; Sherman-Broyles, S.L. Factors influencing levels of genetic diversity in woody plant species. New For. 1992, 6, 95–124. [Google Scholar] [CrossRef]
- Mihretie, Z.; Schueler, S.; Konrad, H.; Bekele, E.; Geburek, T. Patterns of genetic diversity of Prunus africana in Ethiopia: Hot spot but not point of origin for range-wide diversity. Tree Genet. Genomes 2015, 11, 118. [Google Scholar] [CrossRef]
- Boaventura-Novaes, C.R.D.; Novaes, E.; Mota, E.E.S.; Telles, M.P.C.; Coelho, A.S.G.; Chaves, L.J. Genetic drift and uniform selection shape evolution of most traits in Eugenia dysenterica DC. (Myrtaceae). Tree Genet. Genomes 2018, 14, 76. [Google Scholar] [CrossRef]
- Addisalem, A.B.; Duminil, J.; Wouters, D.; Bongers, F.; Smulders, M.J.M. Fine-scale spatial genetic structure in the frankincense tree Boswellia papyrifera (Del.) Hochst. and implications for conservation. Tree Genet. Genomes 2016, 12, 86. [Google Scholar] [CrossRef]
- Hartvig, I.; So, T.; Changtragoon, S.; Tran, H.T.; Bouamanivong, S.; Theilade, I.; Kjær, E.D.; Nielsen, L.R. Population genetic structure of the endemic rosewoods Dalbergia cochinchinensis and D. oliveri at a regional scale reflects the Indochinese landscape and life-history traits. Ecol. Evol. 2018, 8, 530–545. [Google Scholar] [CrossRef] [PubMed]
- Zhai, S.H.; Yin, G.S.; Yang, X.H. Population genetics of the endangered and wild edible plant Ottelia acuminata in southwestern China using novel SSR markers. Biochem. Genet. 2018, 56, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Vu, D.-D.; Bui, T.T.-X.; Nguyen, M.-D.; Shah, S.N.M.; Vu, D.-G.; Zhang, Y.; Nguyen, M.-T.; Huang, X.-H. Genetic diversity and conservation of two threatened dipterocarps (Dipterocarpaceae) in southeast Vietnam. J. For. Res. 2018. [Google Scholar] [CrossRef]
- Mutegi, E.; Snow, A.A.; Rajkumar, M.; Pasquet, R.; Ponniah, H.; Daunay, M.-C.; Davidar, P. Genetic diversity and population structure of wild/weedy eggplant (Solanum insanum, Solanaceae) in southern India: Implications for conservation. Am. J. Bot. 2015, 102, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Institute of Botany, the Chinese Academy of Sciences. Flora Reipublicae Popularis Sinicae; Science Press: Beijing, China, 1994; Volume 40, p. 114. [Google Scholar]
- The, S.N. A review on the medicinal plant Dalbergia odorifera species: Phytochemistry and biological activity. Evid-Based. Complement. Altern. 2017. [Google Scholar] [CrossRef]
- Moritsuka, E.; Chhang, P.; Tagane, S.; Toyama, H.; Sokh, H.; Yahara, T.; Tachida, H. Genetic variation and population structure of a threatened timber tree Dalbergia cochinchinensis in Cambodia. Tree Genet. Genomes 2017, 13, 115. [Google Scholar] [CrossRef]
- Schaal, B.A.; Hayworth, D.A.; Olsen, K.M.; Rauscher, J.T.; Smith, W.A. Phylogeographic studies in plants: Problems and prospects. Mol. Ecol. 1998, 7, 465–474. [Google Scholar] [CrossRef]
- Slatkin, M. Gene flow and the geographic structure of natural populations. Science 1987, 236, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Forest, F.; Grenyer, R.; Rouget, M.; Davies, T.J.; Cowling, R.M.; Faith, D.P.; Balmford, A.; Manning, J.C.; Proches, S.; van der Bank, M.; et al. Preserving the evolutionary potential of floras in biodiversity hotspots. Nature 2007, 445, 757–760. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
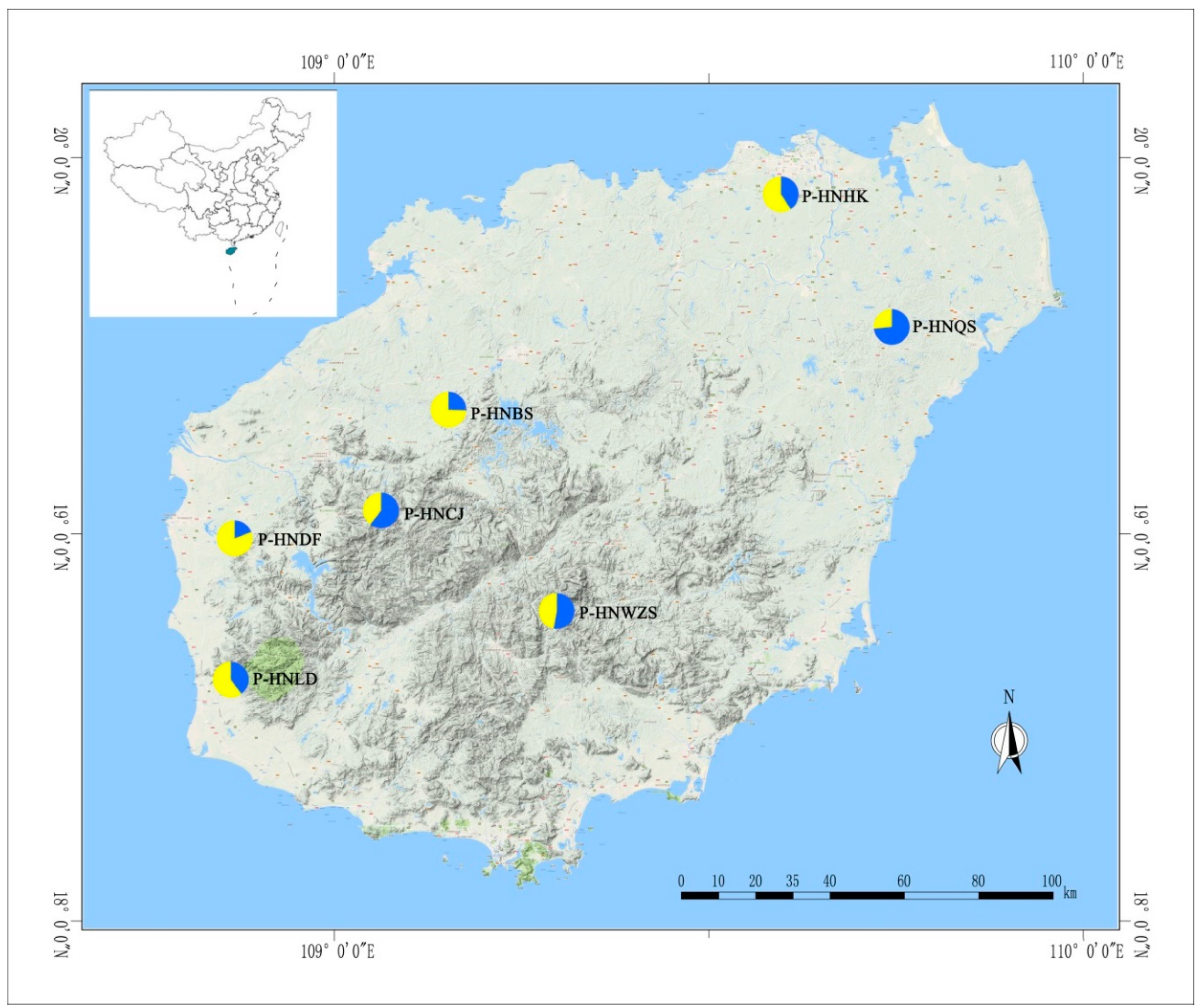
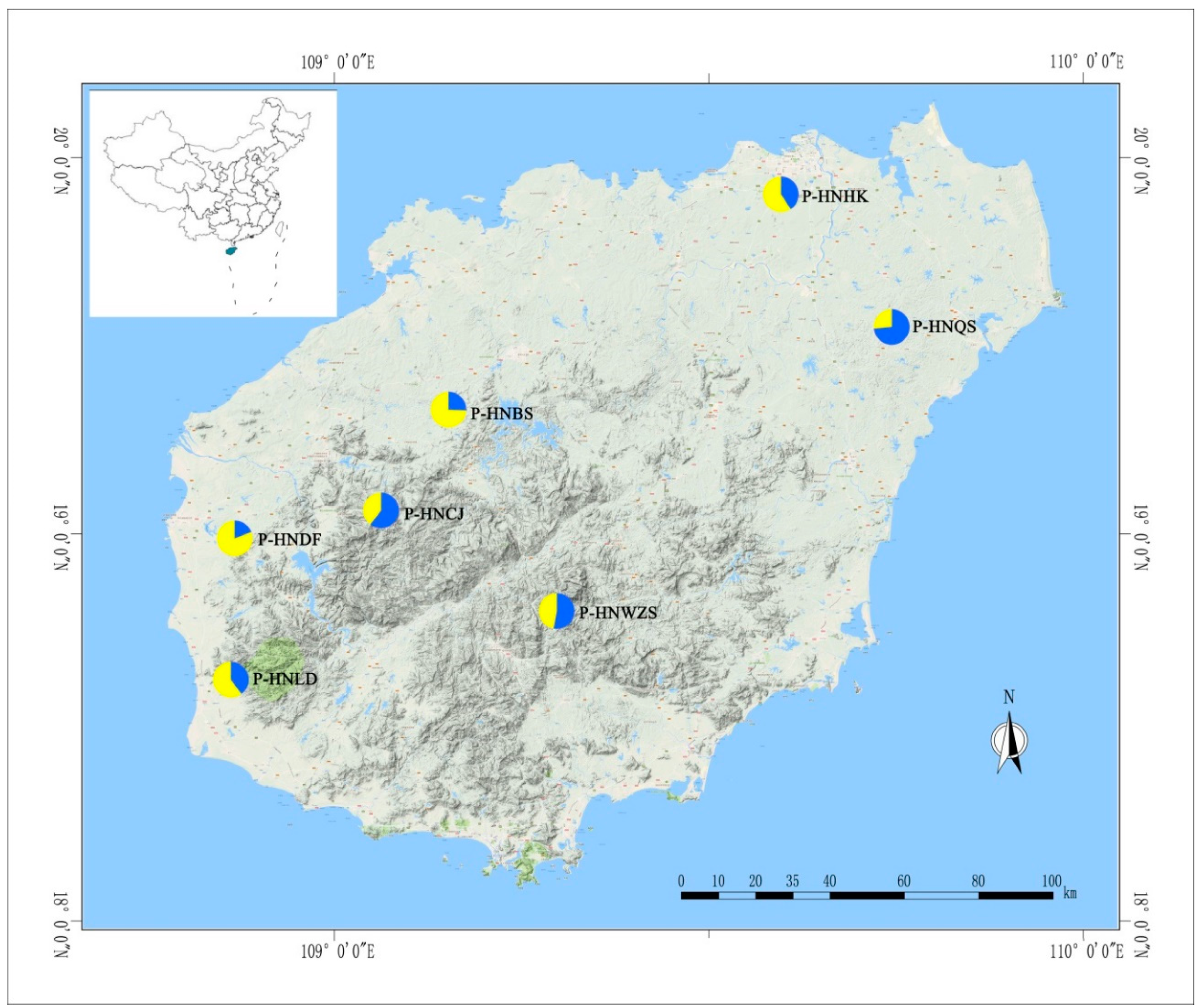
| Population | Size | Origin | Status | Altitude (m) | Latitude (N) | Longitude (E) |
|---|---|---|---|---|---|---|
| HNQS | 7 | Wenchang city and Sansha city | Wild, Qiong mountain | 20–80 | 19°37′–19°53′ | 110°19′–110°23′ |
| HNHK | 3 | Haikou city | Wild, strand plain | 10–80 | 20°04′–20°32′ | 110°21′–109°21′ |
| HNBS | 5 | Baisha autonomous county | Wild, Li mu mountain | 150–250 | 19°12′–19°23′ | 109°7′–109°24′ |
| HNDF | 5 | Dongfang city | Wild, strand plain | 5–100 | 19°6′–19°17′ | 108°37′–108°39′ |
| HNLD | 7 | Ledong autonomous county | Wild, Jianfeng mountain | 35–80 | 18°34′–18°42′ | 108°49′–109°08′ |
| HNCJ | 5 | Changjiang autonomous county | Wild, Bawang mountains | 125–185 | 19°4′–19°08′ | 109°03′–109°12′ |
| HNWZS | 10 | Wuzhisan city | Wild, Wuzhisan mountain | 60–150 | 18°48′–18°53′ | 109°30′–109°67′ |
| Locus | ID | Repeat Motif | Forward Primer (5′-3′) | Reverse Primer (3′-5′) | Product Size (bp) | SSR Position | Tm (°C) |
|---|---|---|---|---|---|---|---|
| S01 | c102105_g1 | (ATA)6 | AGTCCCGCCCACAAAATCAT | CTGGTCAGTCATTCCCCCAC | 252–258 | Unknown | 60 |
| S02 | c11754_g1 | (AAG)6 | GGTCCCTGACTCACTGAAGC | CAACCTCTCTCTGCAGAACCA | 269–275 | 5′UTR | 60 |
| S03 | c1204_g1 | (ATA)6 | GCACGTGGTCAAAGCAATCA | ATGAGCCCCTTCTGCACTTC | 263–266 | Unknown | 60 |
| S04 | c25868_g1 | (GAT)6 | GCTGTGGAGTCACGTTCTCA | TCCCCACAGAATCACAAGCC | 278–287 | 3′UTR | 60 |
| S07 | c29390_g1 | (CCT)6 | GCCAATGACATAATGGGCGG | TGCAGAGAGTCAGGAGCTCT | 226–229 | CDS | 60 |
| S08 | c31361_g1 | (TGT)6 | GGAAGAGAAATGGAGGGTAGCT | TGCCAGACAACCAGAATGCT | 305–320 | Unknown | 60 |
| S09 | c33497_g1 | (CAT)6 | ACCCTCCTCCTCCACCTTTT | ACCGGCTTCAGTGATTGGTT | 230–239 | 5′UTR | 60 |
| S10 | c40172_g1 | (TGC)7 | CACGTACCCAACCGTCAAGA | TCCGACGACCACCTAATCCT | 261–273 | 5′UTR | 60 |
| S11 | c40452_g1 | (ATC)6 | AAAAAGCGAGGACTACGGCA | TGGAGAAGCAGTGCTCGTTT | 224–227 | Unknown | 60 |
| S12 | c34672_g2 | (GAT)7 | GGTGAACAAGCTGGAGTGGA | AAGCCCAGCATCTAAACCCC | 259–271 | CDS | 60 |
| S21 | c56000_g1 | (TCCC)6 | GAGCCTTGAGTTCACCTCCC | TTGGGTGTGAGATTGAGGGC | 238–250 | 5′UTR | 60 |
| S22 | c53146_g1 | (TC)10 | CCACCGATCTTAACCTCCGG | ACTACAAGTGCGTGTGACCC | 254–260 | Unknown | 60 |
| S23 | c56684_g2 | (TC)10 | TGGCGTTGACTTCCAGCATT | GAGCAGTGTCAGCATGATGC | 278–280 | 3′UTR | 60 |
| S24 | c59001_g1 | (CTA)7 | GCTGCAAATGCCAGTGCTTA | CGCTGTTGTCAGTGCATTGG | 232–241 | Unknown | 60 |
| S26 | c60831_g5 | (GTT)7 | CCAATCCCACCAGTGAGGAG | GCAGCACCTCTGAGACAAGT | 241–247 | CDS | 60 |
| S27 | c49315_g1 | (TAC)7 | GAACCTTTCCTTCTGCGCCT | CCTATGAAGCGTGTGCATGC | 266–272 | 3′UTR | 60 |
| S28 | c63495_g1 | (TAC)7 | ACAGCATTTGTGTTTGTGCA | CAGCTGCGCTCTCATTCCTA | 246–249 | Unknown | 60 |
| S29 | c57231_g1 | (TAT)7 | TCCCCGTTCCTCTCTCTCAG | GGACTGTCACATGGCACTCA | 150–168 | 5′UTR | 60 |
| S30 | c48304_g3 | (TGT)7 | TGCCTTGATCCGCTGAGATC | TCCCAAAATCGATGCAAAGCA | 246–258 | 5′UTR | 59 |
| Locus | Na | Ne | Ho | He | GD | PIC | FST | Nm | F | FNA | PHWE a |
|---|---|---|---|---|---|---|---|---|---|---|---|
| S01 | 3 | 1.99 | 0.38 | 0.50 | 0.50 | 0.41 | 0.08 | 2.79 | 0.23 | 0.08 | 0.108 ns |
| S02 | 2 | 1.05 | 0.05 | 0.05 | 0.05 | 0.05 | 0.07 | 3.58 | −0.02 | 0.00 | 0.874 ns |
| S03 | 2 | 1.80 | 0.38 | 0.45 | 0.44 | 0.35 | 0.16 | 1.30 | 0.14 | 0.04 | 0.355 ns |
| S04 | 3 | 1.71 | 0.29 | 0.42 | 0.42 | 0.37 | 0.11 | 2.08 | 0.31 | 0.09 * | 0.002 ** |
| S07 | 2 | 1.83 | 0.36 | 0.46 | 0.45 | 0.35 | 0.12 | 1.85 | 0.21 | 0.07 | 0.173 ns |
| S08 | 4 | 1.38 | 0.24 | 0.28 | 0.27 | 0.26 | 0.05 | 4.68 | 0.13 | 0.03 | 0.001 ** |
| S09 | 3 | 2.57 | 0.36 | 0.62 | 0.61 | 0.54 | 0.17 | 1.22 | 0.42 | 0.16 * | 0.001 *** |
| S10 | 3 | 1.97 | 0.48 | 0.50 | 0.49 | 0.44 | 0.09 | 2.65 | 0.03 | 0.01 | 0.412 ns |
| S11 | 2 | 1.75 | 0.38 | 0.43 | 0.43 | 0.34 | 0.14 | 1.48 | 0.11 | 0.03 | 0.481 ns |
| S12 | 2 | 1.21 | 0.14 | 0.17 | 0.17 | 0.16 | 0.13 | 1.70 | 0.17 | 0.03 | 0.268 ns |
| S21 | 5 | 2.79 | 0.45 | 0.65 | 0.64 | 0.60 | 0.10 | 2.20 | 0.30 | 0.12 * | 0.067 ns |
| S22 | 4 | 1.56 | 0.43 | 0.36 | 0.36 | 0.33 | 0.11 | 1.96 | −0.19 | −0.05 | 0.793 ns |
| S23 | 2 | 1.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.06 | 3.79 | −0.01 | 0.00 | 0.938 ns |
| S24 | 3 | 2.05 | 0.29 | 0.52 | 0.51 | 0.46 | 0.22 | 0.87 | 0.44 | 0.15 * | 0.002 ** |
| S26 | 2 | 1.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.09 | 2.63 | −0.01 | 0.00 | 0.938 ns |
| S27 | 3 | 1.27 | 0.21 | 0.21 | 0.21 | 0.19 | 0.09 | 2.42 | −0.01 | 0.00 | 0.032 * |
| S28 | 2 | 1.80 | 0.26 | 0.42 | 0.44 | 0.35 | 0.10 | 2.19 | 0.25 | 0.08 | 0.105 ns |
| S29 | 4 | 1.71 | 0.26 | 0.42 | 0.42 | 0.35 | 0.12 | 1.75 | 0.37 | 0.11 * | 0.000 *** |
| S30 | 3 | 1.81 | 0.33 | 0.45 | 0.45 | 0.37 | 0.03 | 7.77 | 0.25 | 0.08 | 0.135 ns |
| Mean | 2.84 | 1.70 | 0.28 | 0.37 | 0.36 | 0.31 | 0.11 | 2.58 | 0.16 | 0.03 |
| Population | Size | Alleles | Na | Ne | Np | Ho | He | GD | FIS | PPB % |
|---|---|---|---|---|---|---|---|---|---|---|
| HNQS | 7 | 40 | 2.11 | 1.52 | 1 | 0.25 | 0.31 | 0.28 | 0.09 | 73.68 |
| HNHK | 3 | 35 | 1.84 | 1.54 | 0 | 0.26 | 0.34 | 0.28 | 0.00 | 68.42 |
| HNBS | 5 | 40 | 2.11 | 1.69 | 1 | 0.31 | 0.36 | 0.32 | 0.02 | 78.95 |
| HNDF | 5 | 41 | 2.16 | 1.59 | 3 | 0.38 | 0.37 | 0.33 | −0.11 | 89.47 |
| HNLD | 7 | 46 | 2.42 | 1.64 | 2 | 0.26 | 0.38 | 0.35 | 0.20 | 89.47 |
| HNCJ | 5 | 40 | 2.11 | 1.77 | 0 | 0.36 | 0.40 | 0.36 | −0.03 | 78.95 |
| HNWZS | 10 | 44 | 2.32 | 1.63 | 1 | 0.24 | 0.34 | 0.33 | 0.22 | 78.95 |
| Mean | 5.25 | 40 | 2.15 | 1.63 | - | 0.29 | 0.36 | 0.32 | 0.06 | 79.70 |
| Total a | 42 b | 56 | 2.84 | 1.70 | - | 0.28 | 0.37 | 0.36 | 0.21 | 100.00 |
| Source | d.f. | Sum of Square | Mean of Square | Variance Components | Percentage of Variation | FST | FIS |
|---|---|---|---|---|---|---|---|
| Among populations | 5 | 27.864 | 5.573 | 0.111 | 3% | ||
| Within populations | |||||||
| Among Individuals | 33 | 136.893 | 4.148 | 0.715 | 20% | ||
| Within Individuals | 39 | 106.000 | 2.718 | 2.718 | 77% | ||
| Total | 77 | 270.756 | 3.544 | 100% | 0.03 * | 0.21 *** |
| Population | HNQS | HNHK | HNBS | HNDF | HNLD | HNCJ | HNWZS |
|---|---|---|---|---|---|---|---|
| HNQS | |||||||
| HNHK | 0.088 | ||||||
| HNBS | 0.095 | 0.055 | |||||
| HNDF | 0.115 | 0.090 | 0.062 | ||||
| HNLD | 0.074 | 0.055 | 0.043 | 0.051 | |||
| HNCJ | 0.071 | 0.100 | 0.087 | 0.090 | 0.042 | ||
| HNWZS | 0.061 | 0.078 | 0.051 | 0.051 | 0.044 | 0.054 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, F.; Hong, Z.; Xu, D.; Jia, H.; Zhang, N.; Liu, X.; Yang, Z.; Lu, M. Genetic Diversity of the Endangered Dalbergia odorifera Revealed by SSR Markers. Forests 2019, 10, 225. https://doi.org/10.3390/f10030225
Liu F, Hong Z, Xu D, Jia H, Zhang N, Liu X, Yang Z, Lu M. Genetic Diversity of the Endangered Dalbergia odorifera Revealed by SSR Markers. Forests. 2019; 10(3):225. https://doi.org/10.3390/f10030225
Chicago/Turabian StyleLiu, Fumei, Zhou Hong, Daping Xu, Hongyan Jia, Ningnan Zhang, Xiaojin Liu, Zengjiang Yang, and Mengzhu Lu. 2019. "Genetic Diversity of the Endangered Dalbergia odorifera Revealed by SSR Markers" Forests 10, no. 3: 225. https://doi.org/10.3390/f10030225
APA StyleLiu, F., Hong, Z., Xu, D., Jia, H., Zhang, N., Liu, X., Yang, Z., & Lu, M. (2019). Genetic Diversity of the Endangered Dalbergia odorifera Revealed by SSR Markers. Forests, 10(3), 225. https://doi.org/10.3390/f10030225