Identification and Analysis of microRNAs in the SAM and Leaves of Populus tomentosa

 and
and
Abstract
:1. Introduction
2. Materials and Methods
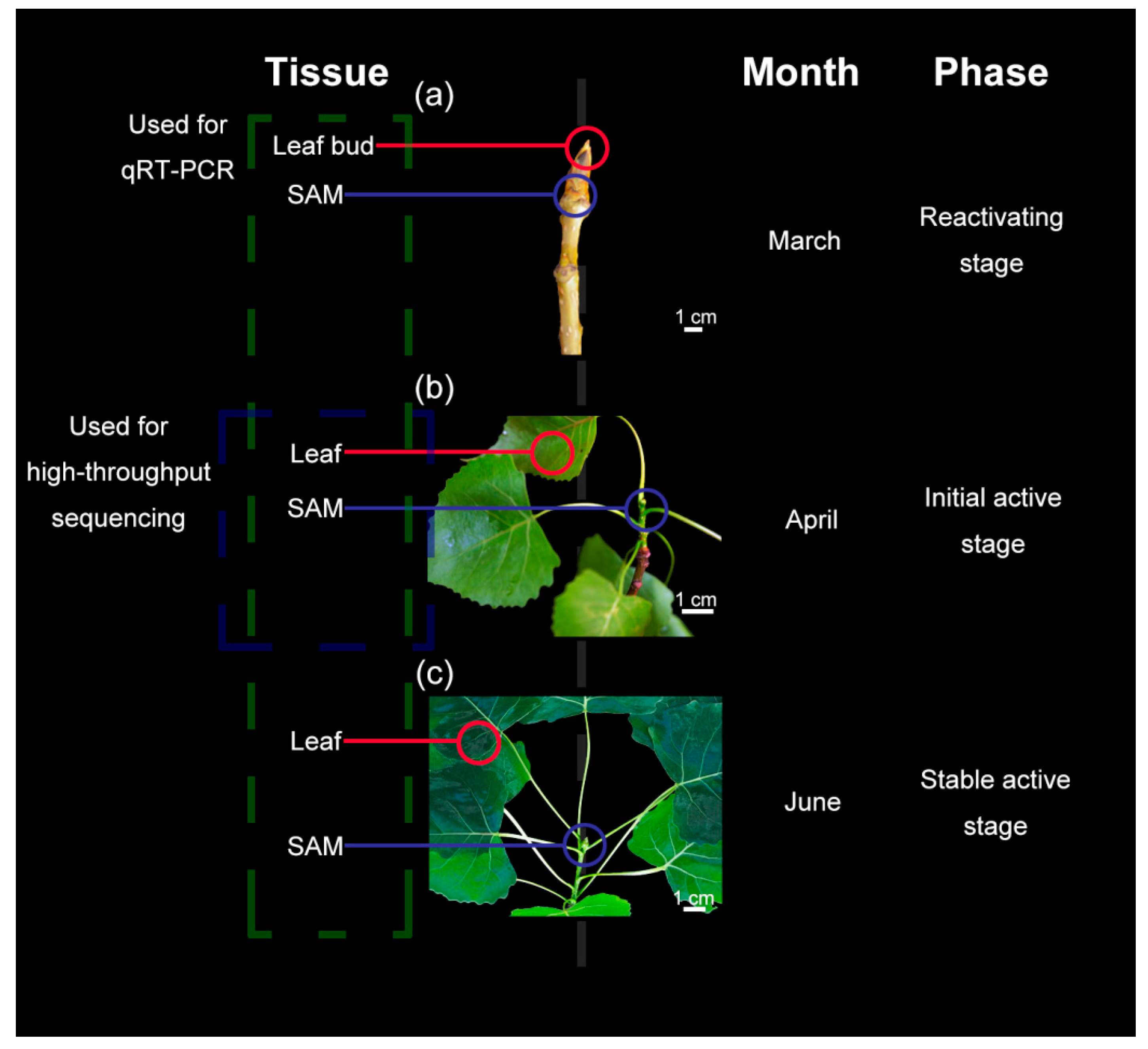
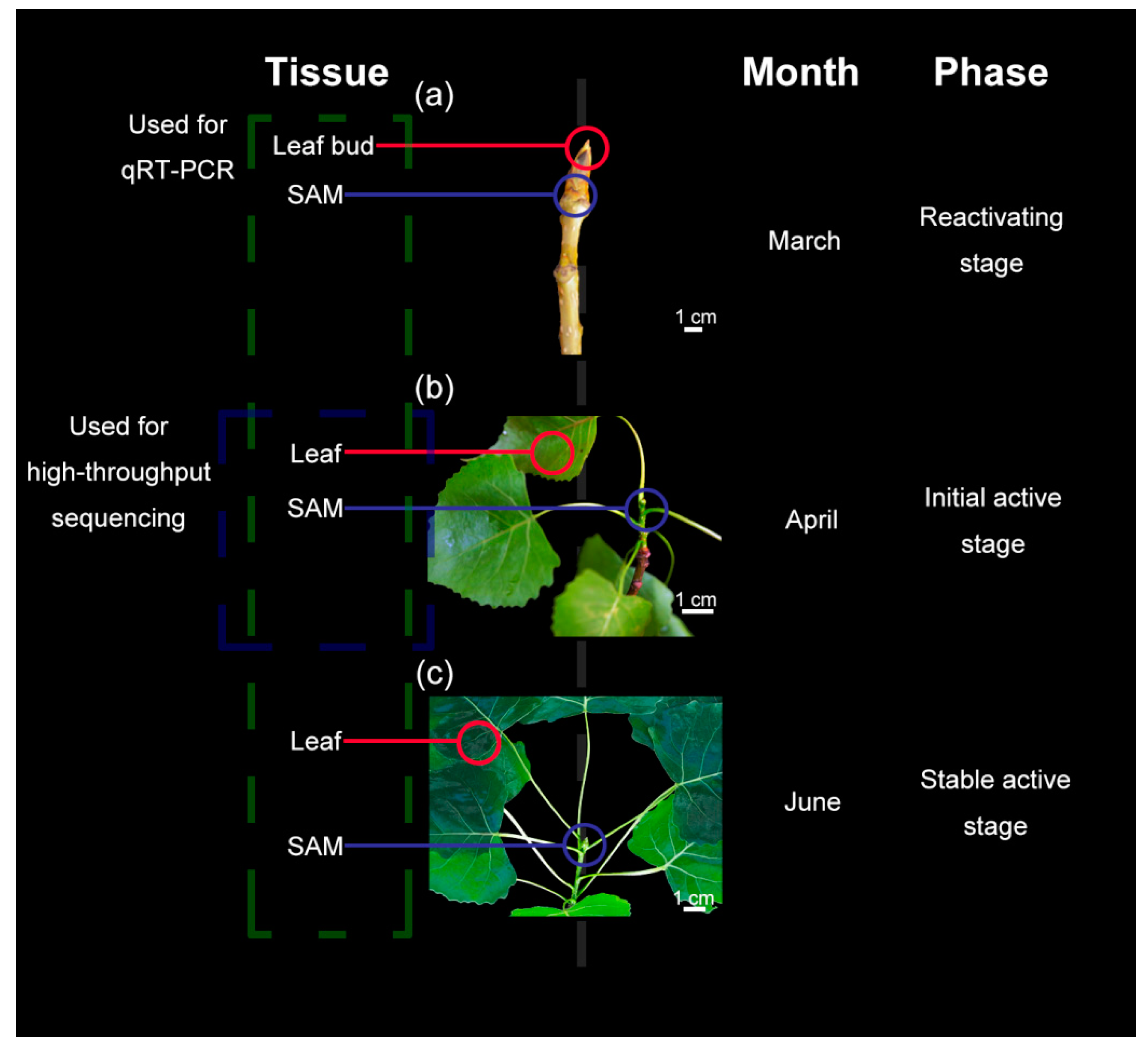
2.1. Plant Materials
2.2. Morphological Observations
2.3. sRNA Library Construction and High-Throughput Sequencing
2.4. Bioinformatic Analysis of Sequencing Data
2.5. Prediction of miRNA Targets
2.6. qRT-PCR Analysis of Differential Gene Expression
3. Results
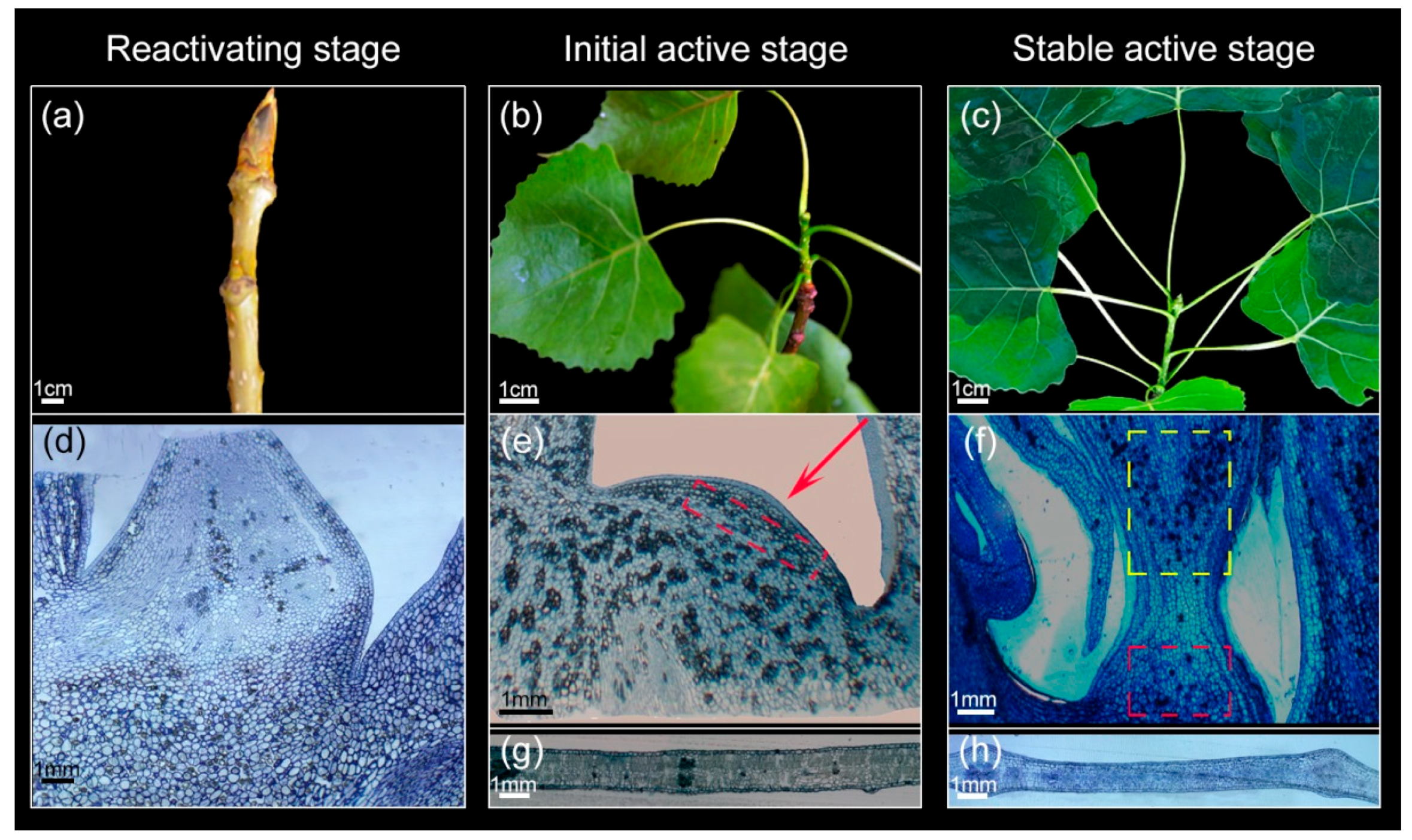
3.1. Morphological and Anatomical Structure Changes of Shoot Tips and Leaves During Growth
3.2. Sequencing and Analysis of sRNAs in the SAM and Leaves
3.3. Identification of Known and Novel miRNAs in P. tomentosa
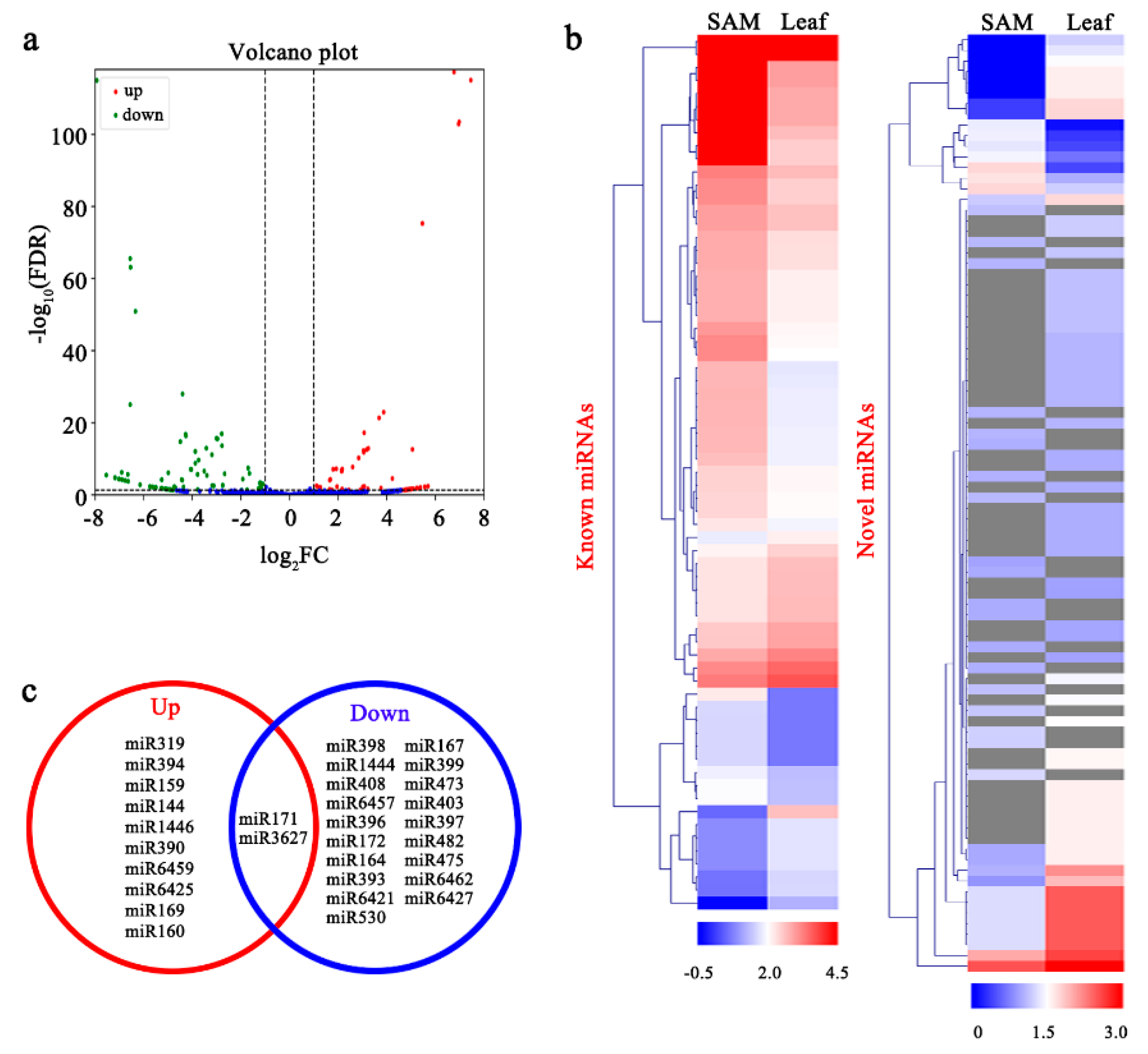
3.4. Analysis of Differentially Expressed miRNAs (DEMs) and their Targets
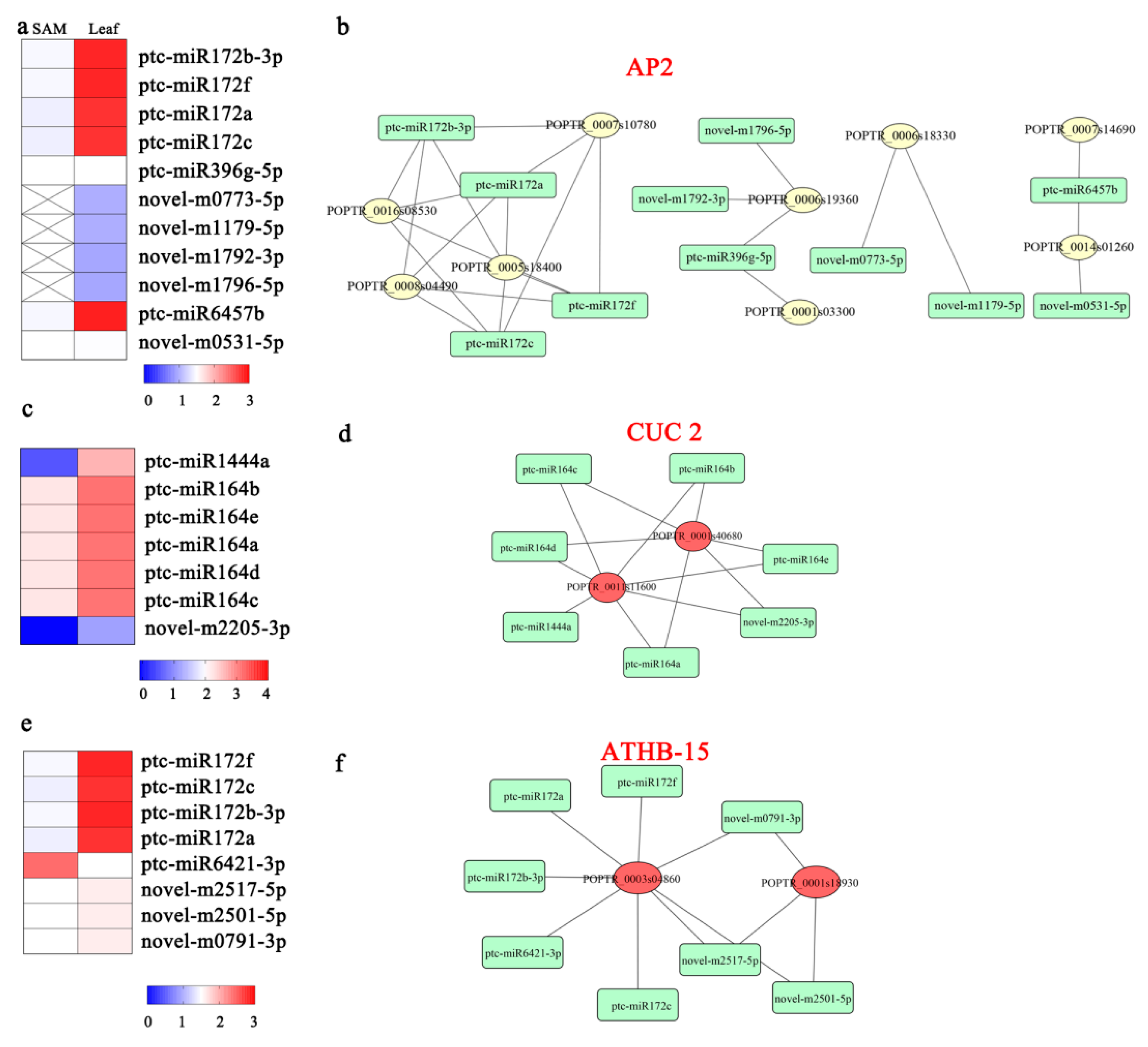
3.5. DEMs Related to Stem Cell Functions
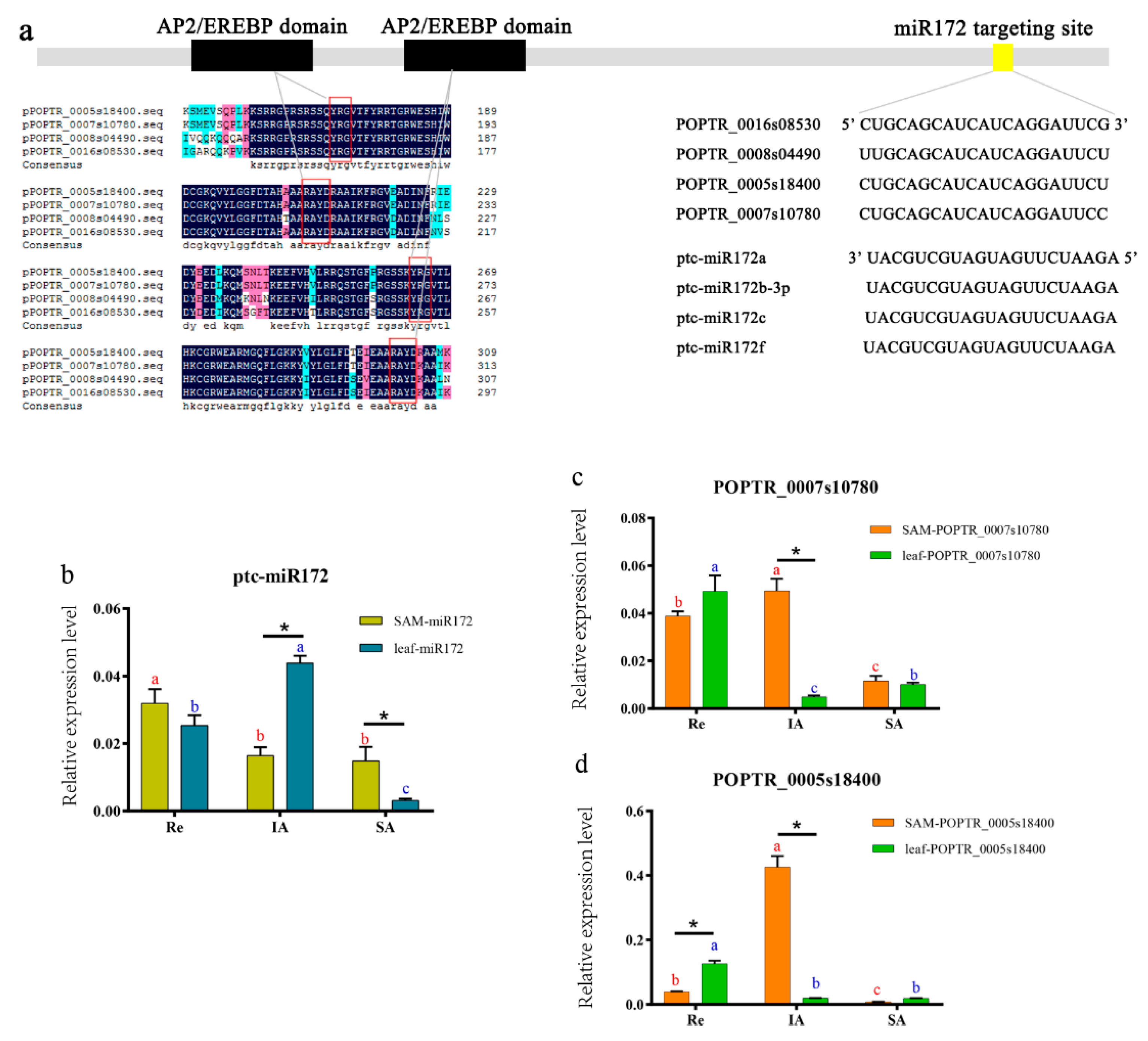
3.6. Expression of miR172 and its Targets
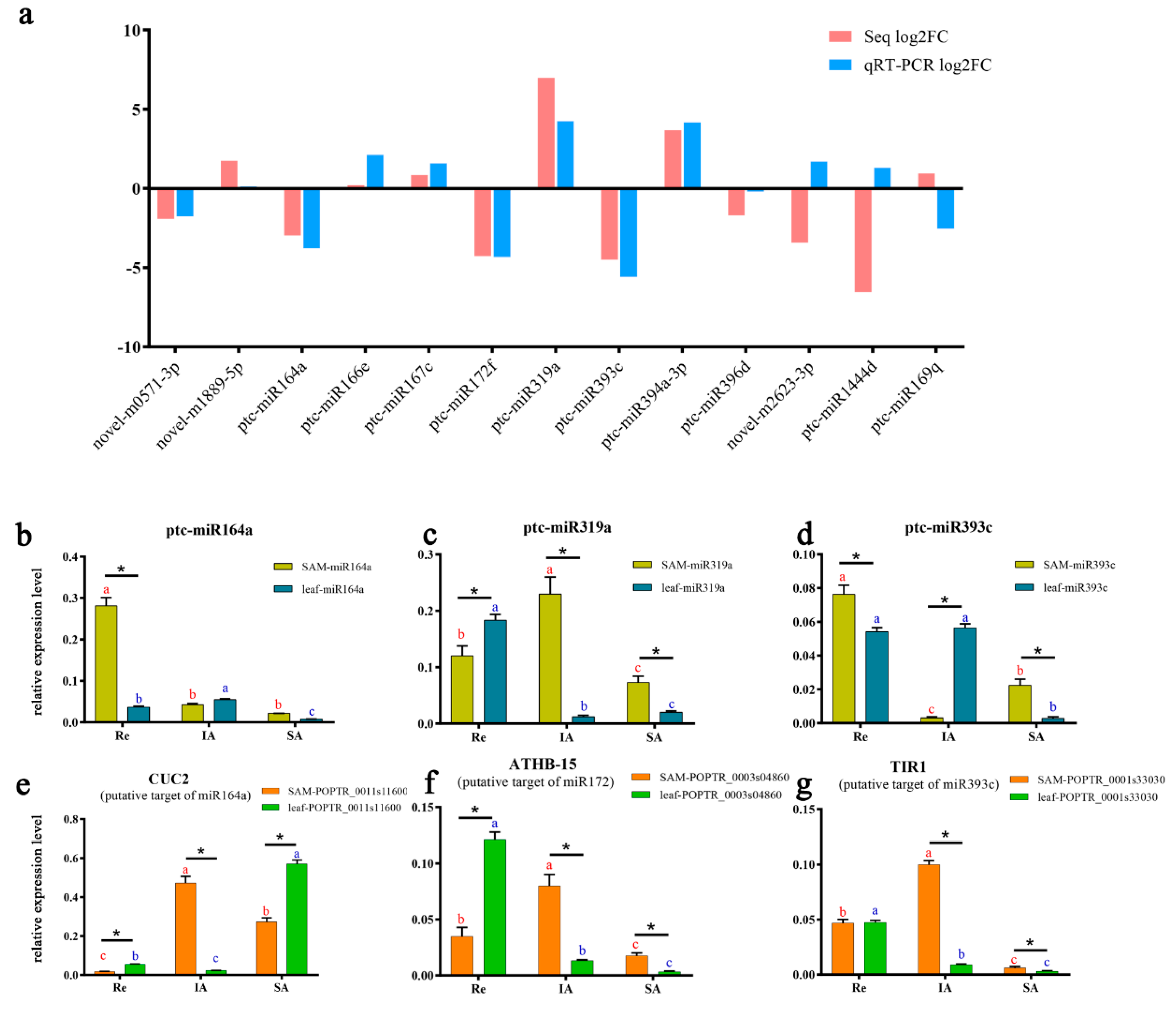
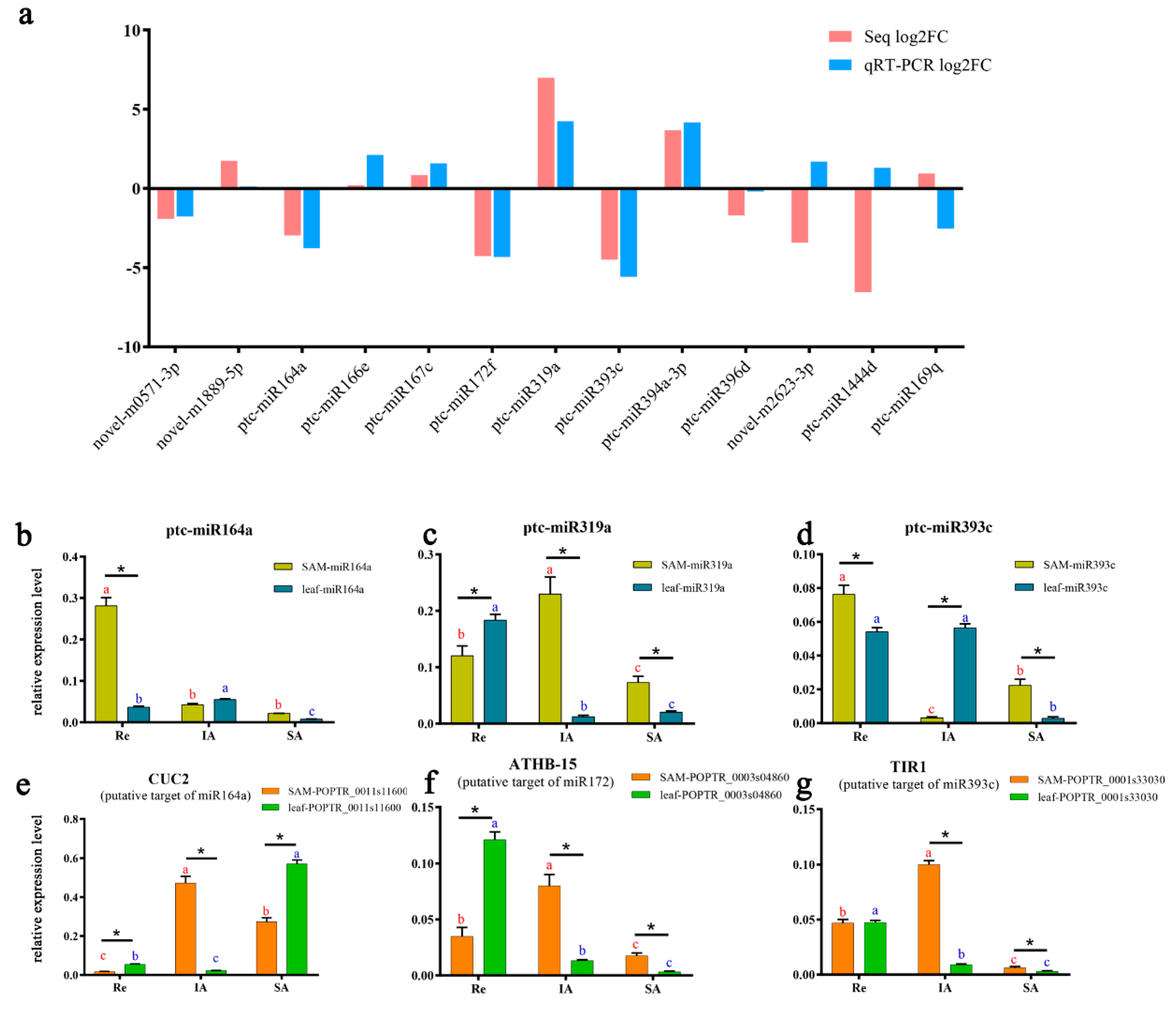
3.7. qRT-PCR Validation of Other Important miRNAs
4. Discussion
4.1. SAM and Leaf Formation
4.2. Complexities of sRNA Generation in P. tomentosa
4.3. miRNA Population Involved in SAM Development and Maintenance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stahl, Y.; Simon, R. Plant stem cell niches. Int. J. Dev. Biol. 2004, 49, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, C.; Ohashi, Y.; Sato, S.; Kato, T.; Tabata, S.; Ueguch, C. Histidine kinase homologs that act as cytokinin receptors possess overlapping functions in the regulation of shoot and root growth in Arabidopsis. Plant Cell 2004, 16, 1365–1377. [Google Scholar] [CrossRef] [PubMed]
- Vernoux, T.; Besnard, F.; Traas, J. Auxin at the shoot apical meristem. CSH Perspect. Biol. 2010, 2, a001487. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, D.; Mandel, T.; Kuhlemeier, C. Auxin regulates the initiation and radial position of plant lateral organs. Plant Cell 2000, 12, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Riefler, M.; Novak, O.; Strnad, M.; Schmülling, T. Arabidopsis cytokinin receptor mutants reveal functions in shoot growth, leaf senescence, seed size, germination, root development, and cytokinin metabolism. Plant Cell 2006, 18, 40–54. [Google Scholar] [CrossRef]
- Shinohara, N.; Taylor, C.; Leyser, O. Strigolactone can promote or inhibit shoot branching by triggering rapid depletion of the auxin efflux protein PIN1 from the plasma membrane. Plos. Biol. 2013, 11, e1001474. [Google Scholar] [CrossRef] [PubMed]
- Jasinski, S.; Piazza, P.; Craft, J.; Hay, A.; Woolley, L.; Rieu, I.; Phillips, A.; Hedden, P.; Tsiantis, M. KNOX action in Arabidopsis is mediated by coordinate regulation of cytokinin and gibberellin activities. Curr. Biol. 2005, 15, 1560–1565. [Google Scholar] [CrossRef]
- Torti, S.; Fornara, F.; Vincent, C.; Andrés, F.; Nordström, K.; Göbel, U.; Knoll, D.; Schoof, H.; Coupland, G. Analysis of the Arabidopsis shoot meristem transcriptome during floral transition identifies distinct regulatory patterns and a leucine-rich repeat protein that promotes flowering. Plant Cell 2012, 24, 444–462. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kim, S.G.; Lee, M.; Lee, I.; Park, H.Y.; Seo, P.J.; Jung, J.H.; Kwon, E.J.; Suh, S.W.; Paek, K.H.; et al. HD-ZIP III activity is modulated by competitive inhibitors via a feedback loop in Arabidopsis shoot apical meristem development. Plant Cell 2008, 20, 920–933. [Google Scholar] [CrossRef]
- Veit, B. Hormone mediated regulation of the shoot apical meristem. Plant Mol. Biol. 2009, 69, 397–408. [Google Scholar] [CrossRef]
- Besnard, F.; Vernoux, T.; Hamant, O. Organogenesis from stem cells in planta: Multiple feedback loops integrating molecular and mechanical signals. Cell Mol. Life Sci. 2011, 68, 2885–2906. [Google Scholar] [CrossRef] [PubMed]
- Scofield, S.; Murison, A.; Jones, A.; Fozard, J.; Aida, M.; Band, L.R.; Bennett, M.; Murray, J.A.H. Coordination of meristem and boundary functions by transcription factors in the SHOOT MERISTEMLESS regulatory network. Development 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, X. Argonautes compete for miR165/166 to regulate shoot apical meristem development. Curr. Opin. Plant Biol. 2012, 15, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.; Fletcher, J.C. Stem cell regulation in the Arabidopsis, shoot apical meristem. Curr. Opin. Plant Biol. 2005, 8, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Honda, M.; Zhu, H.; Zhang, Z.; Guo, X.; Li, T.; Li, Z.; Peng, X.; Nakajima, K.; Duan, L.; et al. Spatiotemporal sequestration of miR165/166 by Arabidopsis Argonaute10 promotes shoot apical meristem maintenance. Cell Rep. 2015, 10, 18–19. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.X.; Li, S.G.; Zhang, L.F.; Han, S.Y.; Li, W.F.; Xu, H.Y.; Yang, W.; Liu, Y.L.; Fan, Y.R.; Qi, L.W. Over-expression of miR166a inhibits cotyledon formation in somatic embryos and promotes lateral root development in seedlings of Larix leptolepis. Plant Cell Tiss. Org. Cult. 2016, 127, 461–473. [Google Scholar] [CrossRef]
- Wang, J.W.; Schwab, R.; Czech, B.; Mica, E.; Weigel, D. Dual effects of miR156-targeted SPL genes and CYP78A5/KLUH on plastochron length and organ size in Arabidopsis thaliana. Plant Cell 2008, 20, 1231–1243. [Google Scholar] [CrossRef]
- Zhao, L.; Kim, Y.J.; Dinh, T.T.; Chen, X. miR172 regulates stem cell fate and defines the inner boundary of APETALA3 and PISTILLATA expression domain in Arabidopsis floral meristems. Plant J. 2007, 51, 840–849. [Google Scholar] [CrossRef]
- Wu, L.; Zhou, H.; Zhang, Q.; Zhang, J.; Ni, F.; Liu, C.; Qi, Y. DNA Methylation mediated by a microRNA pathway. Mol. Cell 2010, 38, 465–475. [Google Scholar] [CrossRef]
- GenBank. Available online: http://www.ncbi.nlm.nih.gov/genbank/ (accessed on 23 June 2018).
- Rfam. Available online: http://rfam.xfam.org/ (accessed on 24 June 2018).
- Gardner, P.P.; Daub, J.; Tate, J.; Moore, B.L.; Osuch, I.H.; Griffiths-Jones, S.; Finn, R.D.; Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R.; et al. Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Res. 2011, 39, D141–D145. [Google Scholar] [CrossRef]
- Populus trichocarpa genome. Available online: ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/002/775/GCA_000002775.2_Poptr2_0/ (accessed on 22 October 2018).
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- miRbase. Available online: http://www.mirbase.org (accessed on 21 October 2018).
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2010, 39, D152–D157. [Google Scholar] [CrossRef] [PubMed]
- MIREAP. Available online: http://sourceforge.net/projects/mireap/ (accessed on 23 October 2018).
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J.; et al. Criteria for annotation of plant microRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef] [PubMed]
- psRobot. Available online: http://omicslab.genetics.ac.cn/psRobot/index.php (accessed on 23 October 2018).
- Cytoscape. Available online: https://cytoscape.org (accessed on 26 October 2018).
- Cao, X.; Zhai, X.; Zhang, Y.; Cheng, Z.; Li, X.; Fan, G. Comparative Analysis of MicroRNA Expression in Three Paulownia Species with Phytoplasma Infection. Forests 2018, 9, 302. [Google Scholar] [CrossRef]
- Pettengill, E.A.; Parmentier-Line, C.; Coleman, G.D. Evaluation of qPCR reference genes in two genotypes of Populus for use in photoperiod and low-temperature studies. BMC Res. Notes 2012, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Allen, P.J.; Bennett, K. PASW statistics by SPSS: A practical guide: Version 18.0; Cengage Learning: South Melbourne, Austria, 2010. [Google Scholar]
- Aichinger, E.; Kornet, N.; Friedrich, T.; Laux, T. Plant stem cell niches. Annu. Rev. Plant Biol. 2012, 63, 615–636. [Google Scholar] [CrossRef]
- Repeat-Repbase. Available online: http://www.girinst.org/repbase (accessed on 23 October 2018).
- Neilsen, C.T.; Goodall, G.J.; Bracken, C.P. IsomiRs–the overlooked repertoire in the dynamic microRNAome. Trends Genet. 2012, 28, 544–549. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic acids res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.C.; Wong, C.E.; Singh, M.B.; Beveridge, C.A.; Phipson, B.; Smyth, G.K.; Bhalla, P.L. Molecular dissection of the pea shoot apical meristem. J. Exp. Bot. 2009, 60, 4201–4213. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.H.; Liu, Y.B.; Zhang, X.S. Auxin-cytokinin interaction regulates meristem development. Mol. Plant 2011, 4, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Chen, Y.Q.; Liu, Q. Sculpting the meristem: The roles of miRNAs in plant stem cells. Biochem. Biophys. Res. Commun. 2011, 409, 363–366. [Google Scholar] [CrossRef]
- Barbier, F.F.; Lunn, J.E.; Beveridge, C.A. Ready, steady, go! A sugar hit starts the race to shoot branching. Curr. Opin. Plant Biol. 2015, 25, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.L.; Eshed, Y. Formation and maintenance of the shoot apical meristem. Trends Plant Sci. 2000, 5, 110–115. [Google Scholar] [CrossRef]
- Wong, C.E.; Zhao, Y.T.; Wang, X.J.; Croft, L.; Wang, Z.H.; Haerizadeh, F.; Mattick, J.S.; Singh, M.B.; Carrol, B.J.; Bhalla, P.L. MicroRNAs in the shoot apical meristem of soybean. J. Exp. Bot. 2011, 62, 2495. [Google Scholar] [CrossRef] [PubMed]
- Park, J.C.M. MIR166/165 genes exhibit dynamic expression patterns in regulating shoot apical meristem and floral development in Arabidopsis. Planta 2007, 225, 1327–1338. [Google Scholar]
- Vazquez, F.; Legrand, S.; Windels, D. The biosynthetic pathways and biological scopes of plant small RNAs. Trends Plant Sci. 2010, 15, 337–345. [Google Scholar] [CrossRef]
- Rajagopalan, R.; Vaucheret, H.; Trejo, J.; Bartel, D.P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Gene Dev. 2006, 20, 3407–3425. [Google Scholar] [CrossRef]
- Zhou, Z.S.; Zeng, H.Q.; Liu, Z.P.; Yang, Z.M. Genome-wide identification of Medicago truncatula microRNAs and their targets reveals their differential regulation by heavy metal. Plant Cell Environ. 2012, 35, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Wall, P.K.; Diloreto, S.; dePamphilis, C.W.; Carlson, J.E. Conservation and divergence of microRNAs in Populus. BMC Genomics. 2007, 8, 481. [Google Scholar] [CrossRef] [PubMed]
- Moxon, S.; Jing, R.; Szittya, G.; Schwach, F.; Pilcher, R.L.R.; Moulton, V.; Dalmay, T. Deep sequencing of tomato short RNAs identifies microRNAs targeting genes involved in fruit ripening. Genome Res. 2008, 18, 1602–1609. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, V.; Szittya, G.; Moxon, S.; Miozzi, L.; Moulton, V.; Dalmay, T.; Burgyan, J. Identification of grapevine microRNAs and their targets using, high-throughput sequencing and degradome analysis. Plant J. 2010, 62, 960–976. [Google Scholar] [PubMed]
- Li, B.; Duan, H.; Li, J.; Deng, X.W.; Yin, W.; Xia, X. Global identification of miRNAs and targets in Populus euphratica under salt stress. Plant Mol. Biol. 2013, 81, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Zeng, J.; He, X.Q. Deep sequencing on a genome-wide scale reveals diverse stage-specific microRNAs in cambium during dormancy-release induced by chilling in poplar. BMC Plant Biol. 2014, 14, 267. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Li, Y.F.; Jagadeeswaran, G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012, 17, 32–41. [Google Scholar] [CrossRef]
- Nova-Franco, B.; Íñiguez, L.P.; Valdés-López, O.; Alvarado-Affantranger, X.; Leija, A.; Fuentes, S.I.; Ramírez, M.; Paul, S.; Reyes, J.L.; Girard, L.; et al. The micro-RNA72c-APETALA2-1 node as a key regulator of the common bean-Rhizobium etli nitrogen fixation symbiosis. Plant Physiol. 2015, 168, 273–291. [Google Scholar] [CrossRef]
- Zhang, B. MicroRNA: A new target for improving plant tolerance to abiotic stress. J. Exp. Bot. 2015, 66, 1749–1761. [Google Scholar] [CrossRef]
- Qin, Y.; Duan, Z.; Xia, X.; Yin, W. Expression profiles of precursor and mature microRNAs under dehydration and high salinity shock in Populus euphratica. Plant Cell Rep. 2011, 30, 1893–1907. [Google Scholar] [CrossRef]
- Puzey, J.R.; Karger, A.; Axtell, M.; Kramer, E.M. Deep annotation of Populus trichocarpa microRNAs from diverse tissue sets. PLoSOne 2012, 7, e33034. [Google Scholar] [CrossRef] [PubMed]
- Koundal, V.; Vinutha, T.; Haq, Q.M.R.; Praveen, S. Modulation of plant development and MYB down regulation: Both during in planta expression of miR159a and in natural ToLCV infection. J. Plant Biochem. Biot. 2010, 19, 171–175. [Google Scholar] [CrossRef]
- Zhu, H.; Hu, F.; Wang, R.; Zhou, X.; Sze, S.H.; Liou, L.W.; Barefoot, A.; Dickman, M.; Zhang, X. Arabidopsis Argonaute10 specifically sequesters miR166/165 to regulate shoot apical meristem development. Cell 2011, 145, 242–256. [Google Scholar] [CrossRef]
- Wang, L.; Gu, X.; Xu, D.; Wang, W.; Wang, H.; Zeng, M.; Chang, Z.; Huang, H.; Cui, X. MiR396-targeted AtGRF transcription factors are required for coordination of cell division and differentiation during leaf development in Arabidopsis. J. Exp. Bot. 2011, 62, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Werner, T.; Motyka, V.; Laucou, V.; Smets, R.; Onckelen, H.V.; Schmulling, T. Cytokinin-deficient ¨ transgenic Arabidopsis plants show multiple developmental alterations indicating opposite functions of cytokinins in the regulation of shoot and root meristem activity. Plant Cell 2003, 15, 2532–2550. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Andersen, S.U.; Ljung, K.; Dolezal, K.; Miotk, A.; Schultheiss, S.J.; Lohmann, J.U. Hormonal control of the shoot stem-cell niche. Nature 2010, 465, 1089–1092. [Google Scholar] [CrossRef]
- Jia, F.; Wu, B.; Li, H.; Huang, J.; Zheng, C. Genome-wide identification and characterisation of F-box family in maize. Mol. Genet. Genomics. 2013, 288, 559–577. [Google Scholar] [CrossRef]
- Chen, Z.H.; Bao, M.L.; Sun, Y.Z.; Yang, Y.J.; Xu, X.H.; Wang, J.H.; Han, N.; Bian, H.W.; Zhu, M.Y. Regulation of auxin response by miR393-targeted transport inhibitor response protein 1 is involved in normal development in Arabidopsis. Plant Mol. Biol. 2011, 77, 619–629. [Google Scholar] [CrossRef]
- Si-Ammour, A.; Windels, D.; Arn-Bouldoires, E.; Kutter, C.; Ailhas, J.; Jr, M.F.; Vazquez, F. miR393 and secondary siRNAs regulate expression of the TIR1/AFB2 auxin receptor clade and auxin-related development of Arabidopsis leaves. Plant Physiol. 2011, 157, 683–691. [Google Scholar] [CrossRef]
- Raman, S.; Greb, T.; Peaucelle, A.; Blein, T.; Laufs, P.; Theres, K. Interplay of miR164, CUP-SHAPED COTYLEDON genes and LATERAL SUPPRESSOR controls axillary meristem formation in Arabidopsis thaliana. Plant J. 2008, 55, 65–76. [Google Scholar] [CrossRef]
- Laufs, P.; Peaucelle, A.; Morin, H.; Traas, J. MicroRNA regulation of the CUC genes is required for boundary size control in Arabidopsis meristems. Development 2004, 131, 4311–4322. [Google Scholar] [CrossRef] [PubMed]
- Hibara, K.; Karim, M.R.; Takada, S.; Taoka, K.; Furutani, M.; Aida, M.; Tasaka, M. Arabidopsis CUP-SHAPED COTYLEDON3 regulates postembryonic shoot meristem and organ boundary formation. Plant Cell 2006, 18, 2946–2957. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.C.; Dugas, D.V.; Bartel, D.P.; Bartel, B. MicroRNA regulation of NAC-domain targets is required for proper formation and separation of adjacent embryonic, vegetative, and floral organs. Curr. Biol. 2004, 14, 1035–1046. [Google Scholar] [CrossRef] [PubMed]

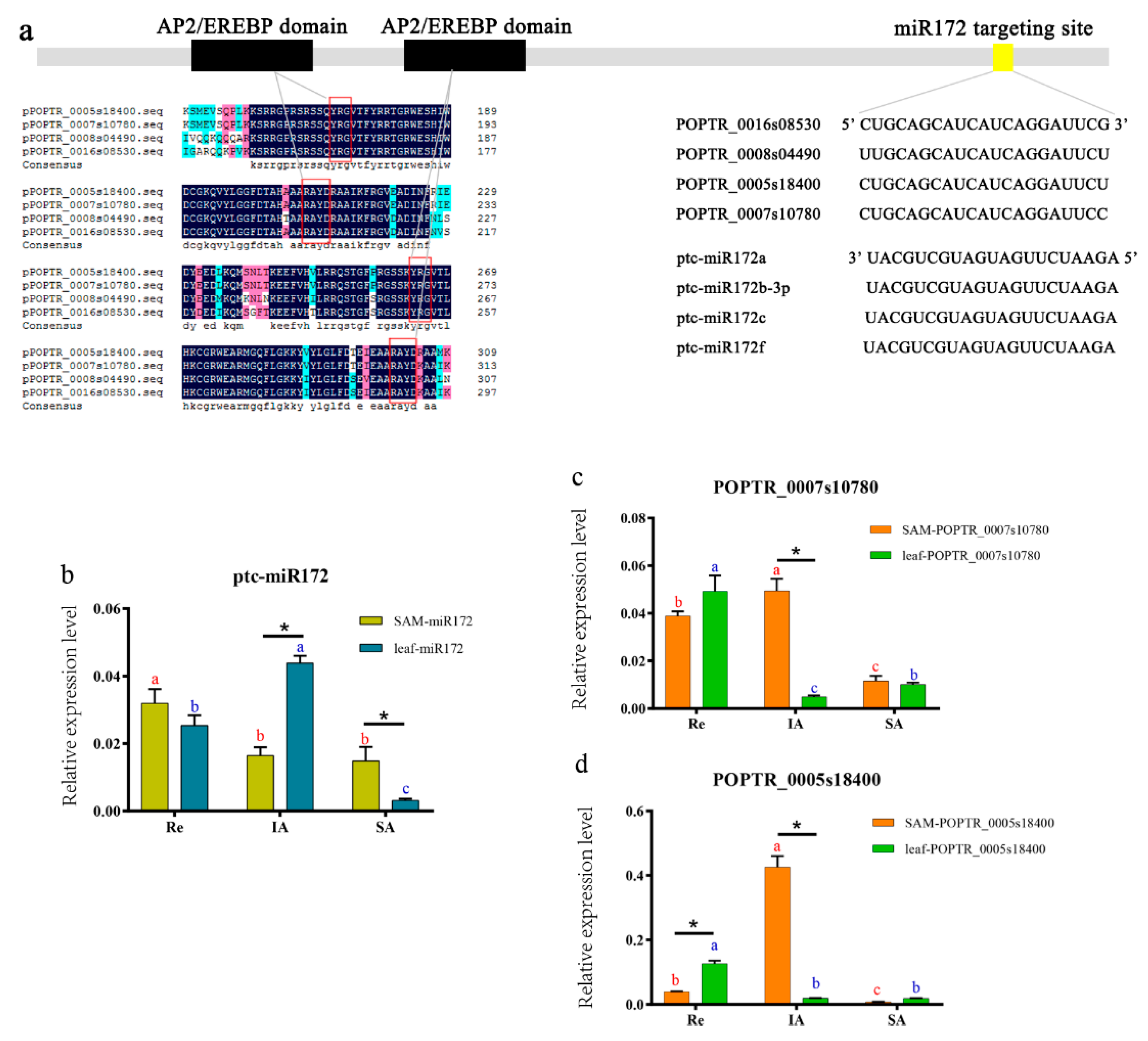

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Leaf 01 | Leaf 02 | SAM 01 | SAM 02 | |
|---|---|---|---|---|
| raw reads | 12,654,992 | 12,248,285 | 12,309,512 | 11,947,552 |
| clean reads | 9,195,827 | 8,775,153 | 6,782,666 | 8,067,503 |
| unique reads | 1,738,201 | 1,653,933 | 1,960,095 | 2,390,698 |
| rRNA | 607,501 | 659,392 | 545,748 | 602,901 |
| snRNA | 6966 | 6521 | 7805 | 9105 |
| snoRNA | 13,082 | 13,579 | 14,493 | 17,325 |
| tRNA | 13,490 | 13,548 | 15,334 | 17,450 |
| repeats | 171,272 | 135,780 | 216,642 | 244,514 |
| unannotated reads | 893,192 | 795,507 | 1,124,667 | 1,462,646 |
| mapped reads | 614,301 | 474,568 | 314,304 | 462,045 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, J.; Lu, W.; Lu, Z.; Ren, S.; Zhao, B.; Wang, L.; Teng, N.; Jin, B. Identification and Analysis of microRNAs in the SAM and Leaves of Populus tomentosa. Forests 2019, 10, 130. https://doi.org/10.3390/f10020130
Cui J, Lu W, Lu Z, Ren S, Zhao B, Wang L, Teng N, Jin B. Identification and Analysis of microRNAs in the SAM and Leaves of Populus tomentosa. Forests. 2019; 10(2):130. https://doi.org/10.3390/f10020130
Chicago/Turabian StyleCui, Jiawen, Weichao Lu, Zhaogeng Lu, Shixiong Ren, Beibei Zhao, Li Wang, Nianjun Teng, and Biao Jin. 2019. "Identification and Analysis of microRNAs in the SAM and Leaves of Populus tomentosa" Forests 10, no. 2: 130. https://doi.org/10.3390/f10020130
APA StyleCui, J., Lu, W., Lu, Z., Ren, S., Zhao, B., Wang, L., Teng, N., & Jin, B. (2019). Identification and Analysis of microRNAs in the SAM and Leaves of Populus tomentosa. Forests, 10(2), 130. https://doi.org/10.3390/f10020130