

A Novel Machine-Learning Approach to Predict Stress-Responsive Genes in Arabidopsis



Abstract
:1. Introduction
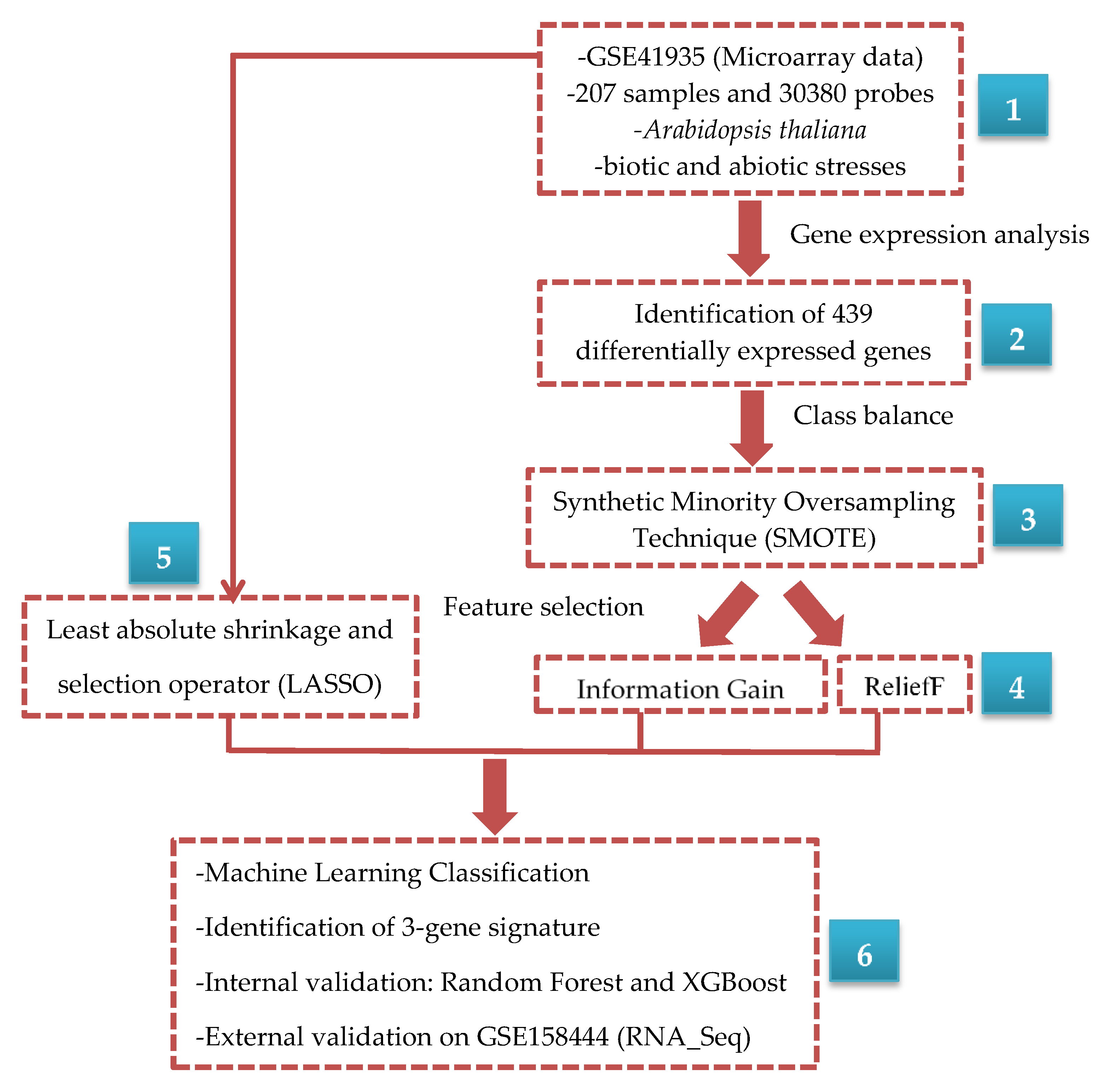
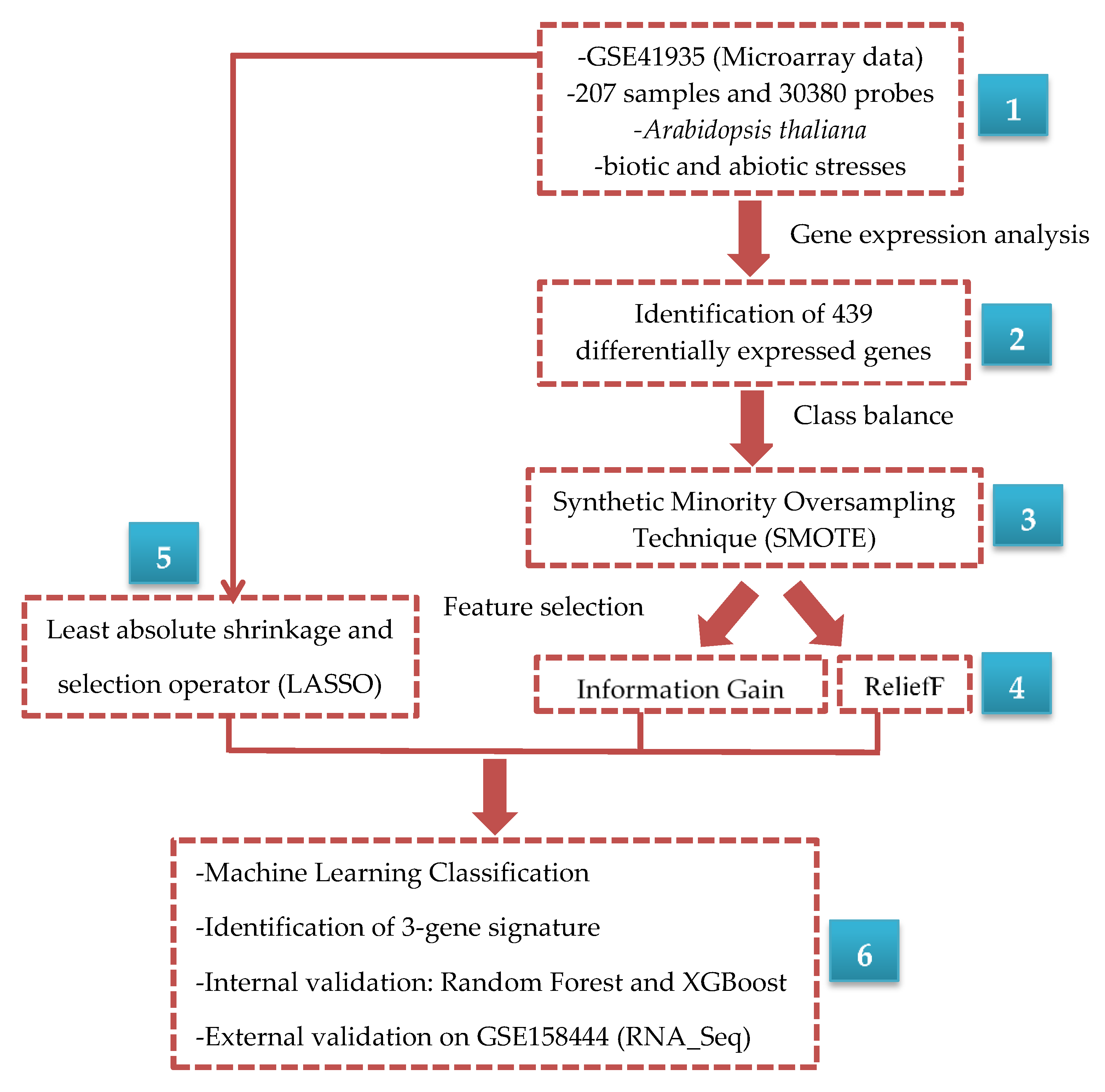
2. Materials and Methods
2.1. Microarray Data
2.2. Class Imbalance
2.3. Feature Selection Methods
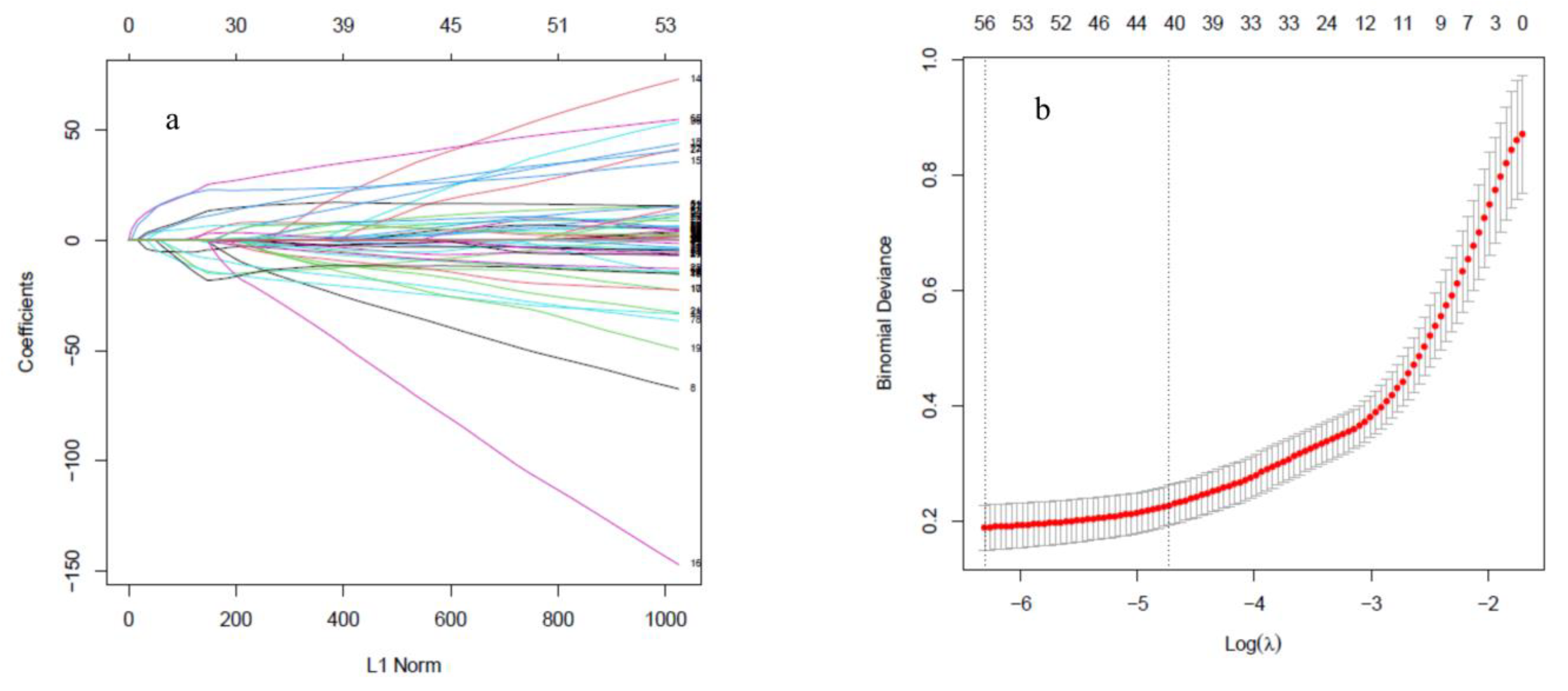
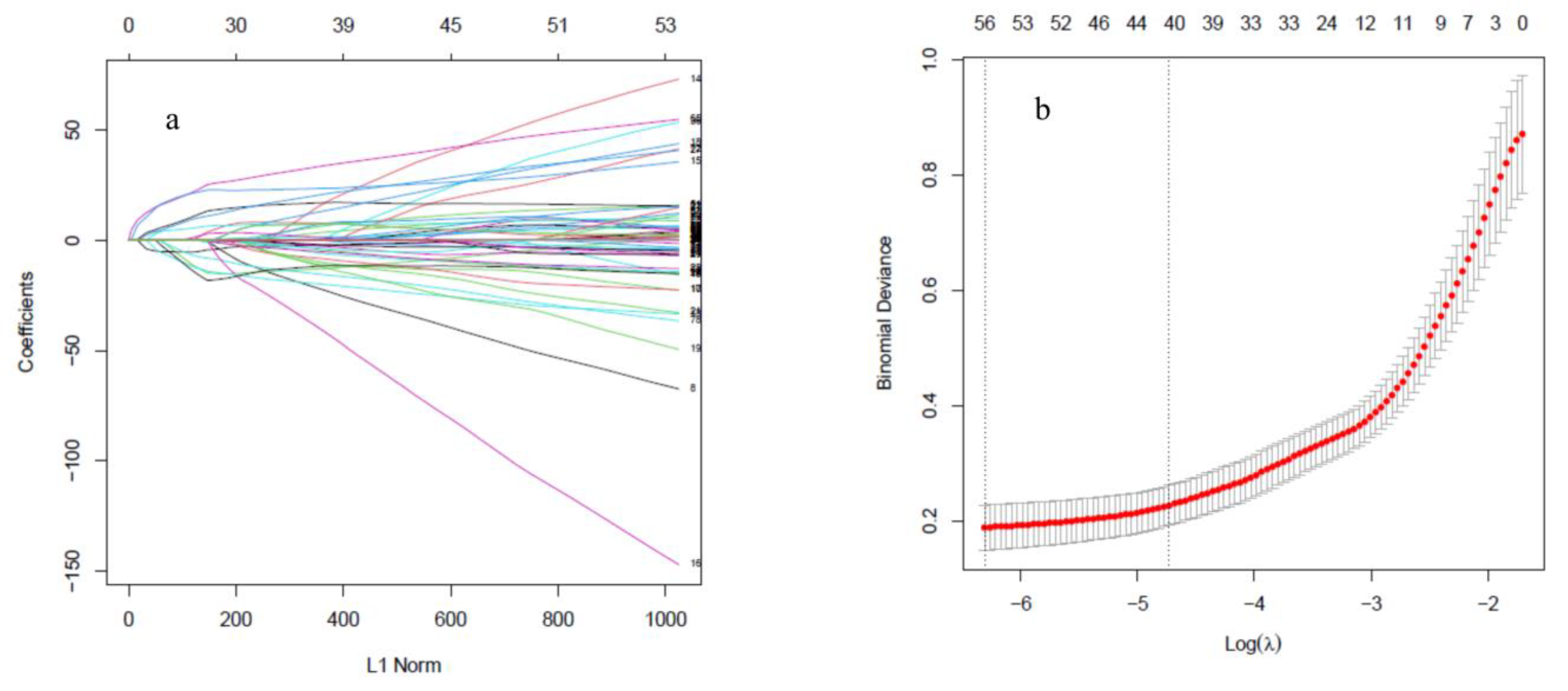
2.3.1. Least Absolute Shrinkage and Selection Operator
2.3.2. Information Gain
2.3.3. ReliefF
2.3.4. Identification and Validation of Gene Signature
2.3.5. Validation of Gene Signature Using External Dataset
2.4. Discrimination Analysis
3. Results and Discussion
3.1. Identification of Differentially Expressed Genes
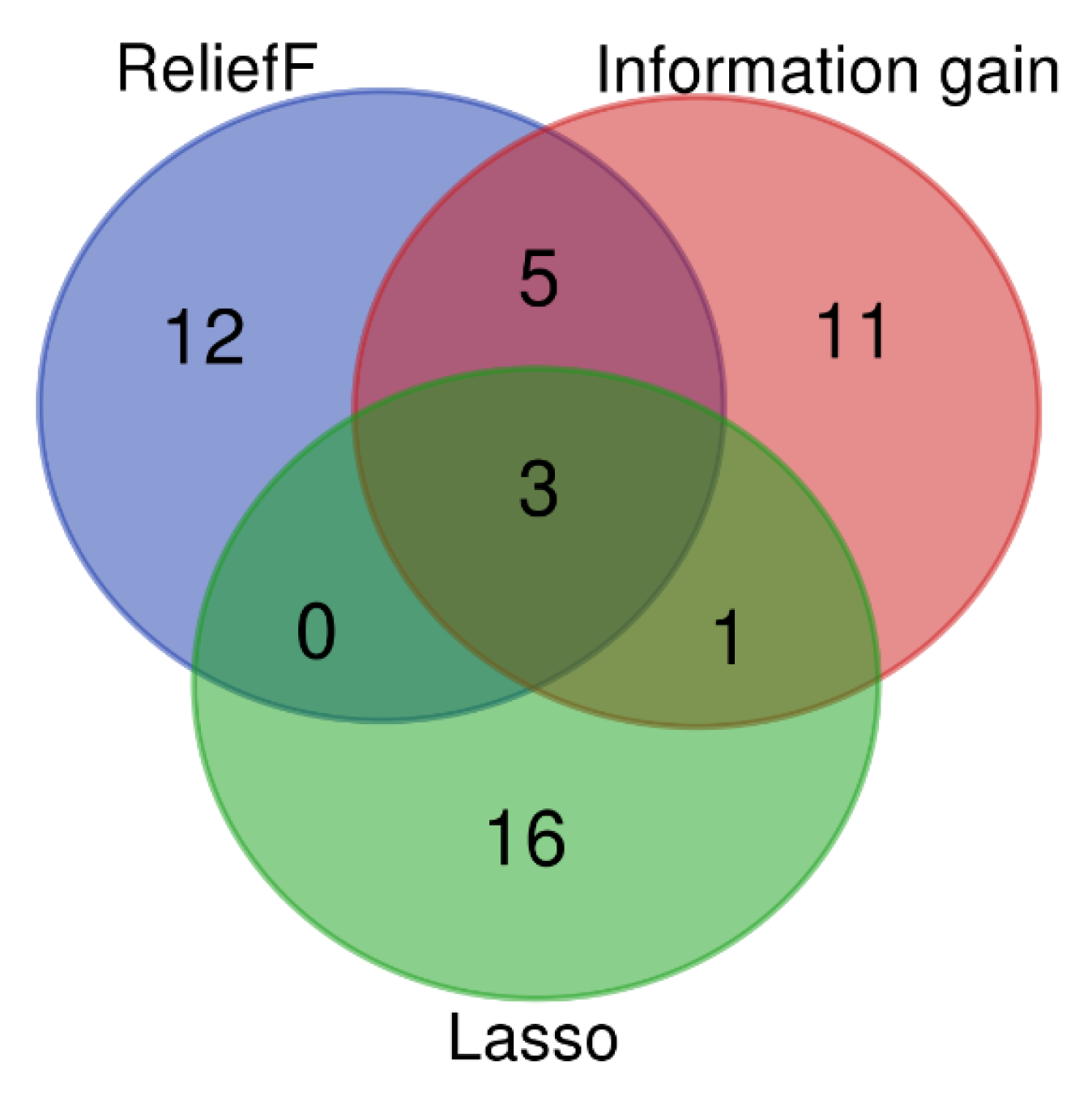
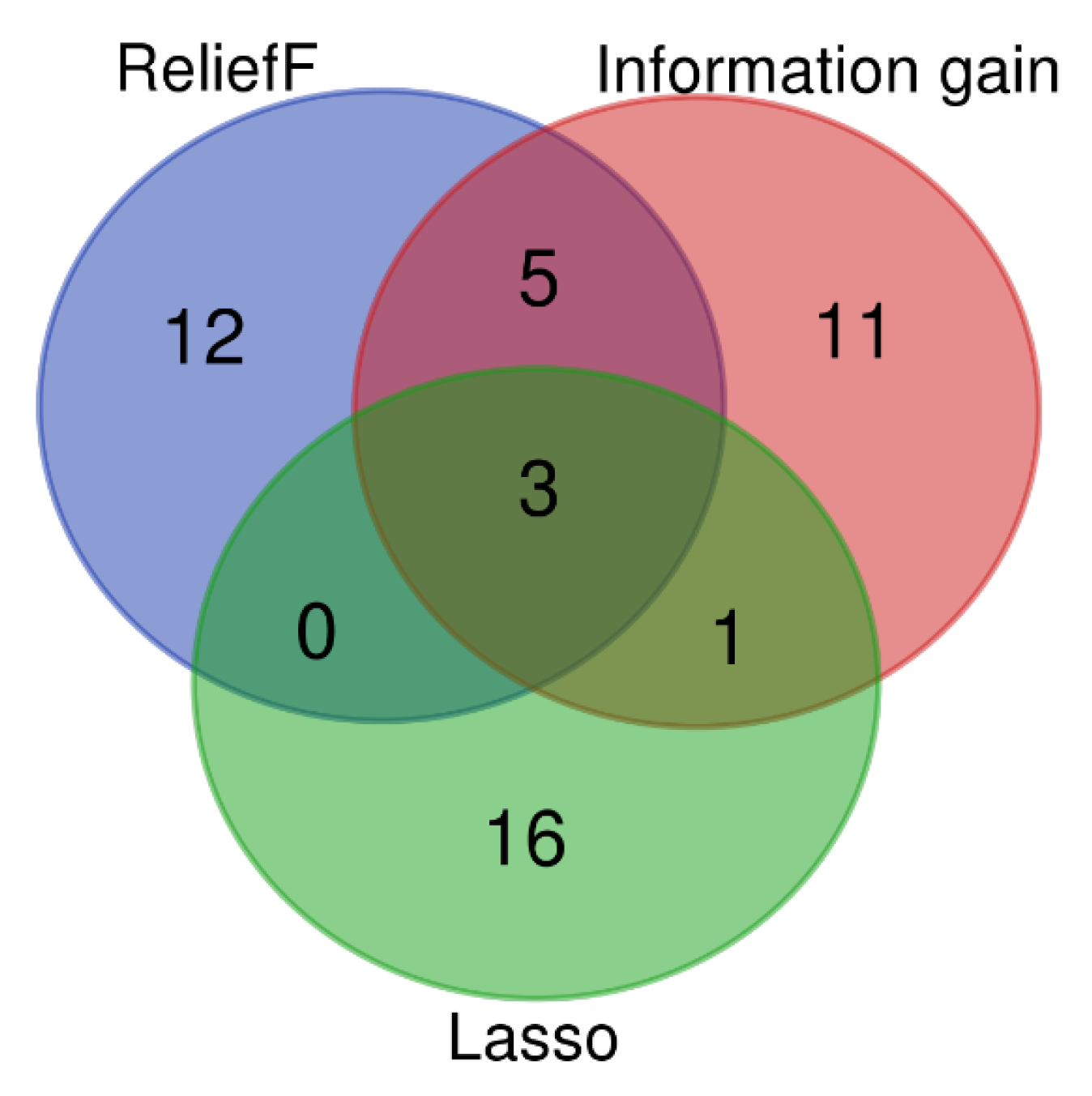
3.2. SMOTE Balancing and Feature Selection
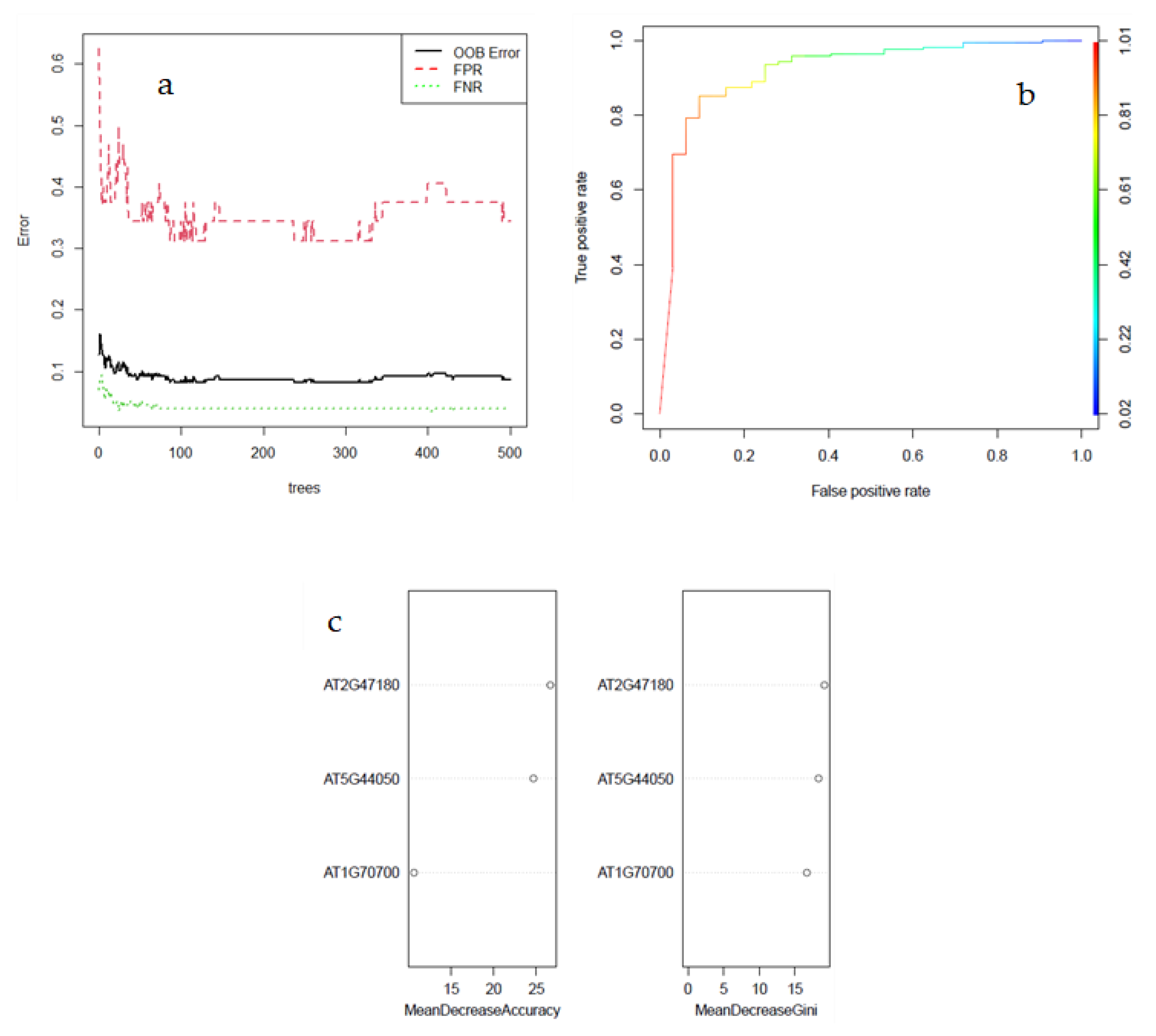
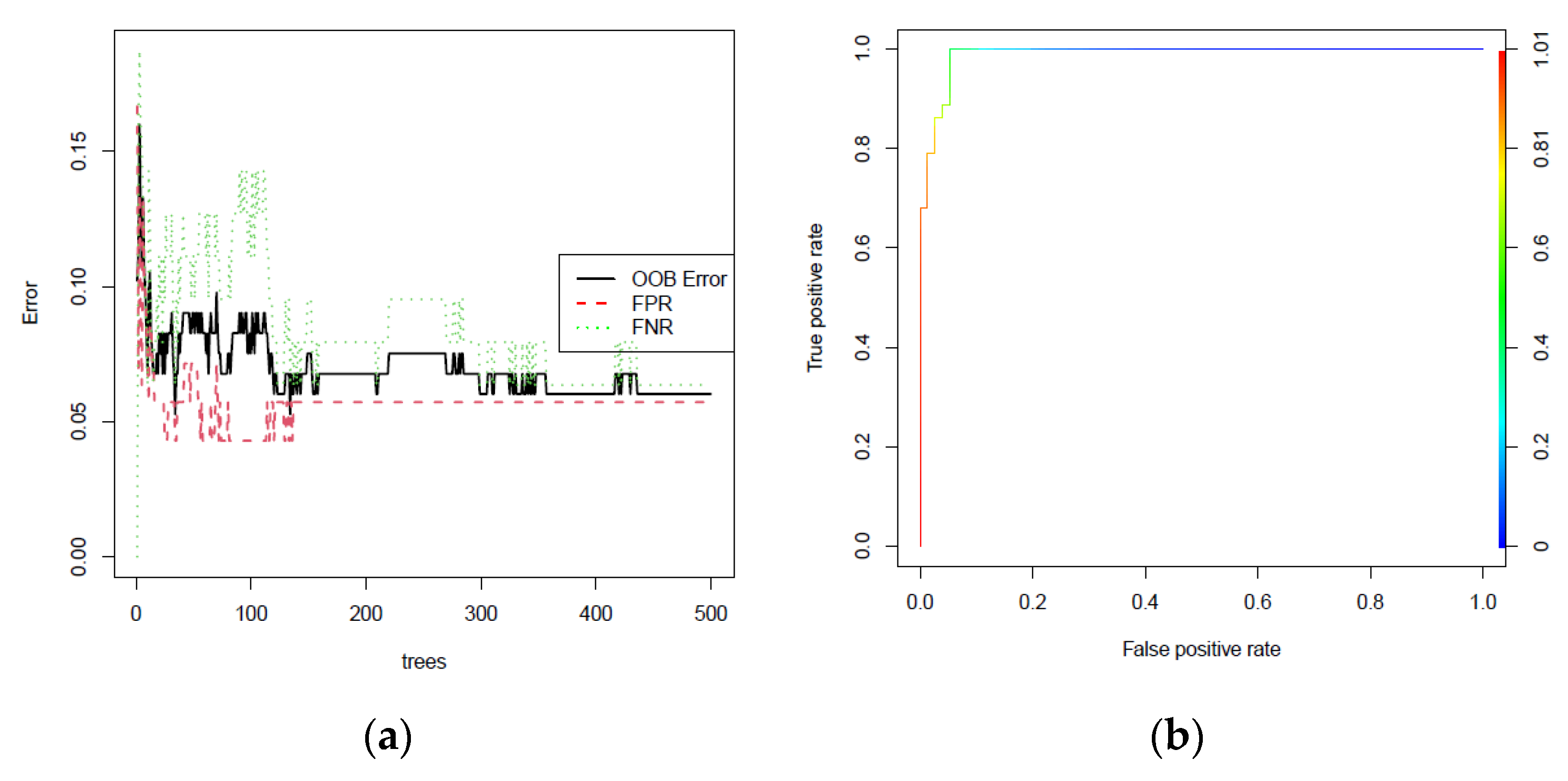
3.3. Machine Learning Classification
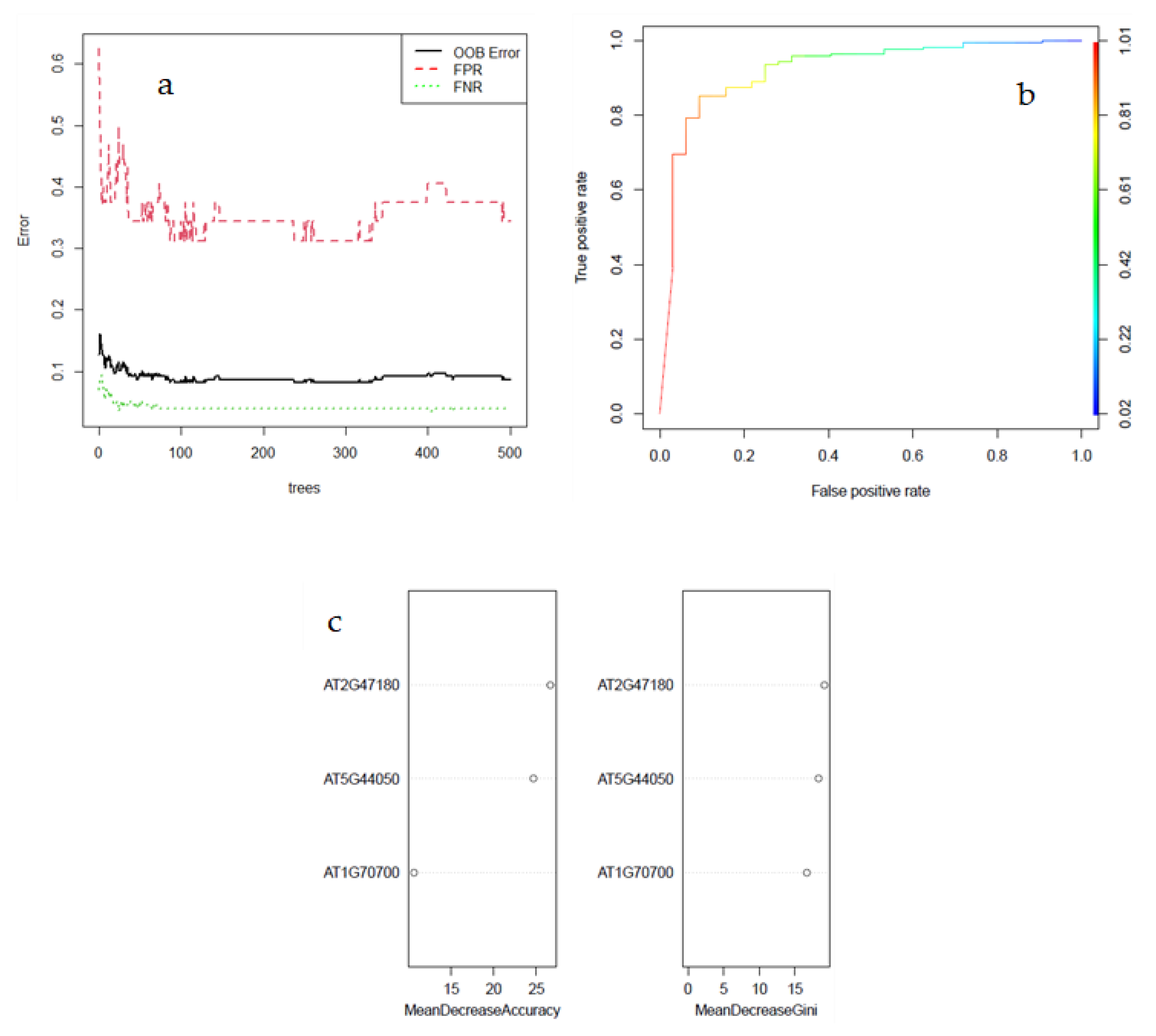
3.4. Selection and Validation of Key Genes
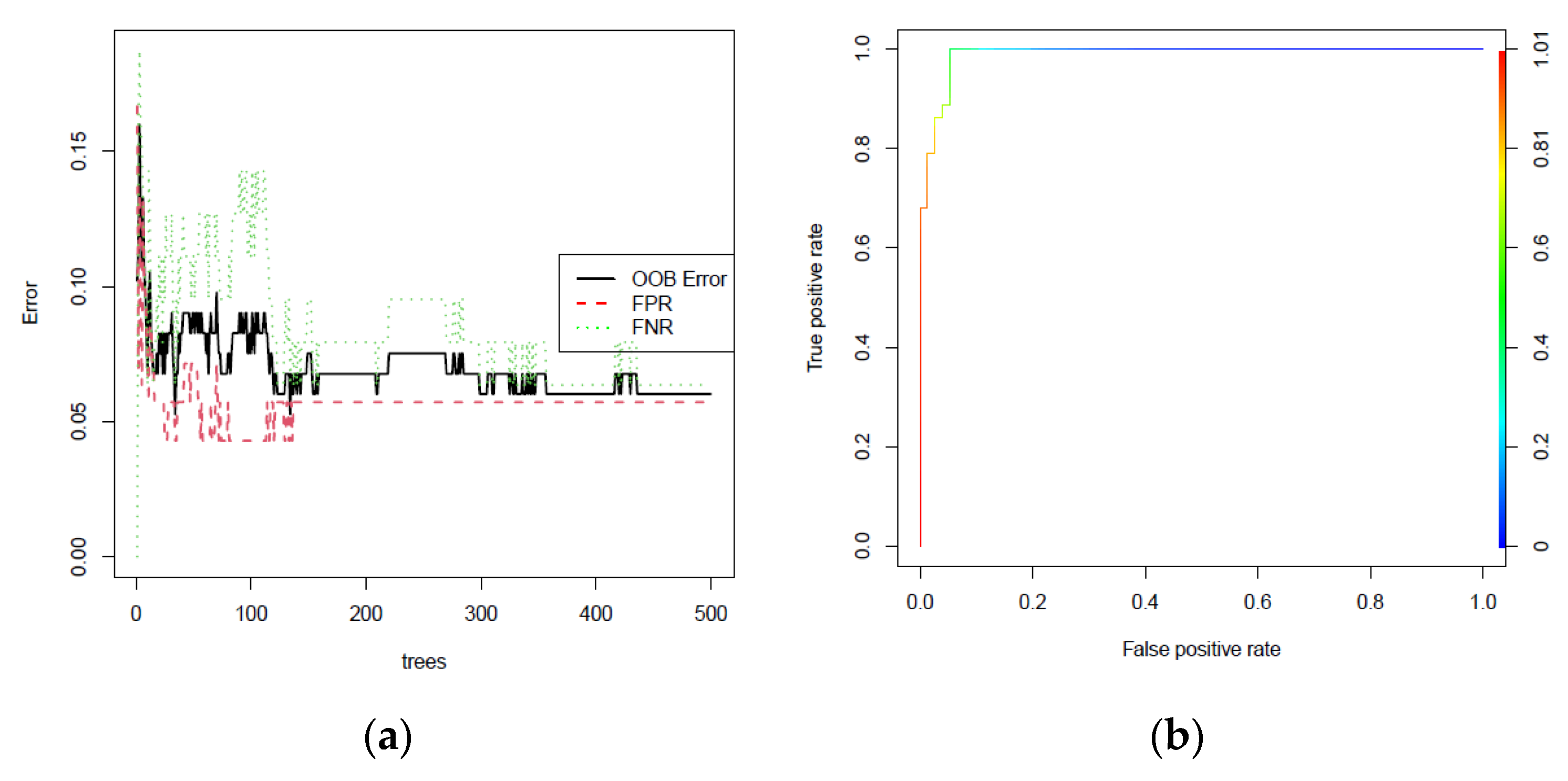
3.5. External Validation of Three-Gene Signature
3.6. Limitation and Future Works
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Barah, P.; Bones, A.M. Multidimensional approaches for studying plant defence against insects: From ecology to omics and synthetic biology. J. Exp. Bot. 2015, 66, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Mosa, K.A.; Ismail, A.; Helmy, M. (Eds.) Introduction to Plant Stresses. In Plant Stress Tolerance: An Integrated Omics Approach; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–19. [Google Scholar]
- Panigrahi, S.C.; Alam, M.S.; Mukhopadhyay, A. Chapter 12—Feature Selection and Analysis of Gene Expression Data Using Low-Dimensional Linear Programming. In Emerging Trends in Computational Biology, Bioinformatics, and Systems Biology; Tran, Q.N., Arabnia, H., Eds.; Morgan Kaufmann: Boston, MA, USA, 2015; pp. 235–264. [Google Scholar]
- Suzuki, N.; Rivero, R.M.; Shulaev, V.; Blumwald, E.; Mittler, R. Abiotic and biotic stress combinations. New Phytol. 2014, 203, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Matters, G.L.; Scandalios, J.G. Changes in plant gene expression during stress. Dev. Genet. 1986, 7, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Moreau, Y.; Aerts, S.; De Moor, B.; De Strooper, B.; Dabrowski, M. Comparison and meta-analysis of microarray data: From the bench to the computer desk. Trends Genet. 2003, 19, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Barah, P.; Jayavelu, N.D.; Sowdhamini, R.; Shameer, K.; Bones, A.M. Transcriptional regulatory networks in Arabidopsis thaliana during single and combined stresses. Nucleic Acids Res. 2016, 44, 3147–3164. [Google Scholar] [CrossRef]
- Coolen, S.; Proietti, S.; Hickman, R.; Olivas, N.H.D.; Huang, P.-P.; Van Verk, M.C.; Van Pelt, J.A.; Wittenberg, A.H.; De Vos, M.; Prins, M.; et al. Transcriptome dynamics of Arabidopsis during sequential biotic and abiotic stresses. Plant J. 2016, 86, 249–267. [Google Scholar] [CrossRef]
- Rasmussen, S.; Barah, P.; Suarez-Rodriguez, M.C.; Bressendorff, S.; Friis, P.; Costantino, P.; Bones, A.M.; Nielsen, H.B.; Mundy, J. Transcriptome Responses to Combinations of Stresses in Arabidopsis. Plant Physiol. 2013, 161, 1783–1794. [Google Scholar] [CrossRef]
- Wang, X.; Li, N.; Li, W.; Gao, X.; Cha, M.; Qin, L.; Liu, L. Advances in Transcriptomics in the Response to Stress in Plants. Glob. Med. Genet. 2020, 07, 30–34. [Google Scholar] [CrossRef]
- Mallik, S.; Zhao, Z. Identification of gene signatures from RNA-seq data using Pareto-optimal cluster algorithm. BMC Syst. Biol. 2018, 12, 126. [Google Scholar] [CrossRef]
- Khalid, S.; Khalil, T.; Nasreen, S. A survey of feature selection and feature extraction techniques in machine learning. In Proceedings of the Science and Information Conference (SAI), London, UK, 27–29 August 2014; pp. 372–378. [Google Scholar]
- Mahendran, N.; Vincent, P.M.D.R.; Srinivasan, K.; Chang, C.-Y. Machine Learning Based Computational Gene Selection Models: A Survey, Performance Evaluation, Open Issues, and Future Research Directions. Front. Genet. 2020, 11, 603808. [Google Scholar] [CrossRef]
- Du, Q.; Campbell, M.; Yu, H.; Liu, K.; Walia, H.; Zhang, Q.; Zhang, C. Network-based feature selection reveals substructures of gene modules responding to salt stress in rice. Plant Direct 2019, 3, e00154. [Google Scholar] [CrossRef]
- Prasetiyowati, M.I.; Maulidevi, N.U.; Surendro, K. Determining threshold value on information gain feature selection to increase speed and prediction accuracy of random forest. J. Big Data 2021, 8, 84. [Google Scholar] [CrossRef]
- Urbanowicz, R.J.; Meeker, M.; La Cava, W.; Olson, R.S.; Moore, J.H. Relief-based feature selection: Introduction and review. J. Biomed. Inform. 2018, 85, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Bechtold, U.; Penfold, C.A.; Jenkins, D.J.; Legaie, R.; Moore, J.D.; Lawson, T.; Matthews, J.S.; Vialet-Chabrand, S.R.; Baxter, L.; Subramaniam, S.; et al. Time-Series Transcriptomics Reveals That AGAMOUS-LIKE22 Affects Primary Metabolism and Developmental Processes in Drought-Stressed Arabidopsis. Plant Cell 2016, 28, 345–366. [Google Scholar] [CrossRef] [PubMed]
- Marais, D.L.D.; McKay, J.K.; Richards, J.H.; Sen, S.; Wayne, T.; Juenger, T.E. Physiological Genomics of Response to Soil Drying in Diverse Arabidopsis Accessions. Plant Cell 2012, 24, 893–914. [Google Scholar] [CrossRef]
- Parkinson, E.; Liberatore, F.; Watkins, W.J.; Andrews, R.; Edkins, S.; Hibbert, J.; Strunk, T.; Currie, A.; Ghazal, P. Gene filtering strategies for machine learning guided biomarker discovery using neonatal sepsis RNA-seq data. Front. Genet. 2023, 14, 1158352. [Google Scholar] [CrossRef]
- Blagus, R.; Lusa, L. SMOTE for high-dimensional class-imbalanced data. BMC Bioinform. 2013, 14, 106. [Google Scholar] [CrossRef]
- Bouckaert, R.R.; Frank, E.; Hall, M.; Kirkby, R.; Reutemann, P.; Seewald, A.; Scuse, D. WEKA Manual for Version 3-9-1; University of Waikato: Hamilton, New Zealand, 2016. [Google Scholar]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Tibshirani, R.J. The lasso problem and uniqueness. Electron. J. Stat. 2013, 7, 1456–1490. [Google Scholar] [CrossRef]
- Lai, C.-M.; Yeh, W.-C.; Chang, C.-Y. Gene selection using information gain and improved simplified swarm optimization. Neurocomputing 2016, 218, 331–338. [Google Scholar] [CrossRef]
- Hall, M.A.; Smith, L.A. Practical feature subset selection for machine learning. In Computer Science ’98 Proceedings of the 21st Australasian Computer Science Conference ACSC’98, Perth, Australia, 4–6 February 1998; McDonald, C., Ed.; Springer: Berlin, Germany, 1998; pp. 181–191. [Google Scholar]
- Robnik-Šikonja, M.; Kononenko, I. Theoretical and Empirical Analysis of ReliefF and RReliefF. Mach. Learn. 2003, 53, 23–69. [Google Scholar] [CrossRef]
- Sing, T.; Sander, O.; Beerenwinkel, N.; Lengauer, T. ROCR: Visualizing classifier performance in R. Bioinformatics 2005, 21, 3940–3941. [Google Scholar] [CrossRef] [PubMed]
- James, G.; Witten, D.; Hastie, T.; Tibshirani, R. An Introduction to Statistical Learning; Springer: New York, NY, USA, 2013. [Google Scholar]
- Kursa, M.B.; Rudnicki, W.R. Feature Selection with the Boruta Package. J. Stat. Softw. 2010, 36, 1–13. [Google Scholar] [CrossRef]
- Bonnot, T.; Nagel, D.H. Time of the day prioritizes the pool of translating mRNAs in response to heat stress. Plant Cell 2021, 33, 2164–2182. [Google Scholar] [CrossRef] [PubMed]
- Biau, G.; Scornet, E. A random forest guided tour. TEST 2016, 25, 197–227. [Google Scholar] [CrossRef]
- Tabl, A.A.; Alkhateeb, A.; ElMaraghy, W.; Rueda, L.; Ngom, A. A Machine Learning Approach for Identifying Gene Biomarkers Guiding the Treatment of Breast Cancer. Front. Genet. 2019, 10, 256. [Google Scholar] [CrossRef]
- Li, Q.; Yang, H.; Wang, P.; Liu, X.; Lv, K.; Ye, M. XGBoost-based and tumor-immune characterized gene signature for the prediction of metastatic status in breast cancer. J. Transl. Med. 2022, 20, 177. [Google Scholar] [CrossRef]
- Tiwari, M.; Sharma, D.; Singh, M.; Tripathi, R.D.; Trivedi, P.K. Expression of OsMATE1 and OsMATE2 alters development, stress responses and pathogen susceptibility in Arabidopsis. Sci. Rep. 2014, 4, 3964. [Google Scholar] [CrossRef]
- Magwanga, R.O.; Lu, P.; Kirungu, J.N.; Lu, H.; Wang, X.; Cai, X.; Zhou, Z.; Zhang, Z.; Salih, H.; Wang, K.; et al. Characterization of the late embryogenesis abundant (LEA) proteins family and their role in drought stress tolerance in upland cotton. BMC Genet. 2018, 19, 6. [Google Scholar] [CrossRef]
- Shah, I.H.; Manzoor, M.A.; Sabir, I.A.; Ashraf, M.; Haq, F.; Arif, S.; Abdullah, M.; Niu, Q.; Zhang, Y. Genome-wide identification and comparative analysis of MATE gene family in Cucurbitaceae species and their regulatory role in melon (Cucumis melo) under salt stress. Hortic. Environ. Biotechnol. 2022, 63, 595–612. [Google Scholar] [CrossRef]
- Taji, T.; Ohsumi, C.; Iuchi, S.; Seki, M.; Kasuga, M.; Kobayashi, M.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Important roles of drought- and cold-inducible genes for galactinol synthase in stress tolerance in Arabidopsis thaliana. Plant J. 2002, 29, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Janse van Rensburg, H.C. The Arabidopsis GolS1 Promotor as a Potential Biosensor for Heat Stress and Fungal Infection? Master’s Thesis, Stellenbosch University, Stellenbosch, South Africa, 2016. [Google Scholar]
- Kahraman, N.; Pehlivan, N. Harboured cation/proton antiporters modulate stress response to integrated heat and salt via up-regulating KIN1 and GOLS1 in double transgenic Arabidopsis. Funct. Plant Biol. 2022, 49, 1070–1084. [Google Scholar] [CrossRef] [PubMed]
- Chini, A.; Ben-Romdhane, W.; Hassairi, A.; Aboul-Soud, M.A.M. Identification of TIFY/JAZ family genes in Solanum lycopersicum and their regulation in response to abiotic stresses. PLoS ONE 2017, 12, e0177381. [Google Scholar] [CrossRef] [PubMed]
- Ebel, C.; BenFeki, A.; Hanin, M.; Solano, R.; Chini, A. Characterization of wheat (Triticum aestivum) TIFY family and role of Triticum Durum TdTIFY11a in salt stress tolerance. PLoS ONE 2018, 13, e0200566. [Google Scholar] [CrossRef]
- Ye, H.; Du, H.; Tang, N.; Li, X.; Xiong, L. Identification and expression profiling analysis of TIFY family genes involved in stress and phytohormone responses in rice. Plant Mol. Biol. 2009, 71, 291–305. [Google Scholar] [CrossRef]
- Erkinbaev, C.; Nadimi, M.; Paliwal, J. A unified heuristic approach to simultaneously detect fusarium and ergot damage in wheat. Meas. Food 2022, 7, 100043. [Google Scholar] [CrossRef]
- Nadimi, M.; Hawley, E.; Liu, J.; Hildebrand, K.; Sopiwnyk, E.; Paliwal, J. Enhancing traceability of wheat quality through the supply chain. Compr. Rev. Food Sci. Food Saf. 2023, 22, 2495–2522. [Google Scholar] [CrossRef] [PubMed]
- Nadimi, M.; Loewen, G.; Bhowmik, P.; Paliwal, J. Effect of Laser Biostimulation on Germination of Sub-Optimally Stored Flaxseeds (Linum usitatissimum). Sustainability 2022, 14, 12183. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classifier | Parameter Adjustment |
|---|---|
| BayesNet | debug: False; estimator: SimpleEstimator; searchAlgorithms: K2; useADTree: False |
| Logistic | debug: False; maxIts: −1; ridge: 10−8 |
| Multilayer Perceptron | debug: False; hiddenLayers: a; learningRate: 0.3; momentum: 0.2; normalizeNumericClass: True; NominalToBinaryFilter: True; normalizeAttributes: True; reset: True; seed: 0; trainingTime: 500; validationThreshold: 20 |
| SMO | buildLogisticModels: False; c: 1.0; checksTurnedOff: False; debug: False; epsilon: 10−12; filterType: Normalize training data; kernel: polyKernel, numFolds: −1; randomSeed: 1; tolerance Parameter: 0.0010 |
| Random Forest | debug: False; maxDepth: 0; numFeatures: 0; numTrees: 10; seed: 1 |
| XGBoost | max_depth = 2, eta = 1, nround = 2, set.seed = 1 |
| Classifier | Predicted Class | Actual Class | Accuracy (%) | TP Rate | FP Rate | Precision | Recall | F-Measure | MCC | ROC Area | PRC Area | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Control | Stress | |||||||||||
| BayesNet | 155 | 5 | control | 96.42 | 0.964 | 0.035 | 0.964 | 0.964 | 0.964 | 0.928 | 0.993 | 0.994 |
| 7 | 168 | stress | ||||||||||
| Logistic | 155 | 5 | control | 95.22 | 0.952 | 0.046 | 0.953 | 0.952 | 0.952 | 0.905 | 0.987 | 0.979 |
| 11 | 164 | stress | ||||||||||
| Multilayer Perceptron | 158 | 2 | control | 96.42 | 0.964 | 0.034 | 0.965 | 0.964 | 0.964 | 0.929 | 0.994 | 0.994 |
| 10 | 165 | stress | ||||||||||
| SMO | 154 | 6 | control | 95.22 | 0.952 | 0.047 | 0.953 | 0.952 | 0.952 | 0.905 | 0.953 | 0.931 |
| 10 | 165 | stress | ||||||||||
| Random Forest | 157 | 3 | control | 97.91 | 0.979 | 0.021 | 0.979 | 0.979 | 0.979 | 0.958 | 0.993 | 0.999 |
| 4 | 171 | stress | ||||||||||
| Classifier | Predicted Class | Actual Class | Accuracy (%) | TP Rate | FP Rate | Precision | Recall | F-Measure | MCC | ROC Area | PRC Area | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Control | Stress | |||||||||||
| BayesNet | 152 | 8 | control | 94.93 | 0.949 | 0.051 | 0.949 | 0.949 | 0.949 | 0.898 | 0.993 | 0.993 |
| 9 | 166 | stress | ||||||||||
| Logistic | 156 | 4 | control | 95.52 | 0.955 | 0.043 | 0.956 | 0.955 | 0.955 | 0.911 | 0.976 | 0.965 |
| 11 | 164 | stress | ||||||||||
| Multilayer Perceptron | 158 | 2 | control | 98.51 | 0.985 | 0.015 | 0.985 | 0.985 | 0.985 | 0.97 | 0.998 | 0.998 |
| 3 | 172 | stress | ||||||||||
| SMO | 159 | 1 | control | 97.61 | 0.976 | 0.022 | 0.977 | 0.976 | 0.976 | 0.953 | 0.977 | 0.965 |
| 7 | 168 | stress | ||||||||||
| Random Forest | 160 | 0 | control | 98.51 | 0.985 | 0.014 | 0.986 | 0.985 | 0.985 | 0.971 | 0.998 | 0.999 |
| 5 | 170 | stress | ||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nazari, L.; Ghotbi, V.; Nadimi, M.; Paliwal, J. A Novel Machine-Learning Approach to Predict Stress-Responsive Genes in Arabidopsis. Algorithms 2023, 16, 407. https://doi.org/10.3390/a16090407
Nazari L, Ghotbi V, Nadimi M, Paliwal J. A Novel Machine-Learning Approach to Predict Stress-Responsive Genes in Arabidopsis. Algorithms. 2023; 16(9):407. https://doi.org/10.3390/a16090407
Chicago/Turabian StyleNazari, Leyla, Vida Ghotbi, Mohammad Nadimi, and Jitendra Paliwal. 2023. "A Novel Machine-Learning Approach to Predict Stress-Responsive Genes in Arabidopsis" Algorithms 16, no. 9: 407. https://doi.org/10.3390/a16090407
APA StyleNazari, L., Ghotbi, V., Nadimi, M., & Paliwal, J. (2023). A Novel Machine-Learning Approach to Predict Stress-Responsive Genes in Arabidopsis. Algorithms, 16(9), 407. https://doi.org/10.3390/a16090407