


Optimized Tensor Decomposition and Principal Component Analysis Outperforming State-of-the-Art Methods When Analyzing Histone Modification Chromatin Immunoprecipitation Profiles



Abstract
1. Introduction
- In contrast to variable gene expression, histone modification is more stable [4]; thus, it may be used to characterize the state of the genome.
- In contrast to DNA methylation, which has only two states, methylated or not, histones are modified in various ways. Thus, histone modification is related to a more detailed functionality of the genome [5].
- Since histone modification may activate or suppress transcription, combinations of various histone modifications may have more complicated transcriptional roles [6].
- To identify histone modification throughout the genome, the detection of antibody binding to the entire genome is required [7]. In contrast, gene expression can be detected only if transcription start sites are sequenced.
- In contrast to gene expression that can be measured only when exons are considered or DNA methylation that is meaningful only if promoter regions are considered, specific regions of the genome cannot be used due to limited knowledge of the position-specific functionality of histone modification [8].
- Because of these two reasons, HTS for histone modification requires more depth, which is both time-consuming and expensive [9].
- Similarly, the number of datasets and computational resources required to identify histone modification is greater than that required to measure gene expression and DNA methylation [10].
2. Materials and Methods
2.1. Histone Modification Profiles
2.2. Histone Modification Profile Preprocessing
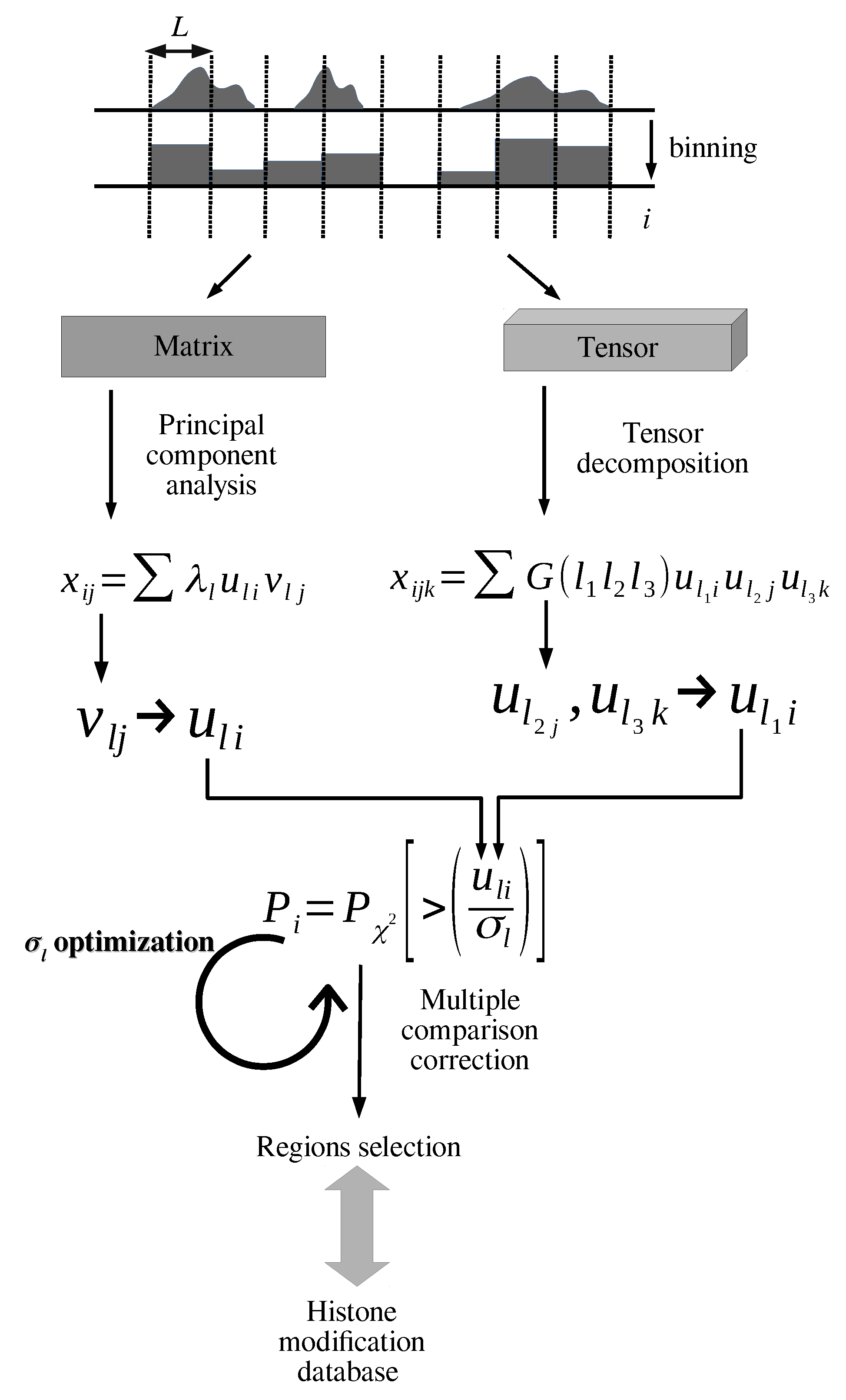
2.3. PCA-Based Unsupervised FE with Optimized SD
2.4. TD-Based Unsupervised FE with Optimized SD
2.5. Performance Evaluation by Enrichr
2.6. Methods for Comparison
- The method must accept bed or bigWig file formats as input, since the proposed method cannot accept the sam/bam format as input, which is used by the most popular methods.
- The method must be stand-alone (not web-based).
- The method must be performed on the Linux platform.
- The method must be implemented as free (open) software.
3. Results
3.1. GSE24850
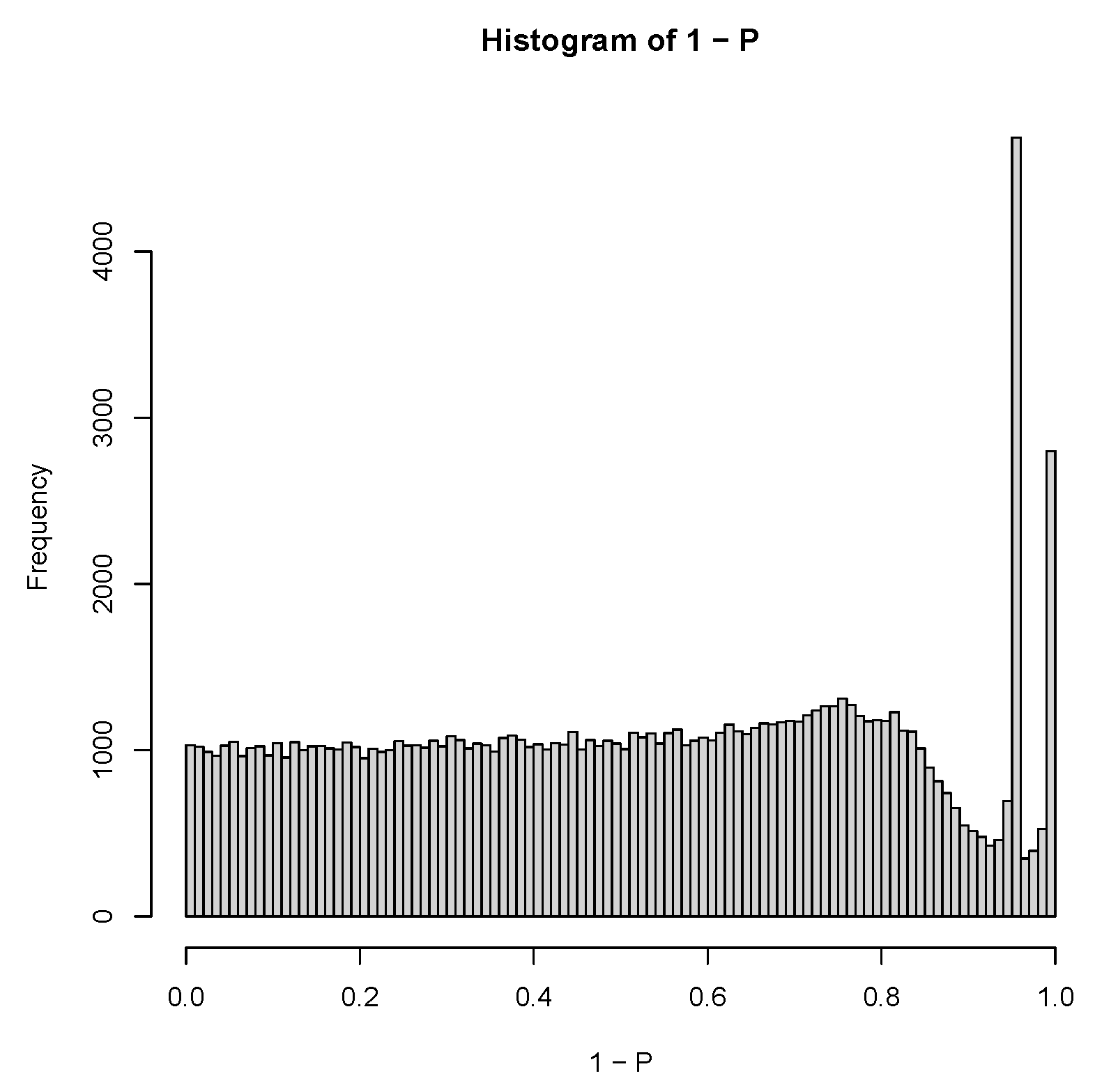
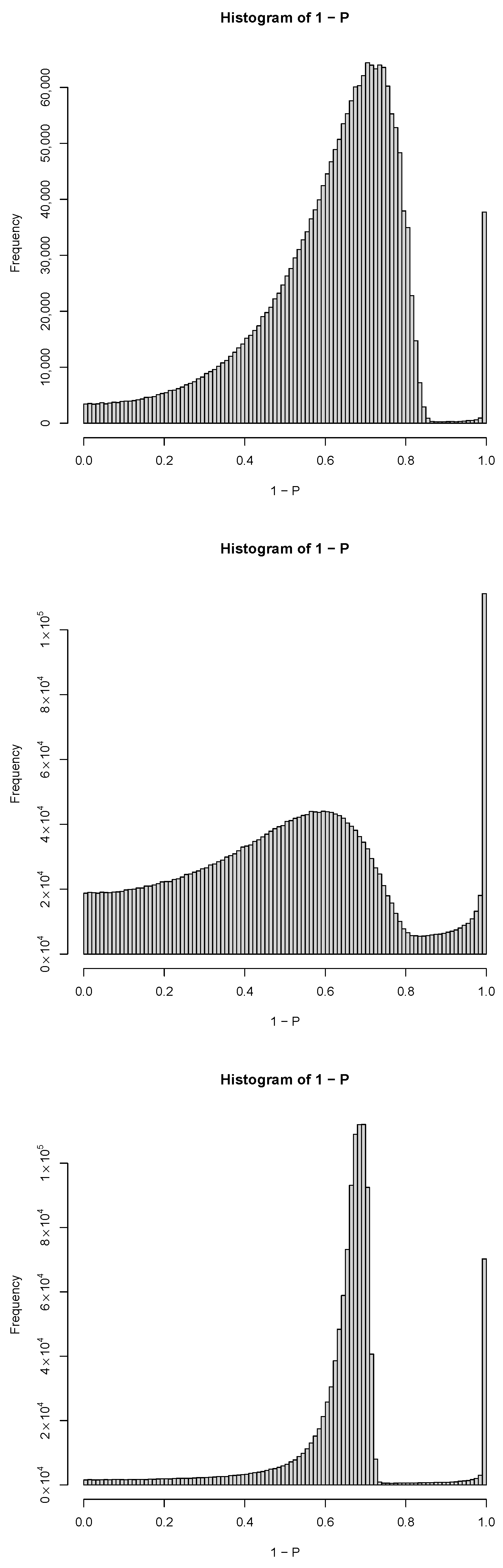
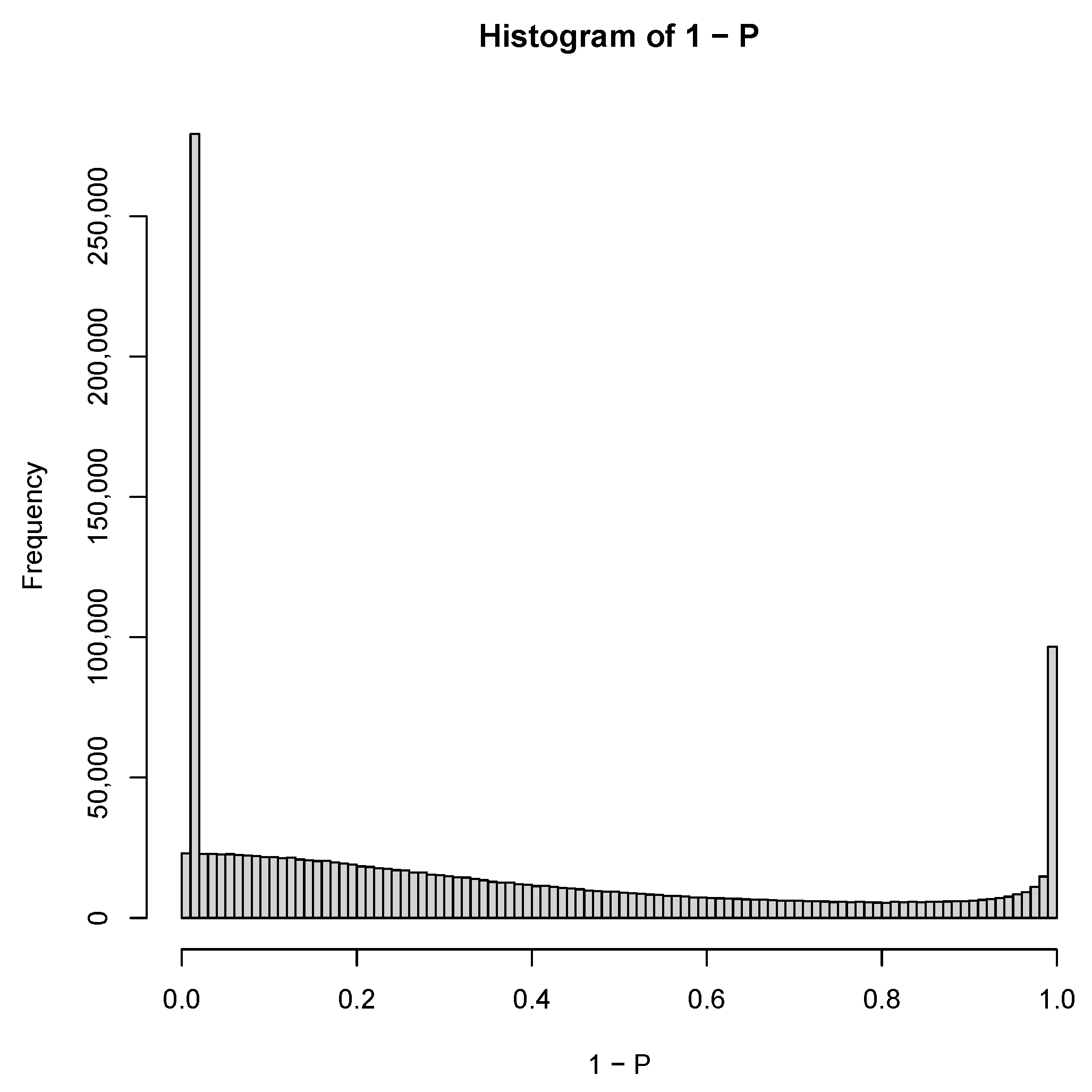
3.1.1. PCA-Based Unsupervised FE with Optimized SD
3.1.2. Comparisons with State-of-the-Art Methods
MOSAiCS
DFilter
F-Seq2
HOMER
RSEG
3.2. Histone Modification Other than H3K9me3
3.2.1. GSE159075
3.2.2. GSE74055
3.2.3. GSE124690
3.2.4. GSE188173
3.2.5. GSE159022
3.2.6. GSE168971
3.2.7. GSE159411
3.2.8. GSE181596
3.3. TD-Based Unsupervised FE with Optimized SD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nakato, R.; Sakata, T. Methods for ChIP-seq analysis: A practical workflow and advanced applications. Methods 2021, 187, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 2002, 12, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Gruppuso, P.A.; Boylan, J.M.; Zabala, V.; Neretti, N.; Abshiru, N.A.; Sikora, J.W.; Doud, E.H.; Camarillo, J.M.; Thomas, P.M.; Kelleher, N.L.; et al. Stability of histone post-translational modifications in samples derived from liver tissue and primary hepatic cells. PLoS ONE 2018, 13, e0203351. [Google Scholar] [CrossRef]
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone post-translational modifications—Cause and consequence of genome function. Nat. Rev. Genet. 2022, 23, 563–580. [Google Scholar] [CrossRef]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications - writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef]
- Bock, I.; Dhayalan, A.; Kudithipudi, S.; Brandt, O.; Rathert, P.; Jeltsch, A. Detailed specificity analysis of antibodies binding to modified histone tails with peptide arrays. Epigenetics 2011, 6, 256–263. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, F.; van Steensel, B. Histone modifications: From genome-wide maps to functional insights. Genome Biol. 2005, 6, 113. [Google Scholar] [CrossRef] [PubMed][Green Version]
- O’Geen, H.; Echipare, L.; Farnham, P.J. Using ChIP-Seq Technology to Generate High-Resolution Profiles of Histone Modifications. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2011; pp. 265–286. [Google Scholar] [CrossRef]
- Shah, S.G.; Mandloi, T.; Kunte, P.; Natu, A.; Rashid, M.; Reddy, D.; Gadewal, N.; Gupta, S. HISTome2: A database of histone proteins, modifiers for multiple organisms and epidrugs. Epigene. Chromatin 2020, 13, 31. [Google Scholar] [CrossRef]
- Thomas, R.; Thomas, S.; Holloway, A.K.; Pollard, K.S. Features that define the best ChIP-seq peak calling algorithms. Briefings Bioinform. 2016, 18, 441–450. [Google Scholar] [CrossRef]
- Flensburg, C.; Kinkel, S.A.; Keniry, A.; Blewitt, M.E.; Oshlack, A. A comparison of control samples for ChIP-seq of histone modifications. Front. Genet. 2014, 5, 329. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, Y.h.; Turki, T. Adapted tensor decomposition and PCA based unsupervised feature extraction select more biologically reasonable differentially expressed genes than conventional methods. Sci. Rep. 2022, 12, 17438. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, Y.H.; Turki, T. Principal component analysis- and tensor decomposition-based unsupervised feature extraction to select more suitable differentially methylated cytosines: Optimization of standard deviation versus state-of-the-art methods. Genomics 2023, 115, 110577. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Maze, I.; Feng, J.; Wilkinson, M.B.; Sun, H.; Shen, L.; Nestler, E.J. Cocaine dynamically regulates heterochromatin and repetitive element unsilencing in nucleus accumbens. Proc. Natl. Acad. Sci. USA 2011, 108, 3035–3040. [Google Scholar] [CrossRef] [PubMed]
- Kanki, Y.; Muramatsu, M.; Miyamura, Y.; Kikuchi, K.; Higashijima, Y.; Nakaki, R.; Suehiro, J.; Sasaki, Y.; Kubota, Y.; Koseki, H.; et al. Bivalent-histone-marked immediate-early gene regulation is vital for VEGF-responsive angiogenesis. Cell Rep. 2022, 38, 110332. [Google Scholar] [CrossRef]
- Yan, J.; Chen, S.A.A.; Local, A.; Liu, T.; Qiu, Y.; Dorighi, K.M.; Preissl, S.; Rivera, C.M.; Wang, C.; Ye, Z.; et al. Histone H3 lysine 4 monomethylation modulates long-range chromatin interactions at enhancers. Cell Res. 2018, 28, 204–220. [Google Scholar] [CrossRef]
- Kaya-Okur, H.S.; Wu, S.J.; Codomo, C.A.; Pledger, E.S.; Bryson, T.D.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019, 10, 1930. [Google Scholar] [CrossRef]
- Wei, X.; Lienhard, M.; Murgai, A.; Franke, J.; Pöhle-Kronawitter, S.; Kotsaris, G.; Wu, H.; Börno, S.; Timmermann, B.; Glauben, R.; et al. Neurofibromin 1 controls metabolic balance and Notch-dependent quiescence of juvenile myogenic progenitors. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sarode, G.V.; Neier, K.; Shibata, N.M.; Shen, Y.; Goncharov, D.A.; Goncharova, E.A.; Mazi, T.A.; Joshi, N.; Settles, M.L.; LaSalle, J.M.; et al. Wilson Disease: Intersecting DNA Methylation and Histone Acetylation Regulation of Gene Expression in a Mouse Model of Hepatic Copper Accumulation. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1457–1477. [Google Scholar] [CrossRef]
- Gonzalez-Teran, B.; Pittman, M.; Felix, F.; Thomas, R.; Richmond-Buccola, D.; Hüttenhain, R.; Choudhary, K.; Moroni, E.; Costa, M.W.; Huang, Y.; et al. Transcription factor protein interactomes reveal genetic determinants in heart disease. Cell 2022, 185, 794–814.e30. [Google Scholar] [CrossRef]
- Yuan, H.; Suzuki, S.; Terui, H.; Hirata-Tsuchiya, S.; Nemoto, E.; Yamasaki, K.; Saito, M.; Shiba, H.; Aiba, S.; Yamada, S. Loss of IκBζ Drives Dentin Formation via Altered H3K4me3 Status. J. Dent. Res. 2022, 101, 220345221075968. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, Y.H. Unsupervised Feature Extraction Applied to Bioinformatics; Springer International Publishing: Berlin/Heidelberg, Germany, 2020. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2008, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2008, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Chung, D.; Liang, K.; Keleş, S. Statistical Analysis of ChIP-seq Data with MOSAiCS. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2013; pp. 193–212. [Google Scholar] [CrossRef]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Kumar, V.; Muratani, M.; Rayan, N.A.; Kraus, P.; Lufkin, T.; Ng, H.H.; Prabhakar, S. Uniform, optimal signal processing of mapped deep-sequencing data. Nat. Biotechnol. 2013, 31, 615–622. [Google Scholar] [CrossRef]
- Zhao, N.; Boyle, A.P. F-Seq2: Improving the feature density based peak caller with dynamic statistics. NAR Genom. Bioinform. 2021, 3, lqab012. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Song, Q.; Smith, A.D. Identifying dispersed epigenomic domains from ChIP-Seq data. Bioinformatics 2011, 27, 870–871. [Google Scholar] [CrossRef]
- Morales, J.; Pujar, S.; Loveland, J.E.; Astashyn, A.; Bennett, R.; Berry, A.; Cox, E.; Davidson, C.; Ermolaeva, O.; Farrell, C.M.; et al. A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature 2022, 604, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Kim, Y.W.; Kang, J.; Kim, A. Histone H3K4me1 and H3K27ac play roles in nucleosome eviction and eRNA transcription, respectively, at enhancers. FASEB J. 2021, 35, e21781. [Google Scholar] [CrossRef] [PubMed]
- Scrucca, L.; Fop, M.; Murphy, T.B.; Raftery, A.E. mclust 5: Clustering, Classification and Density Estimation Using Gaussian Finite Mixture Models. R J. 2016, 8, 289–317. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO IDs and descriptions |
|---|
| GSE24850 This study contained 11 H3K9me3 ChIP-seq mouse nucleus accumbens experiments comprising six controls, three saline-treated samples, and two cocaine samples [16]. Among them, five controls and five treated samples were employed. Ten bed files that were provided as a Supplementary File in GEO were downloaded. One control sample with a GEO identification (ID) of GSM612984 was discarded, and the other ten samples were used. |
| GSE159075 This study contained various histone modification ChIP-seq experiments using human umbilical vein endothelial cell lines [17]. Among them, three H3K4me3 ChIP-seq, three H3K27me3 ChIP-seq, and three H3K27ac ChIP-seq, as well as one input ChIP-seq profile, were employed. The corresponding 10 bigWig files were downloaded from the GEO Supplementary File. |
| GSE74055 This study contained various histone modification ChIP-seq experiments using mouse E14 or DKO cell lines [18]. Among them, 16 H3K4me1 ChIP-seq profiles and the corresponding 16 input profiles (32 profiles in total) were downloaded in the bigWig format provided in the GEO Supplementary File. |
| GSE124690 This study comprised various histone modification CUT&Tag experiments using human H1 or K562 cell lines [19]. Among them, six bulk H3K4me1 profiles (GSM3536499_H1_K4me1_Rep1.bed.gz, GSM3536499_H1_K4me1_Rep2.bed.gz, GSM3536516_K562_K4me1_Rep1.bed.gz, GSM3536516_K562_K4me1_Rep2.bed.gz, GSM3680223_K562_H3K4me1_Abcam_8895.bed.gz, GSM3680224_K562_H3K4me1 _ActMot_39113.bed.gz), four bulk H3K3me3 profiles (GSM3536501_H1_K4me3 _Rep1.bed.gz, GSM3536501_H1_K4me3_Rep2.bed.gz, GSM3536518_K562_K4me3 _Rep1.bed.gz, GSM3536518_K562_K4me3_Rep2.bed.gz), and four bulk H3K27ac profiles (GSM3536497_H1_K27ac_Rep1.bed.gz, GSM3536497_H1 _K27ac_Rep2.bed.gz, GSM3536514_K562_K27ac_Rep1.bed.gz, GSM3536514_K562_K27ac _Rep2.bed.gz) were downloaded from the GEO Supplementary File. |
| GSE188173 This study contained nine ChIP-seq H3K27ac profiles (with one control and one treated with SPT) using patient–derived xenografts of human castration-resistant prostate cancer (18 profiles in total). The corresponding 18 bigWig files were extracted from the file GSE188173_RAW.tar retrieved from GEO Supplementary File. |
| GSE159022 This study comprised four H3K4me3 ChIP-seq profiles, four H3K27me3 ChIP-seq profiles, and four H4K16ac ChIP-seq profiles using mouse progenitor cells (two wild type (WT) and two neurofibromin knockouts) [20]. Among them, four H3K27me3 profiles were used and four bigWig files were downloaded from the GEO Supplementary File. |
| GSE168971 This study contained H3K27ac and H3K9ac ChIP-seq profiles taken from various experimental conditions [21], six H3K9ac profiles using C3H-WT mouse liver, and two corresponding inputs were used. The corresponding eight bigWig files were downloaded from the GEO Supplementary File. |
| GSE159411 This study comprised various ChIP-seq profiles [22]. Among them, four H3K36me3 ChIP-seq profiles (two hiPSC cardiomyocytes and two WT hiPSCs) were used. The four corresponding bigWig files were downloaded from the GEO Supplementary File. |
| GSE181596 This study consisted of four H3K27me3 ChIP-seq profiles (two controls and two treatments) and four H3K4me3 ChIP-seq profiles (two controls and two treatments) in addition to two input profiles that used cells as odontoblasts (treatment was siRNA: si-IKBz) [23]. Among them, four H3K27me3 ChIP-seq profiles were downloaded from the GEO Supplementary File. |
| Names of methods and descriptions |
|---|
| MOSAiCS MOSAiCS [28] was implemented as a bioconductor package [29]. Version 2.32.0 was installed in R [30] and applied to GSE24850. MOSAiCS provides biologically motivated statistical models for reads that arise under both non-enrichment (background) and enrichment (signal). Furthermore, MOSAiCS builds a parametric background model that accounts for biases such as GC content and mappability that are inherent to ChIP-seq data. The MOSAiCS model does not assume punctuated or broad peak structures, but instead quantifies whether the ChIP reads show enrichment compared to the background reads for every genomic interval (e.g., bin) of user-defined size in the genome. |
| DFilter DFilter [31] was implemented as a Linux command-line program. Ver. 1.6 was downloaded from https://reggenlab.github.io/DFilter/ (accessed on 20 August 2023). DFilter takes as input a set of sequence tags mapped to a reference genome. Based on the genomic distribution of tags, the algorithm classifies individual n-base-pair bins as positive (signal) or negative (noise) regions. DFilter implements linear finite-impulse-response detection, that is, a windowed linear filter h of user-specified width, followed by the standard thresholding step. |
| F-Seq2 F-Seq2 [32] was implemented as a Linux command-line program. It was downloaded from https://github.com/Boyle-Lab/F-Seq2 (accessed on 20 August 2023). F-Seq2 employed the Gaussian kernel density function to quantify the amount of protein binding. The total control read count was linearly scaled to be equal to the total treatment read count at the individual chromosome level, as the ratios of total reads fluctuated between different chromosomes. |
| HOMER HOMER [33] was implemented as a Linux command-line program. The latest version, HOMER 4.11, which was released on 24 October 2019, was downloaded from http://homer.ucsd.edu/homer/ (accessed on 20 August 2023). For each ChIP-seq experiment, ChIP-enriched regions (peaks) were found by first identifying significant clusters of ChIP-seq tags and then filtering these clusters for those that were significantly enriched relative to background sequencing and local ChIP-seq signal. |
| RSEG RSEG [34] was implemented as a Linux command-line program. The latest version, 0.4.9, was downloaded from http://smithlabresearch.org/software/rseg/ (accessed on 20 August 2023). The negative binomial distribution is assumed to quantify the amount of protein binding between control and treated samples using the NBDiff distribution, which is the discrete distribution of the difference between two independent negative binomial random variables. |
| Term | Overlap | p-Value | Adjusted p-Value |
|---|---|---|---|
| H3K9me3 Brain Mid Frontal Lobe | 22/217 | ||
| H3K9me3 Brain Inferior Temporal Lobe | 18/170 | ||
| H3K9me3 CD4 Naive Primary Cells | 10/64 | ||
| H3K9me3 Brain Anterior Caudate | 15/141 | ||
| H3K9me3 IMR90 | 45/814 | ||
| H3K9me3 Brain Hippocampus Middle | 15/170 | ||
| H3K9me3 Stomach Smooth Muscle | 17/217 | ||
| H3K9me3 Colon Smooth Muscle | 10/92 | ||
| H3K9me3 CD8 Naive Primary Cells | 11/110 | ||
| H3K9me3 Brain Cingulate Gyrus | 11/115 |
| Enriched | ||||||
|---|---|---|---|---|---|---|
| Methods | Pair No. | Regions | Entrez | Gene | Histone | H3K9me3 |
| /Peaks | Genes | Symbols | Modification | |||
| Proposed method | — | 1302 | 894 | 641 | 10 | 10 |
| 1 | 4367 | 1833 | 994 | 3 | 1 | |
| 2 | 3648 | 1599 | 851 | 0 | 0 | |
| MOSAiCS | 3 | 2096 | 1136 | 567 | 0 | 0 |
| 4 | 1985 | 1018 | 532 | 2 | 2 | |
| 5 | 5556 | 2223 | 1184 | 2 | 0 | |
| 1 | 25,080 | 6286 | 2621 | 1 | 0 | |
| 2 | 22,863 | 5721 | 2524 | 1 | 0 | |
| DFilter | 3 | 21,371 | 5470 | 2499 | 1 | 0 |
| 4 | 23,811 | 5987 | 2631 | 1 | 0 | |
| 5 | 23,369 | 5902 | 2544 | 1 | 0 | |
| F-Seq2 | — | — | — | — | — | — |
| HOMER | — | 114,727 | 6771 | 6747 | 1 | 0 |
| RSEG | — | — | — | — | — | — |
| Histone | |||||||
|---|---|---|---|---|---|---|---|
| Modification | |||||||
| GEO ID | Histone | Regions | Entrez | Gene | All | Targeted | Species |
| Modification | /Peaks | Genes | Symbols | ||||
| H3K4me3 | 34,538 | 13,692 | 13,671 | 198 | 54 | ||
| GSE159075 | H3K27me3 | 62,141 | 5217 | 5208 | 83 | 56 | Human |
| H3K27ac | 61,306 | 11,604 | 11,590 | 175 | 24 | ||
| GSE74055 | H3K4me1 | 61,329 | 11,890 | 11,858 | 58 * | 6 * | Mouse |
| (when PCA is replaced with TD) | 70,187 | 14,220 | 14,187 | 102 * | 10 * | ||
| H3K4me1 | 164,466 | 14,893 | 14,866 | 3 * | 0 * | ||
| GSE124690 | H3K4me3 | 37,534 | 14,972 | 14,946 | 200 | 54 | Human |
| (CUT&Tag) | H3K27ac | 81,249 | 13,086 | 13,061 | 139 | 24 | |
| GSE188173 | H3K27ac | 105,438 | 15,579 | 15,548 | 155 | 24 | Human |
| GSE159022 | H3K27me3 | 55,923 | 5022 | 4996 | 70 | 56 | Mouse |
| GSE168971 | H3K9ac | 58,490 | 15,460 | 15,452 | 81 * | 6 * | Mouse |
| GSE159411 | H3K36me3 | 253,326 | 12,282 | 12,270 | 201 | 32 | Human |
| GSE181596 | H3K27me3 | 36,972 | 3543 | 3545 | 72 | 56 | Human |
| Number of Assumed Clusters | |||||||
|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | |||||
| Proposed Method | |||||||
| Adjusted -Value | Adjusted -Value | Adjusted-Value | |||||
| Cluster | >0.01 | ≤0.01 | >0.01 | ≤0.01 | >0.01 | ≤0.01 | |
| Gaussian Mixture | 1 | 104,902 | 428 | 99,285 | 0 | 39,555 | 0 |
| 2 | 0 | 874 | 5605 | 1222 | 61,985 | 0 | |
| 3 | — | — | 0 | 92 | 3350 | 1240 | |
| 4 | — | — | — | — | 0 | 74 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turki, T.; Roy, S.S.; Taguchi, Y.-H. Optimized Tensor Decomposition and Principal Component Analysis Outperforming State-of-the-Art Methods When Analyzing Histone Modification Chromatin Immunoprecipitation Profiles. Algorithms 2023, 16, 401. https://doi.org/10.3390/a16090401
Turki T, Roy SS, Taguchi Y-H. Optimized Tensor Decomposition and Principal Component Analysis Outperforming State-of-the-Art Methods When Analyzing Histone Modification Chromatin Immunoprecipitation Profiles. Algorithms. 2023; 16(9):401. https://doi.org/10.3390/a16090401
Chicago/Turabian StyleTurki, Turki, Sanjiban Sekhar Roy, and Y.-H. Taguchi. 2023. "Optimized Tensor Decomposition and Principal Component Analysis Outperforming State-of-the-Art Methods When Analyzing Histone Modification Chromatin Immunoprecipitation Profiles" Algorithms 16, no. 9: 401. https://doi.org/10.3390/a16090401
APA StyleTurki, T., Roy, S. S., & Taguchi, Y.-H. (2023). Optimized Tensor Decomposition and Principal Component Analysis Outperforming State-of-the-Art Methods When Analyzing Histone Modification Chromatin Immunoprecipitation Profiles. Algorithms, 16(9), 401. https://doi.org/10.3390/a16090401