
Identification of Intrinsically Disordered Protein Regions Based on Deep Neural Network-VGG16


Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Pre-Processing the Features of Proteins for the Input of Our Model
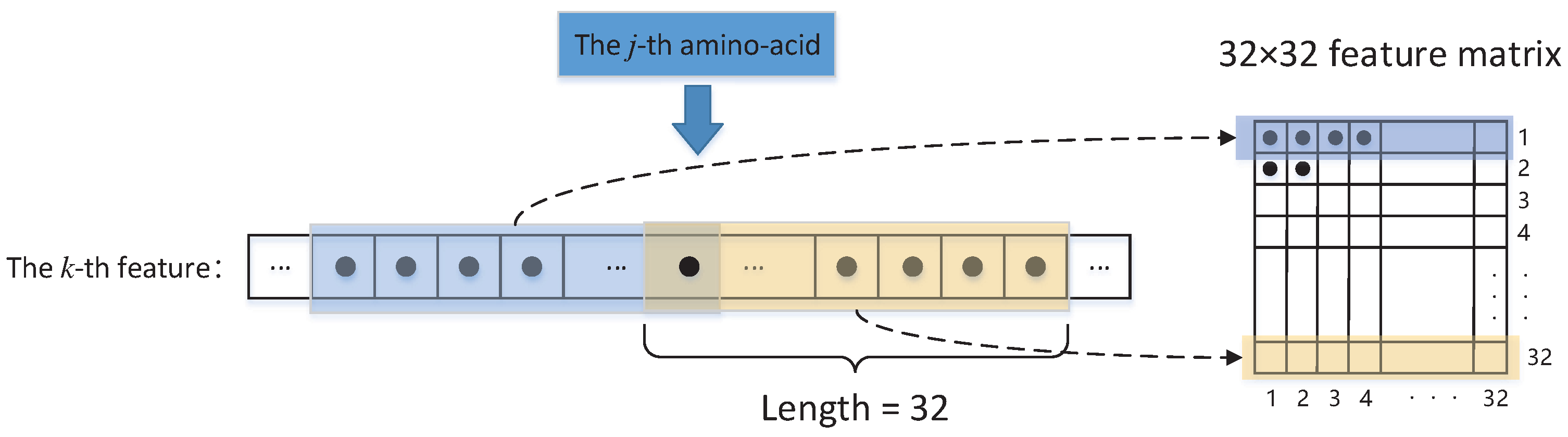
- Given an amino-acid sequence of a protein of size L, we use a sliding window of size 21 and, for each amino-acid in the sliding window, compute the physicochemical properties, evolutionary information, and amino-acid propensities that are defined in the above paragraph. These computed feature values of amino-acids are then averaged over our sliding window and used to represent the feature value of the amino-acid in the center of our sliding window. For two terminal amino-acids of a protein, we append 10 zeros on the left and right side of the amino-acid sequence of a protein, respectively. Thus, for each amino-acid sequence of a protein, we can associate it with a feature matrixwhere the entries of vector represents the feature values (seven physicochemical properties, twenty entries of a column in the PSSM matrix, and 3 amino-acid propensities) associated with the j-th amino-acid. Assume that a vector can be denoted bywhere is the k-th feature value that is associated with the j-th amino-acid averaged over the sliding window of size 21.
- Thus, for the j-th amino-acid, its thirty input features yield a sequence of matrices . Therefore, at each step, we choose three consecutive from the sequence of matrices as the input of VGG16 network.
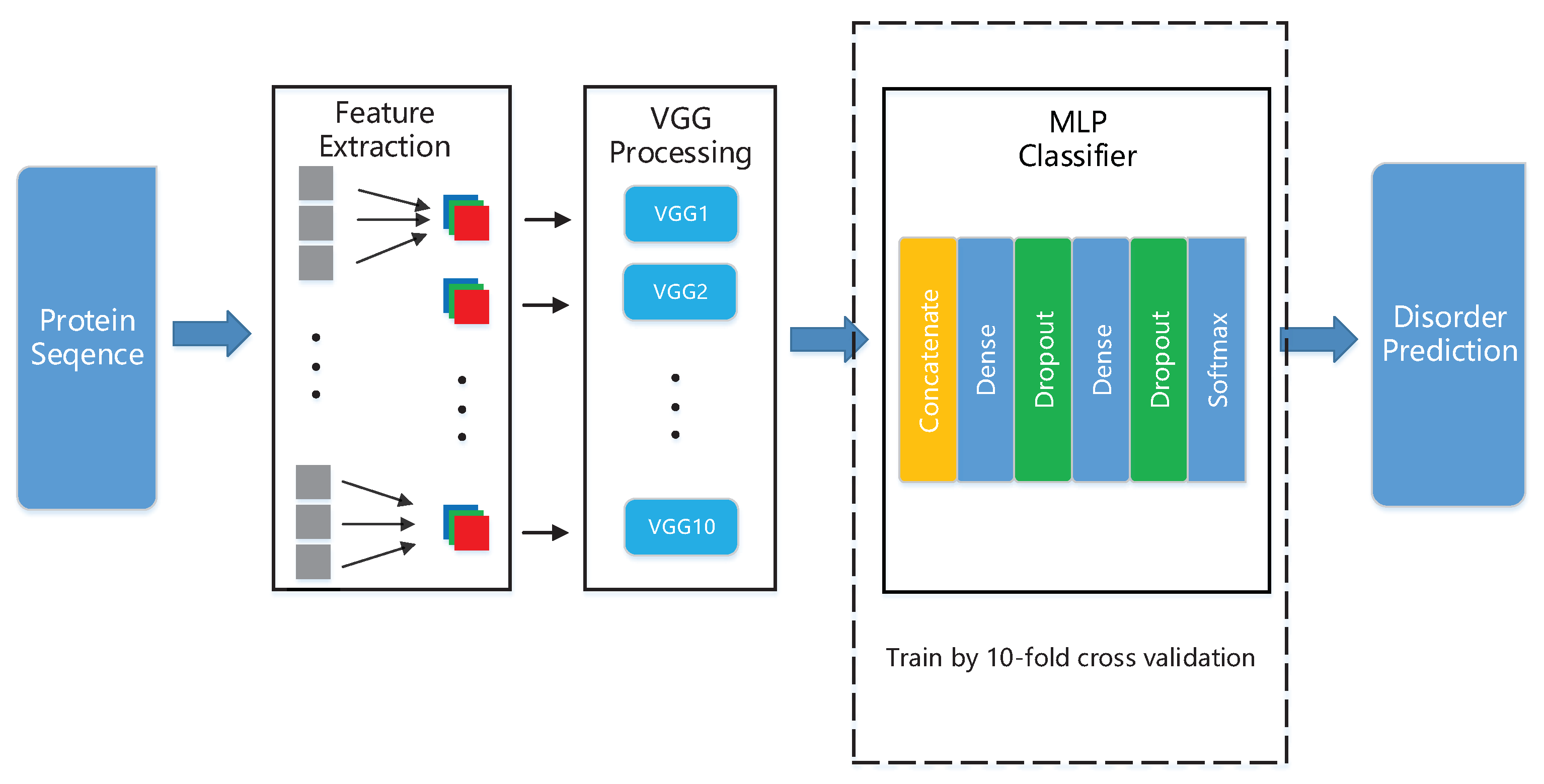
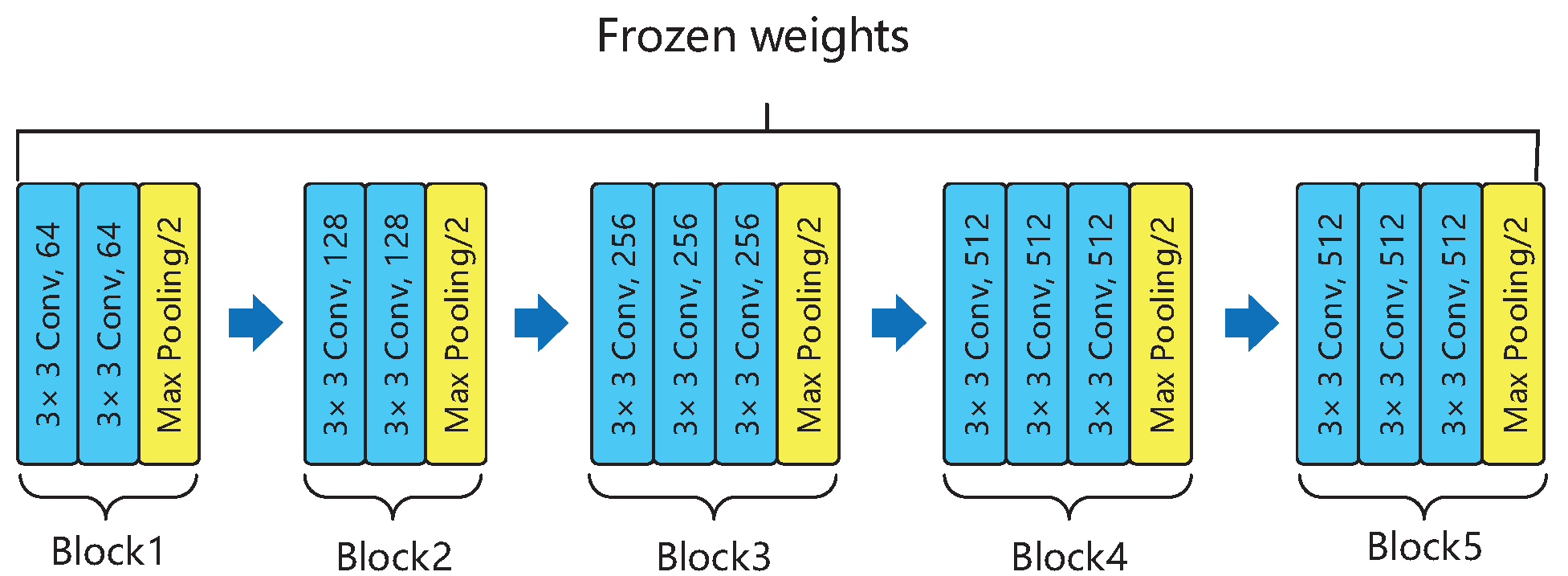
2.3. Designing and Training Our Deep Neural Network
2.4. Performance Evaluation
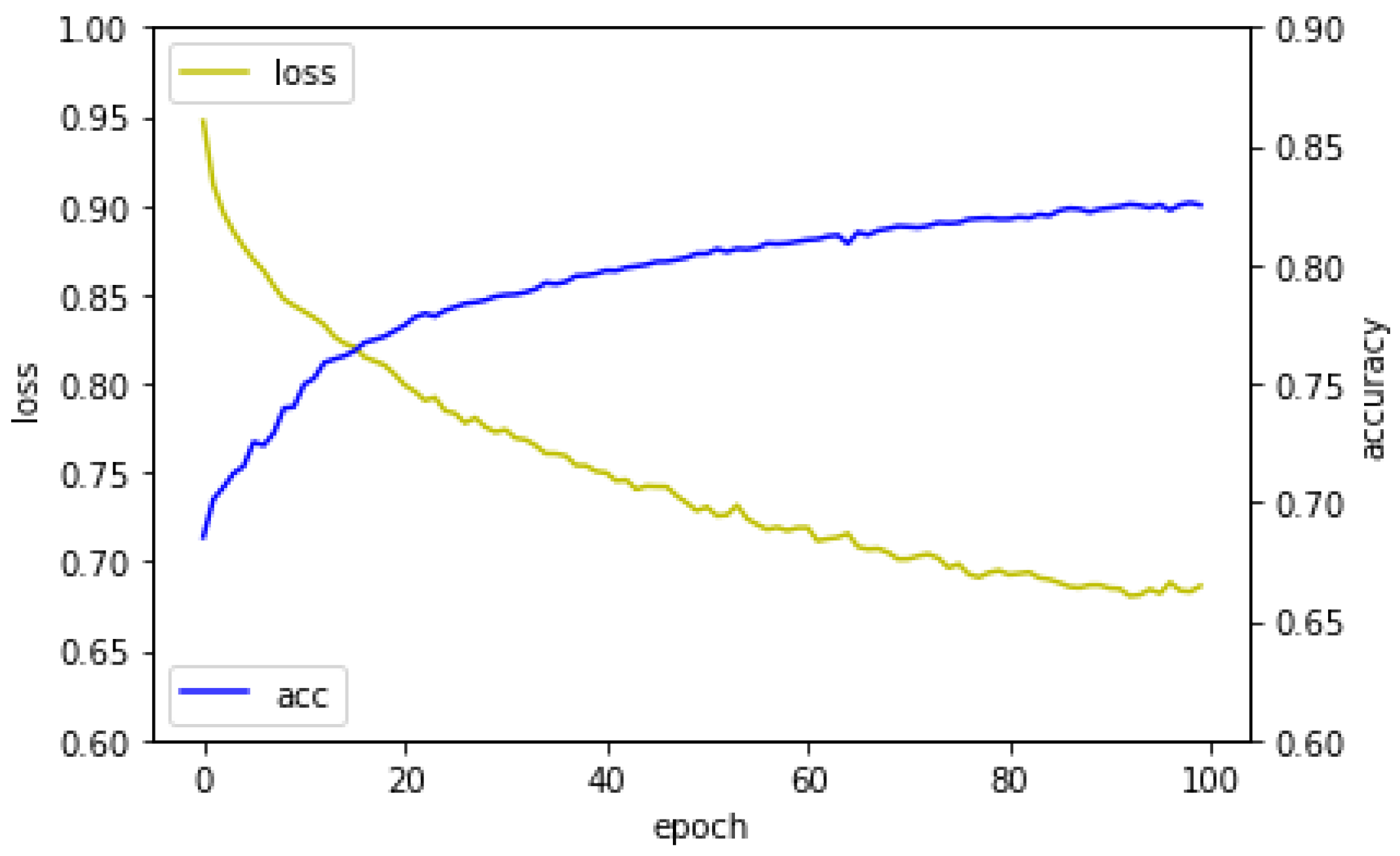
3. Results and Discussion
The Performance Comparison on the Test Dataset DIS166 and Blind Test Datasets
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uversky, V.N. The mysterious unfoldome: Structureless, underappreciated, yet vital part of any given proteome. J. Biomed. Biotechnol. 2010, 2010, 568068. [Google Scholar] [CrossRef] [PubMed]
- Lieutaud, P.; Ferron, F.; Uversky, A.V.; Kurgan, L.; Uversky, V.N.; Longhi, S. How disordered is my protein and what is its disorder for? A guide through the “dark side” of the protein universe. Intrinsically Disord. Proteins 2016, 4, e1259708. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, X.; Liu, B. A comprehensive review and comparison of existing computational methods for intrinsically disordered protein and region prediction. Brief. Bioinform. 2019, 20, 330–346. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Uversky, V.; Kurgan, L. Computational prediction of intrinsic disorder in proteins. Curr. Protoc. Protein Sci. 2017, 88, 2–16. [Google Scholar] [CrossRef]
- Uversky, V.N. Functional roles of transiently and intrinsically disordered regions within proteins. FEBS J. 2015, 282, 1182–1189. [Google Scholar] [CrossRef]
- Holmstrom, E.D.; Liu, Z.; Nettels, D.; Best, R.B.; Schuler, B. Disordered RNA chaperones can enhance nucleic acid folding via local charge screening. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, V.; Kulkarni, P. Intrinsically disordered proteins and phenotypic switching: Implications in cancer. Prog. Mol. Biol. Transl. Sci. 2019, 166, 63–84. [Google Scholar] [PubMed]
- Buljan, M.; Chalancon, G.; Dunker, A.K.; Bateman, A.; Balaji, S.; Fuxreiter, M.; Babu, M.M. Alternative splicing of intrinsically disordered regions and rewiring of protein interactions. Curr. Opin. Struct. Biol. 2013, 23, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Konrat, R. NMR contributions to structural dynamics studies of intrinsically disordered proteins. J. Magn. Reson. 2014, 241, 74–85. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Ulrich, E.L.; Cheng, Y.; Dunker, A.K.; Markley, J.L. Addressing the intrinsic disorder bottleneck in structural proteomics. Proteins: Struct. Funct. Bioinform. 2005, 59, 444–453. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Galzitskaya, O.V. The Ising model for prediction of disordered residues from protein sequence alone. Phys. Biol. 2011, 8, 035004. [Google Scholar] [CrossRef] [PubMed]
- Linding, R.; Russell, R.B.; Neduva, V.; Gibson, T.J. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003, 31, 3701–3708. [Google Scholar] [CrossRef] [PubMed]
- Galzitskaya, O.V.; Garbuzynskiy, S.O.; Lobanov, M.Y. FoldUnfold: Web server for the prediction of disordered regions in protein chain. Bioinformatics 2006, 22, 2948–2949. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, X.; Liu, B. RFPR-IDP: Reduce the false positive rates for intrinsically disordered protein and region prediction by incorporating both fully ordered proteins and disordered proteins. Brief. Bioinform. 2020, 22, 2000–2011. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.J.; Pang, Y.H.; Liu, B. IDP-Seq2Seq: Identification of intrinsically disordered regions based on sequence to sequence learning. Bioinformatics 2020, 36, 5177–5186. [Google Scholar] [CrossRef]
- Hanson, J.; Yang, Y.; Paliwal, K.; Zhou, Y. Improving protein disorder prediction by deep bidirectional long short-term memory recurrent neural networks. Bioinformatics 2017, 33, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Hanson, J.; Paliwal, K.K.; Litfin, T.; Zhou, Y. SPOT-Disorder2: Improved Protein Intrinsic Disorder Prediction by Ensembled Deep Learning. Genom. Proteom. Bioinform. 2019, 17, 645–656. [Google Scholar] [CrossRef]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Faraggi, E.; Xue, B.; Dunker, A.K.; Uversky, V.N.; Zhou, Y. SPINE-D: Accurate prediction of short and long disordered regions by a single neural-network based method. J. Biomol. Struct. Dyn. 2012, 29, 799–813. [Google Scholar] [CrossRef]
- Walsh, I.; Martin, A.J.; Di Domenico, T.; Tosatto, S.C. ESpritz: Accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Mizianty, M.J.; Stach, W.; Chen, K.; Kedarisetti, K.D.; Disfani, F.M.; Kurgan, L. Improved sequence-based prediction of disordered regions with multilayer fusion of multiple information sources. Bioinformatics 2010, 26, i489–i496. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, L.P.; Bujnicki, J.M. MetaDisorder: A meta-server for the prediction of intrinsic disorder in proteins. BMC Bioinform. 2012, 13, 1–11. [Google Scholar] [CrossRef]
- Schlessinger, A.; Punta, M.; Yachdav, G.; Kajan, L.; Rost, B. Improved disorder prediction by combination of orthogonal approaches. PLoS ONE 2009, 4, e4433. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.S.; Woo, J.; Lee, S.; Kang, A.R. Malware Detection of Hangul Word Processor Files Using Spatial Pyramid Average Pooling. Sensors 2020, 20, 5265. [Google Scholar] [CrossRef] [PubMed]
- Anwer, R.M.; Khan, F.S.; van de Weijer, J.; Molinier, M.; Laaksonen, J. Binary patterns encoded convolutional neural networks for texture recognition and remote sensing scene classification. ISPRS J. Photogramm. Remote Sens. 2018, 138, 74–85. [Google Scholar] [CrossRef]
- Simonyan, K.; Zisserman, A. Very deep convolutional networks for large-scale image recognition. arXiv 2014, arXiv:1409.1556. [Google Scholar]
- Hatos, A.; Hajdu-Soltész, B.; Monzon, A.M.; Palopoli, N.; Álvarez, L.; Aykac-Fas, B.; Bassot, C.; Benítez, G.I.; Bevilacqua, M.; Chasapi, A.; et al. DisProt: Intrinsic protein disorder annotation in 2020. Nucleic Acids Res. 2020, 48, D269–D276. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.R.; Thomson, R.; Mcneil, P.; Esnouf, R.M. RONN: The bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 2005, 21, 3369–3376. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.L.; Kurgan, L. Comprehensive comparative assessment of in-silico predictors of disordered regions. Curr. Protein Pept. Sci. 2012, 13, 6–18. [Google Scholar] [CrossRef]
- Meiler, J.; Müller, M.; Zeidler, A.; Schmäschke, F. Generation and evaluation of dimension-reduced amino acid parameter representations by artificial neural networks. Mol. Model. Annu. 2001, 7, 360–369. [Google Scholar] [CrossRef]
- Jones, D.T.; Ward, J.J. Prediction of disordered regions in proteins from position specific score matrices. Proteins Struct. Funct. Bioinform. 2003, 53, 573–578. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Klimke, W.; Maglott, D.R. NCBI Reference Sequences: Current status, policy and new initiatives. Nucleic Acids Res. 2009, 37, D32–D36. [Google Scholar] [CrossRef] [PubMed]
- Ketkar, N. Introduction to keras. In Deep Learning with Python; Springer: Berlin/Heidelberg, Germany, 2017; pp. 97–111. [Google Scholar]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Sens | Spec | BACC | MCC |
|---|---|---|---|---|
| DISvgg | 0.6713 | 0.8828 | 0.7771 | 0.5132 |
| RFPR-IDP | 0.7557 | 0.7817 | 0.7687 | 0.4406 |
| SPOT-Disorder2 | 0.7103 | 0.8084 | 0.7594 | 0.4952 |
| Methods | Sens | Spec | BACC | MCC |
|---|---|---|---|---|
| DISvgg | 0.5993 | 0.9429 | 0.7711 | 0.5270 |
| RFPR-IDP | 0.5464 | 0.9546 | 0.7505 | 0.5139 |
| SPOT-Disorder2 | 0.4941 | 0.9439 | 0.7190 | 0.4486 |
| Methods | Sens | Spec | BACC | MCC |
|---|---|---|---|---|
| DISvgg | 0.7160 | 0.7956 | 0.7558 | 0.4577 |
| RFPR-IDP | 0.7490 | 0.7580 | 0.7540 | 0.4420 |
| SPOT-Disorder2 | 0.6380 | 0.8200 | 0.7290 | 0.4482 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, P.; Zhao, J.; Zhang, J. Identification of Intrinsically Disordered Protein Regions Based on Deep Neural Network-VGG16. Algorithms 2021, 14, 107. https://doi.org/10.3390/a14040107
Xu P, Zhao J, Zhang J. Identification of Intrinsically Disordered Protein Regions Based on Deep Neural Network-VGG16. Algorithms. 2021; 14(4):107. https://doi.org/10.3390/a14040107
Chicago/Turabian StyleXu, Pengchang, Jiaxiang Zhao, and Jie Zhang. 2021. "Identification of Intrinsically Disordered Protein Regions Based on Deep Neural Network-VGG16" Algorithms 14, no. 4: 107. https://doi.org/10.3390/a14040107
APA StyleXu, P., Zhao, J., & Zhang, J. (2021). Identification of Intrinsically Disordered Protein Regions Based on Deep Neural Network-VGG16. Algorithms, 14(4), 107. https://doi.org/10.3390/a14040107