Appendix A. All Constraints in an MILP Formulation for Rank-2 Chemical Graphs
To formulate an MILP that represents a chemical graph
, we distinguish a tuple
from a tuple
. For a tuple
, let
denote the tuple
. Let
. We call a tuple
proper if
where the latter is assumed because otherwise
G must consist of two atoms of
. Assume that each tuple
is proper. Let
be a fictitious chemical element that represents null, call a tuple
with
fictitious, and define
to be the set of all fictitious tuples; i.e.,
. To represent chemical elements
in an MILP, we encode these elements
into some integers denoted by
. Assume that, for each element
,
is a positive integer and that
.
Appendix A.1. Applicability Domain
We use the range-based method to define an applicability domain for our method. For this, we find the range (the minimum and maximum) of each descriptor over all relevant chemical compounds and represent each range as a set of linear constraints in the constraint set of our MILP formulation. Recall that stands for a set of chemical graphs used for constructing a prediction function. However, the number of examples in may not be large enough to capture a general feature on the structure of chemical graphs. For this, we also use some data set from the whole set of chemical graphs in a database. Let denote the set of chemical graphs such that for each integer . Formally the set of variables and constraints is given as follows.
AD constraints in :
constants:
Integers and ; An integer ;
An integer ;
variables for descriptors in x:
A real variable : represents ;
: represents the number of vertices of degree i in H;
: represents ;
, : represents the number of vertices of chemical element
a in the core of H;
, : represents the number of vertices of chemical element
a in the non-core of H;
, : represents the number of k-bonds in the core of H;
, : represents the number of k-bonds in the non-core of H;
, : represents the number of core edges
in H that are assigned tuple ;
, : represents the number of non-core edges in
H that are assigned tuple ;
In the following, we derive an MILP that satisfies the condition in Theorem 2. Let , , and be given integers. We describe the set with several sets of constraints.
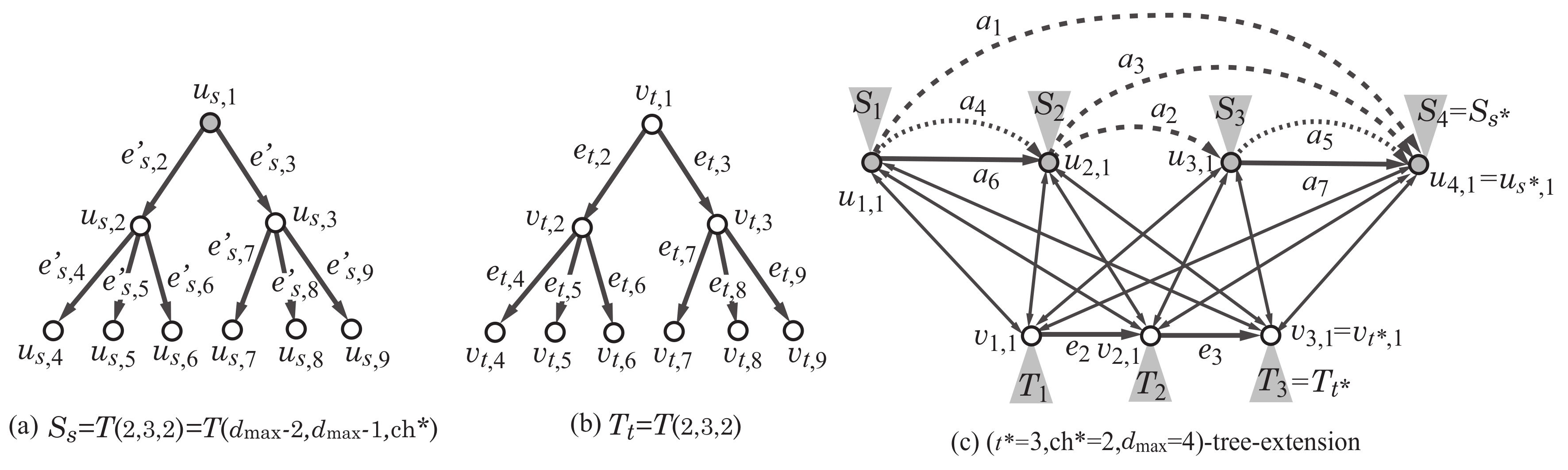
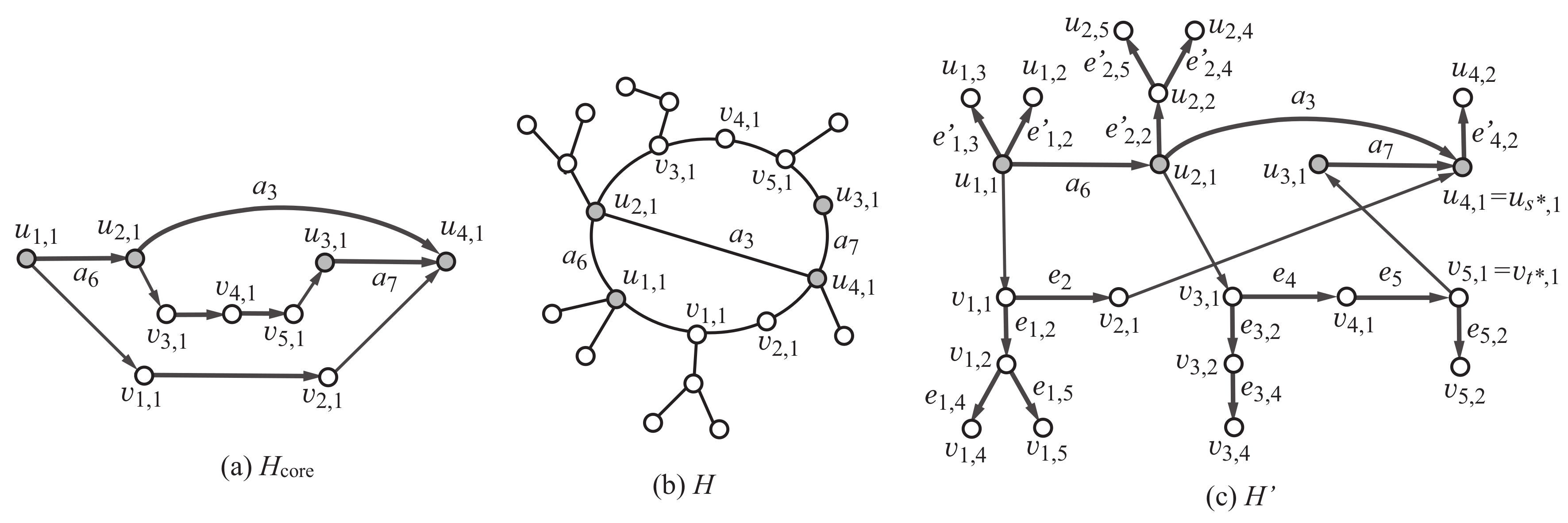
Appendix A.2. Construction of Scheme Graph and Tree-Extension
We infer a subgraph H such that the maximum degree is , , and . For this, we first construct the -tree-extension of the scheme graph . We use the following notations: For and , let (resp., ) denote the set of indices i of edges such that the tail (resp., head) of is . Let , , and .
As described in
Section 2.3.1, some edge
may be replaced with a subpath
of
, which consists of the roots of trees
. We assign color
i to the vertices in such a subpath
by setting a variable
of each vertex
to be
i. For each edge
, we prepare a binary variable
to denote that edge
is used (resp., not used) in a selected graph
H when
(resp.,
). We also include constraints necessary for the variables to satisfy a degree condition at each of the vertices
,
and
,
.
constants:
Integers , , , and ;
, : a lower bound on the out-degree of vertex in H;
, : a lower bound on the in-degree of vertex in H;
, : an upper bound on the out-degree of vertex in H;
, : an upper bound on the in-degree of vertex in H;
variables:
, : represents edge (, )
( ⇔ edge is used in H);
, , : (resp., ) represents
direction (resp., ), where (resp., ) ⇔
edge is used in H and direction (resp., ) is assigned
to edge ;
, : represents the color assigned to vertex
( ⇔ vertex is assigned color c);
, : the number of vertices with color c;
, : the out-degree of vertex in the core of H;
, : the in-degree of vertex in the core of H;
, , ( ⇔ );
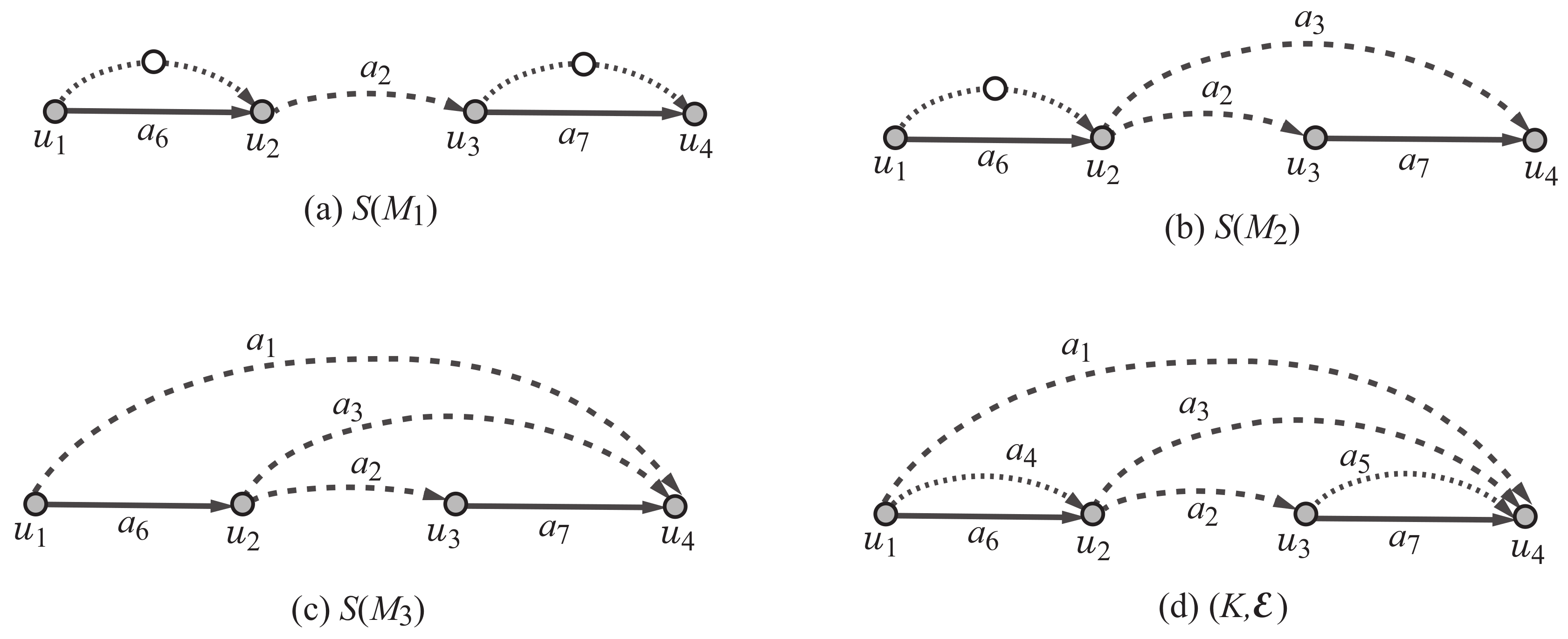
Appendix A.3. Specification for Chemical Graphs with Rank 2
To generate any of the three rank-2 polymer topologies in
, we use the scheme graph
,
in
Figure 2d, where
,
,
,
and
. Recall that each color
is assigned to edge
. We impose some more constraints on the degree of each of the vertices
,
and
,
so that the core of a selected graph
H satisfies one of the three least simple graphs in
Figure 2a–c. We also let a variable
mean the topological parameter
of a selected subgraph
H.
constants:
, ,
, , , , , ,
, , , , , ,
, , , , , ,
, , , , , ,
, , , ,
, , , ,
, , , ,
, , , ,
variables:
: The topology-parameter for rank 2;
Appendix A.4. Selecting A Subgraph
We prepare a binary variable (resp., ) for each vertex in tree (resp., in tree ). We include constraints so that the path is partitioned into subpaths , , where possibly some is empty, and the resulting subgraph H becomes a connected rank-2 graph with , , and .
constants:
Integers , ;
Prepare the set of the indices of children of a vertex
the index of the parent of a non-root vertex , and
the set of indices i such that the height of a vertex is h
in the rooted tree ;
variables:
, , : represents vertex
( ⇔ vertex is used in H and edge is used in H);
, , : represents vertex
( ⇔ vertex is used in H and edge is used in H);
, : represents edge ,
where and are fictitious edges ( ⇔ edge is used in H);
Appendix A.5. Assigning Multiplicity
We prepare an integer variable or for each edge e in the -tree-extension of the scheme graph to denote the multiplicity of e in a selected graph H and include necessary constraints for the variables to satisfy in H.
variables:
, : represents the multiplicity of edge ,
where if edge is not in H;
, , : with (resp., ) represents
the multiplicity of edge (resp., );
, : represents the multiplicity of edge ;
, , : represents the multiplicity of edge ;
Appendix A.6. Assigning Chemical Elements and Valence Condition
We include constraints so that each vertex v in a selected graph H satisfies the valence condition; i.e., . With these constraints, a rank-2 chemical graph on a selected subgraph H will be constructed.
constants:
A set of chemical elements, where denotes null;
A coding , such that ; , ; and if ;
Let and denote and , respectively;
A valence function: ;
variables:
, , :
with (resp., ) represents (resp., );
, , , :
⇔ for and for ;
, , , :
⇔ the multiplicity of edge in H is k;
, , , :
⇔ the multiplicity of edge , (or , ) in H is k;
, , :
⇔ the multiplicity of edge in H is k;
, , , :
⇔ the multiplicity of edge in H is k;
Appendix A.7. Descriptors for Mass, the Numbers of Elements and Bonds
We include constraints to compute descriptors
,
(
,
(
) and
according to the definitions in
Section 2.1.2.
constants:
A function ; Let denote the observed mass of a chemical element , and
define ;
variables:
, ;
, ;
;
, ;
, ;
: the number of hydrogen atoms to be included in G;
Appendix A.8. Descriptor for the Number of Specified Degree
We include constraints to compute descriptors
(
) according to the definitions in
Section 2.1.2. We also add constraints so that the maximum degree of a non-core vertex in
H is at most 3 (resp., equal to 4) when
(resp.,
.
variables:
, , :
represents for or for ;
, , , :
⇔ ;
, ;
Appendix A.9. Descriptor for the Number of Adjacency-Configurations
We include constraints to compute descriptors
and
(
) according to the definitions in
Section 2.1.2.
constants:
A set of proper tuples ;
The set ;
variables:
, , :
⇔ edge is assigned tuple ; i.e., ;
, , :
⇔ edge is assigned tuple ; i.e., ;
, , , :
⇔ edge , (or , ) is assigned tuple ; i.e.,
;
, , , :
⇔ edge is assigned tuple ; i.e., ;
, ;
, ;
Appendix A.10. Descriptor for 1-Path Connectivity
We include constraints to compute descriptor according to the definition.
variables:
A real variable ;
, , , :
⇔ and ,
where is in H if and only if ;
, , : ⇔
and where is in H if and only if ;
, , , : ⇔
and for
(or and for ),
where edge or is in H if and only if ;
, , , , :
⇔ and ,
where is in H if and only if ;
constraints:
where a tolerance
is set to be
.
Appendix A.11. Constraints for Left-Heavy Trees
To reduce the number of rank-2 chemical graphs G that are isomorphic to each other, we include in some additional constraints so that each subtree selected from tree or satisfies the following property:
for any two siblings and , in , the number of descendants of is not smaller than that of .
For this, we define to be the number of descendants of a vertex (or ) in a selected graph H and , , . We include constraints that compute the values of recursively.
variables:
, , : the number of descendants of vertex
in tree for and vertex in tree for ;

 ,
, 













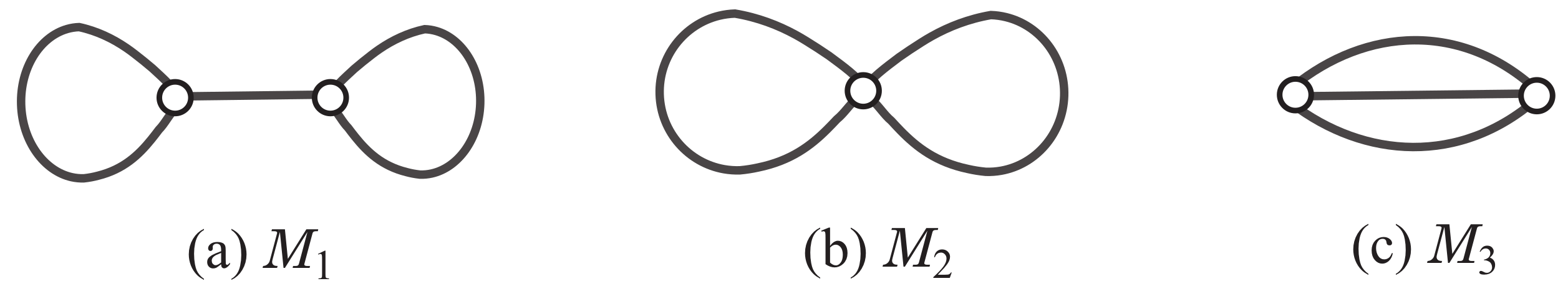{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}