
A Comparison of Classical Force-Fields for Molecular Dynamics Simulations of Lubricants

 ,
, 

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methodology
2.1. Density and Viscosity Benchmarking
2.1.1. Setup
2.1.2. Procedure
2.2. Structure and Friction in Multicomponent Tribological Systems
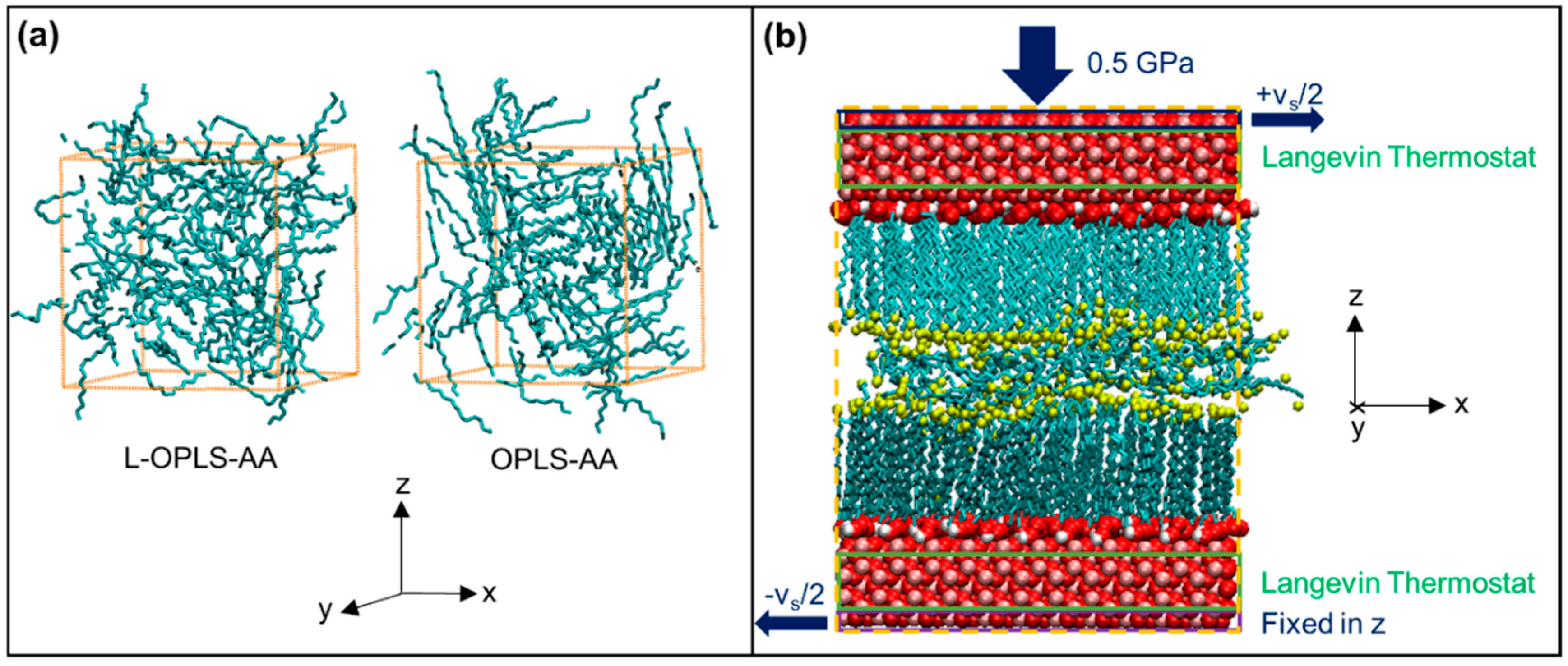
2.2.1. Setup
2.2.2. Procedure
3. Results and Discussion
3.1. Density and Viscosity Benchmarking
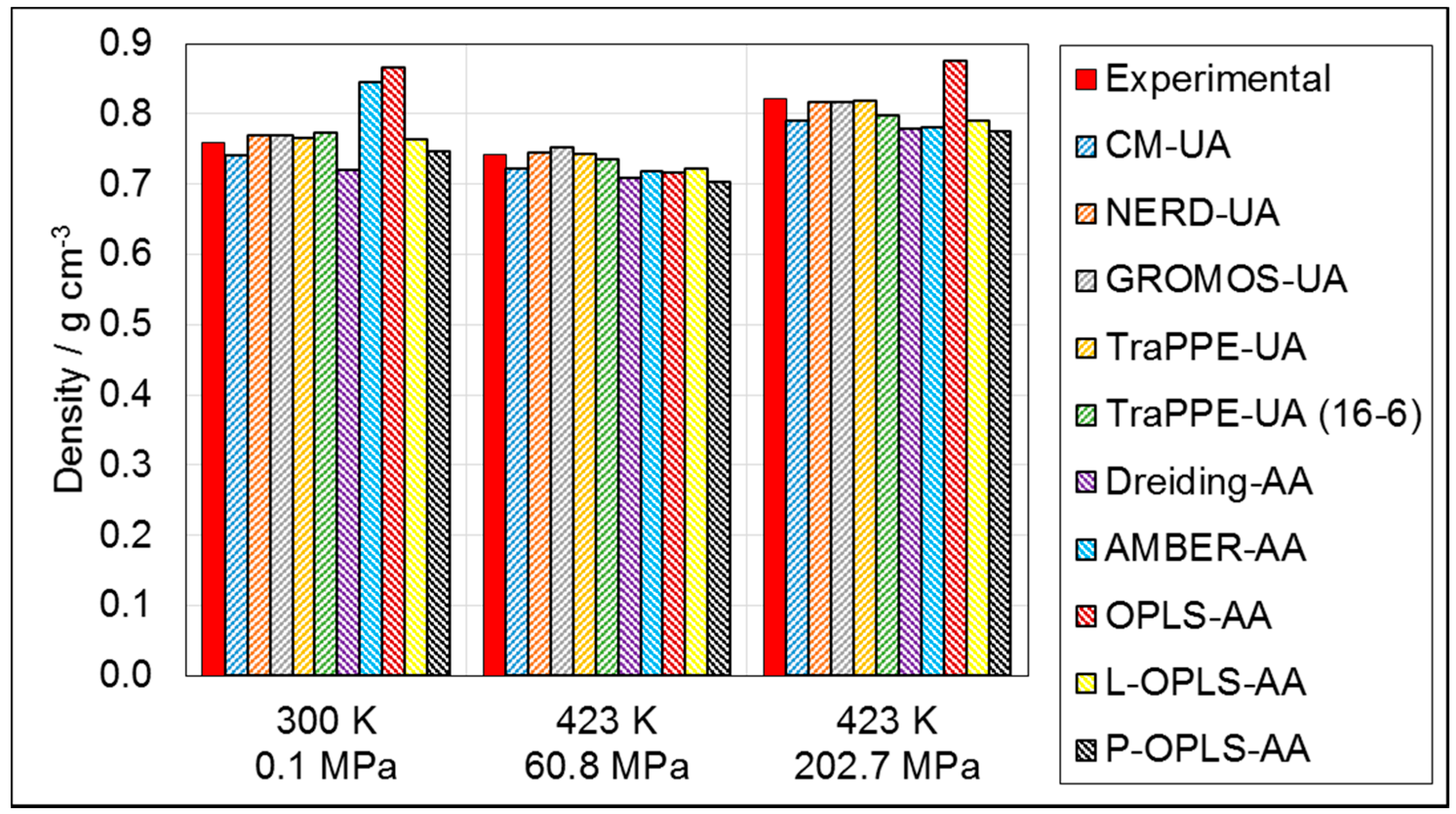
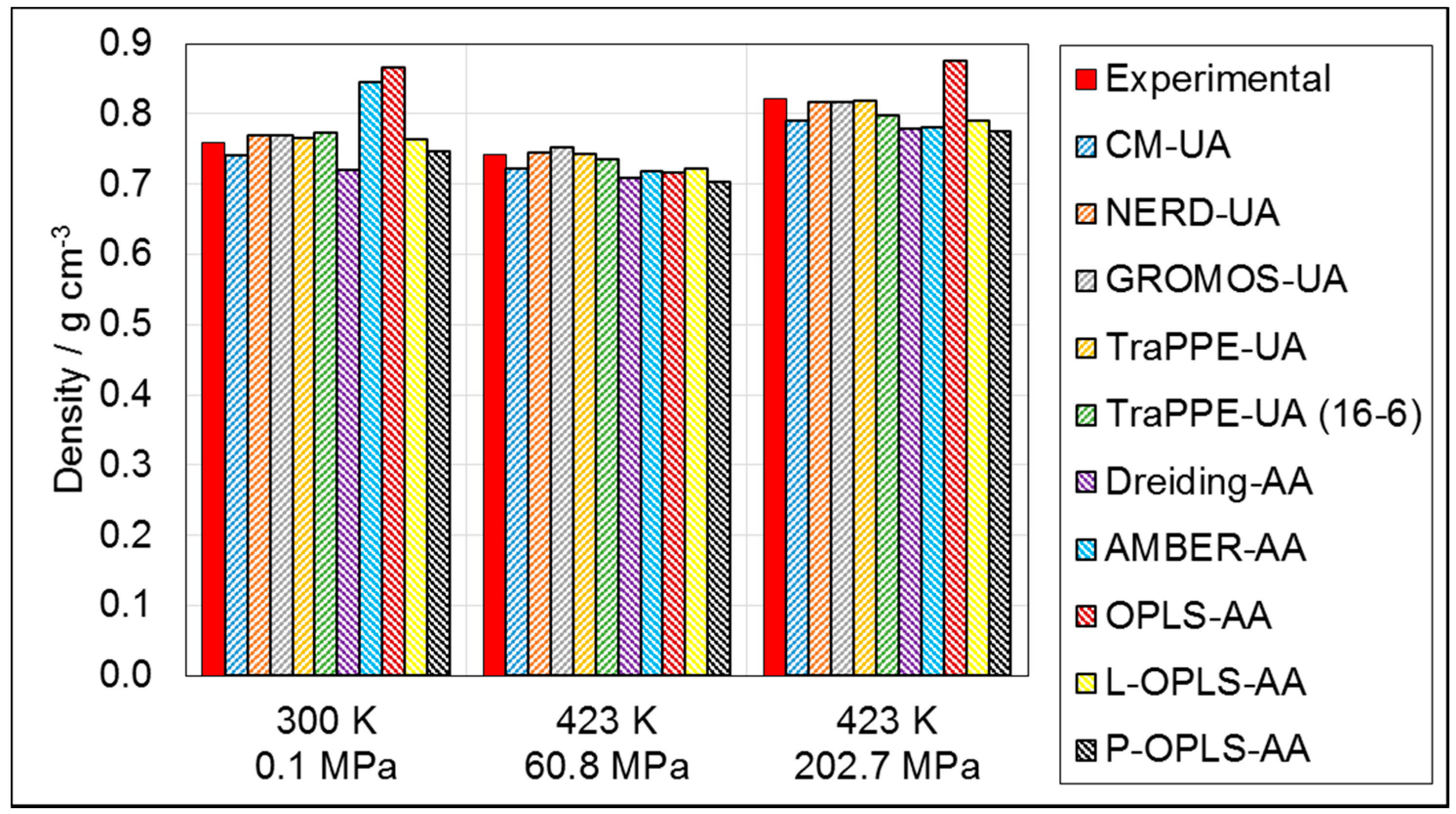
3.1.1. Density
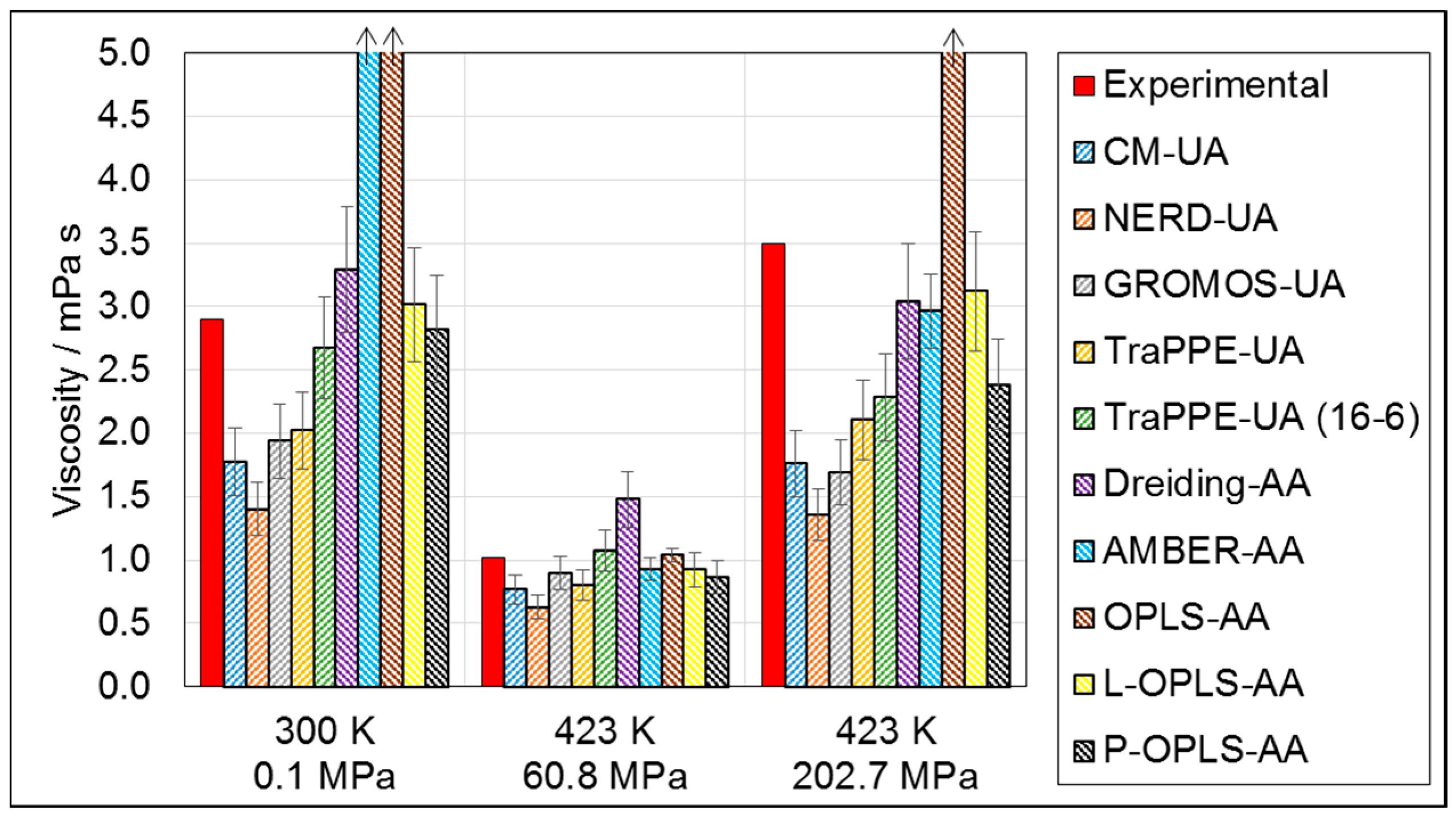
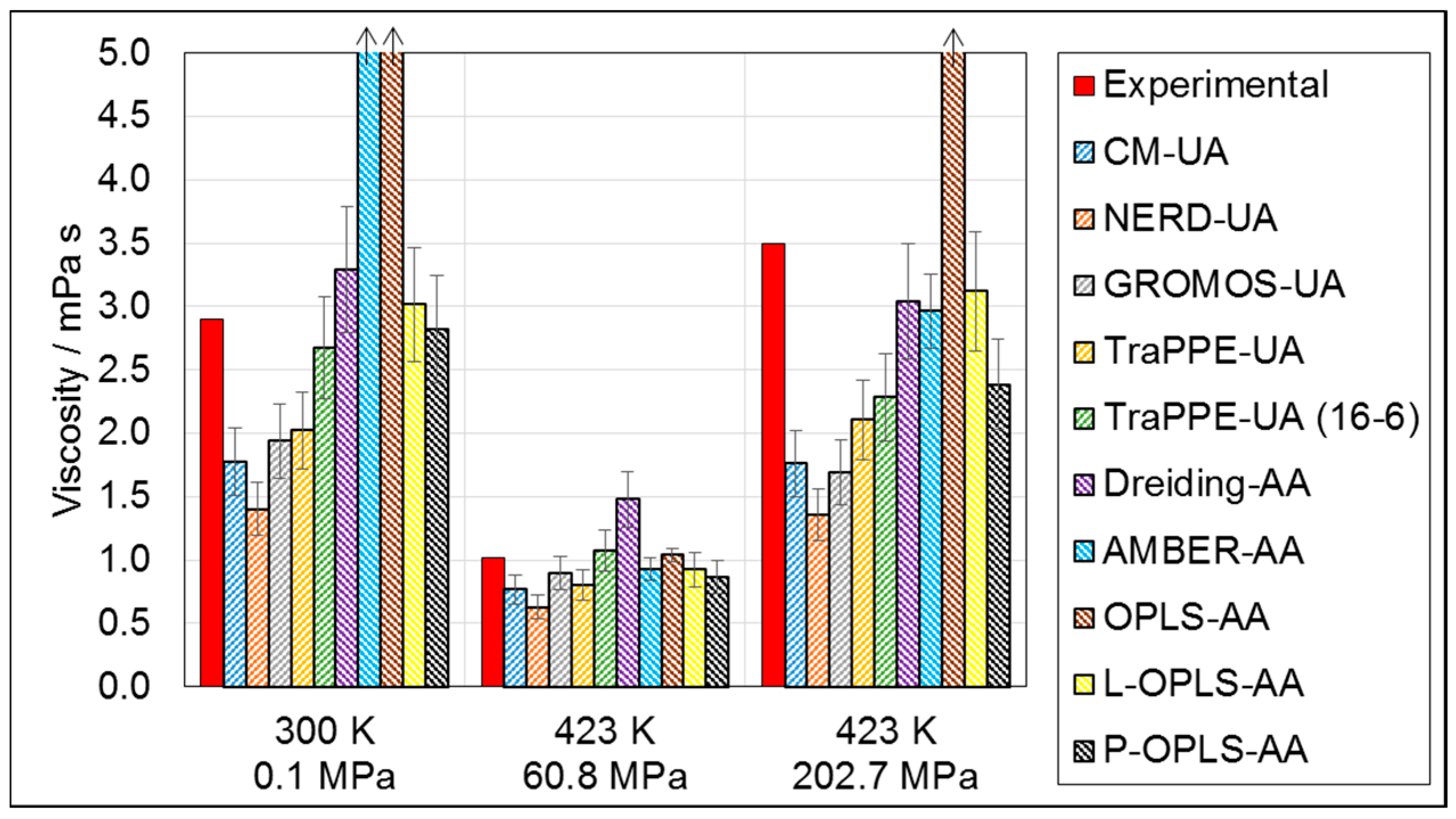
3.1.2. Viscosity
3.2. Structure and Friction in Complex Tribological Systems
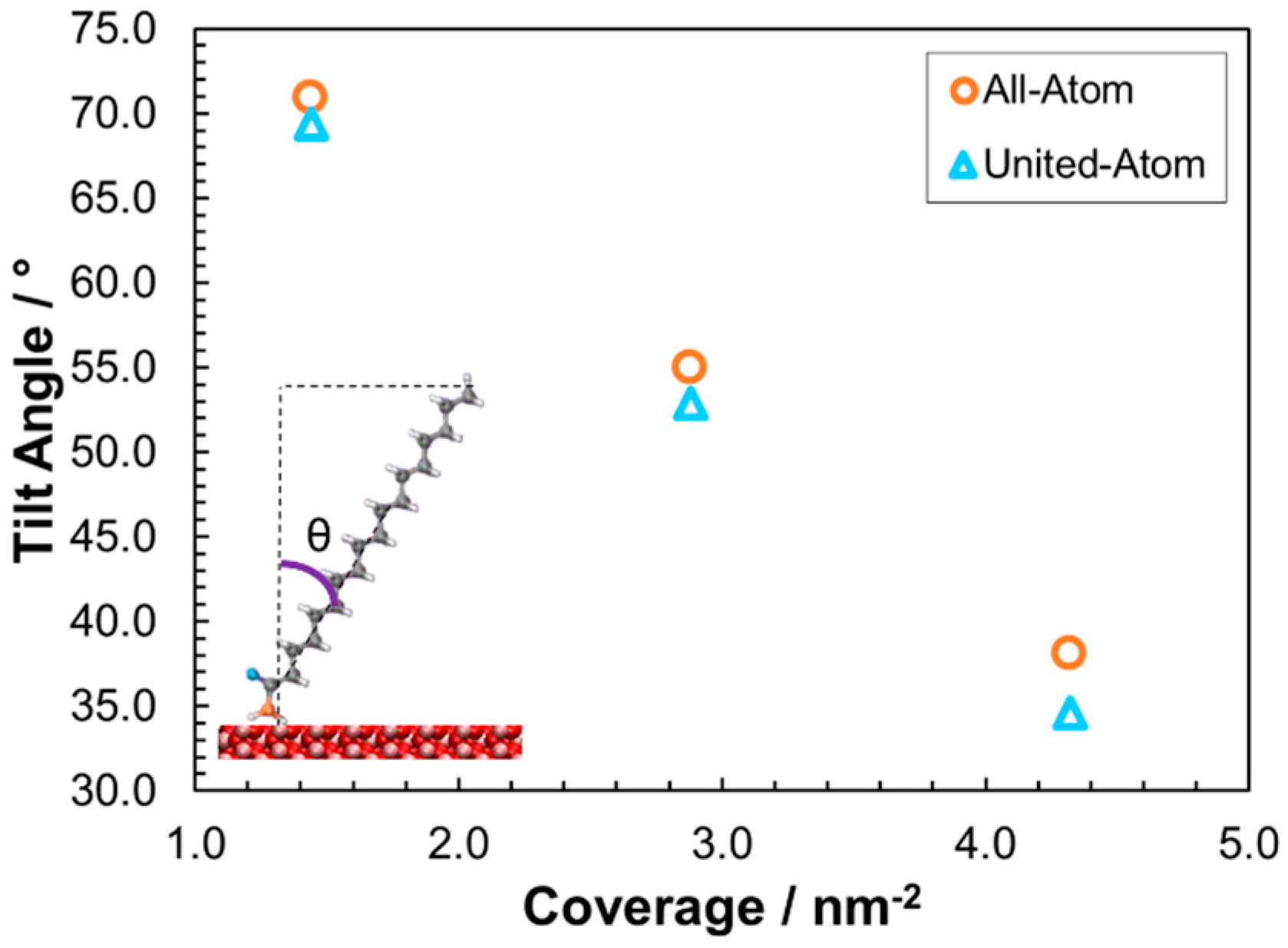
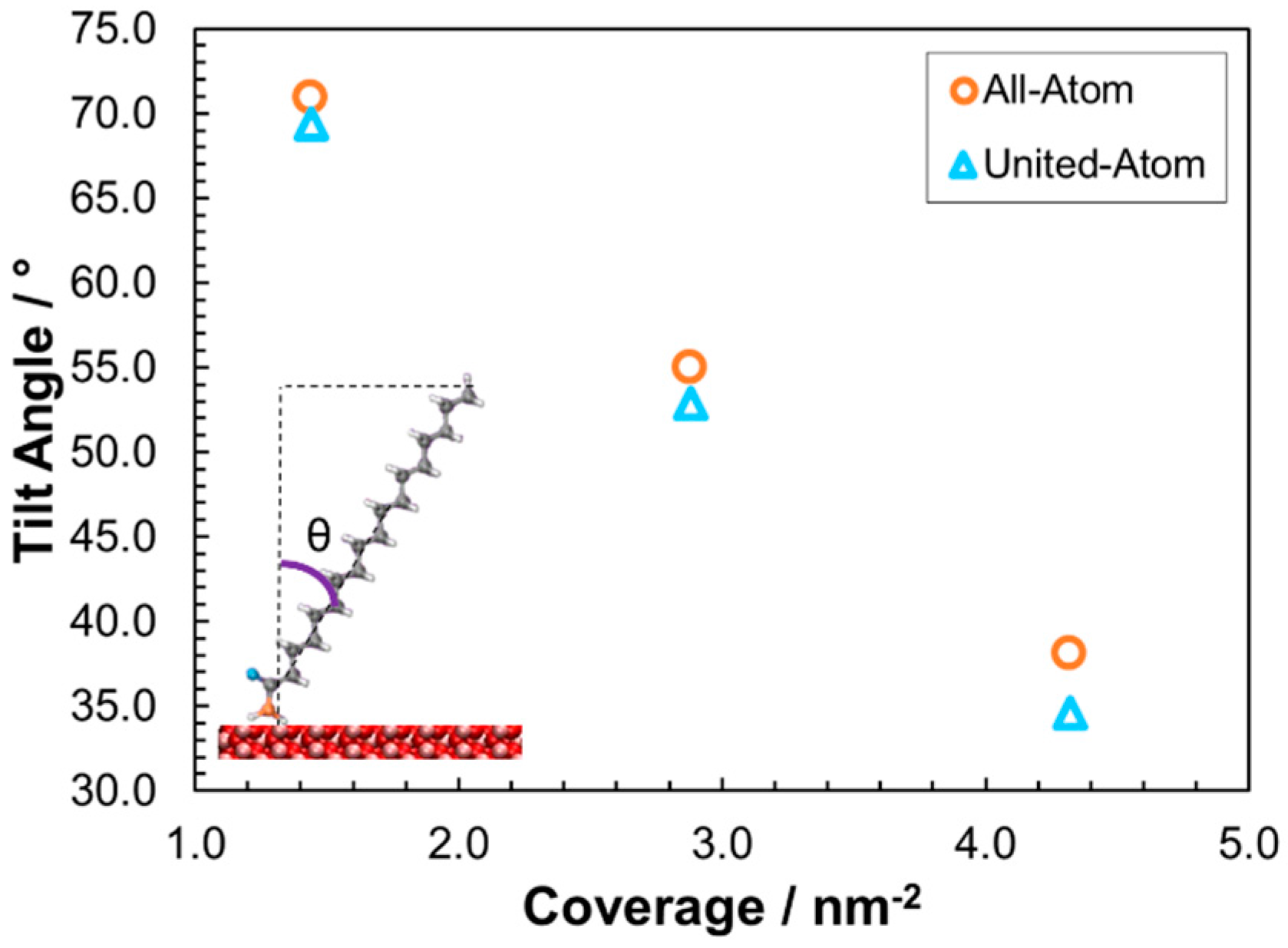
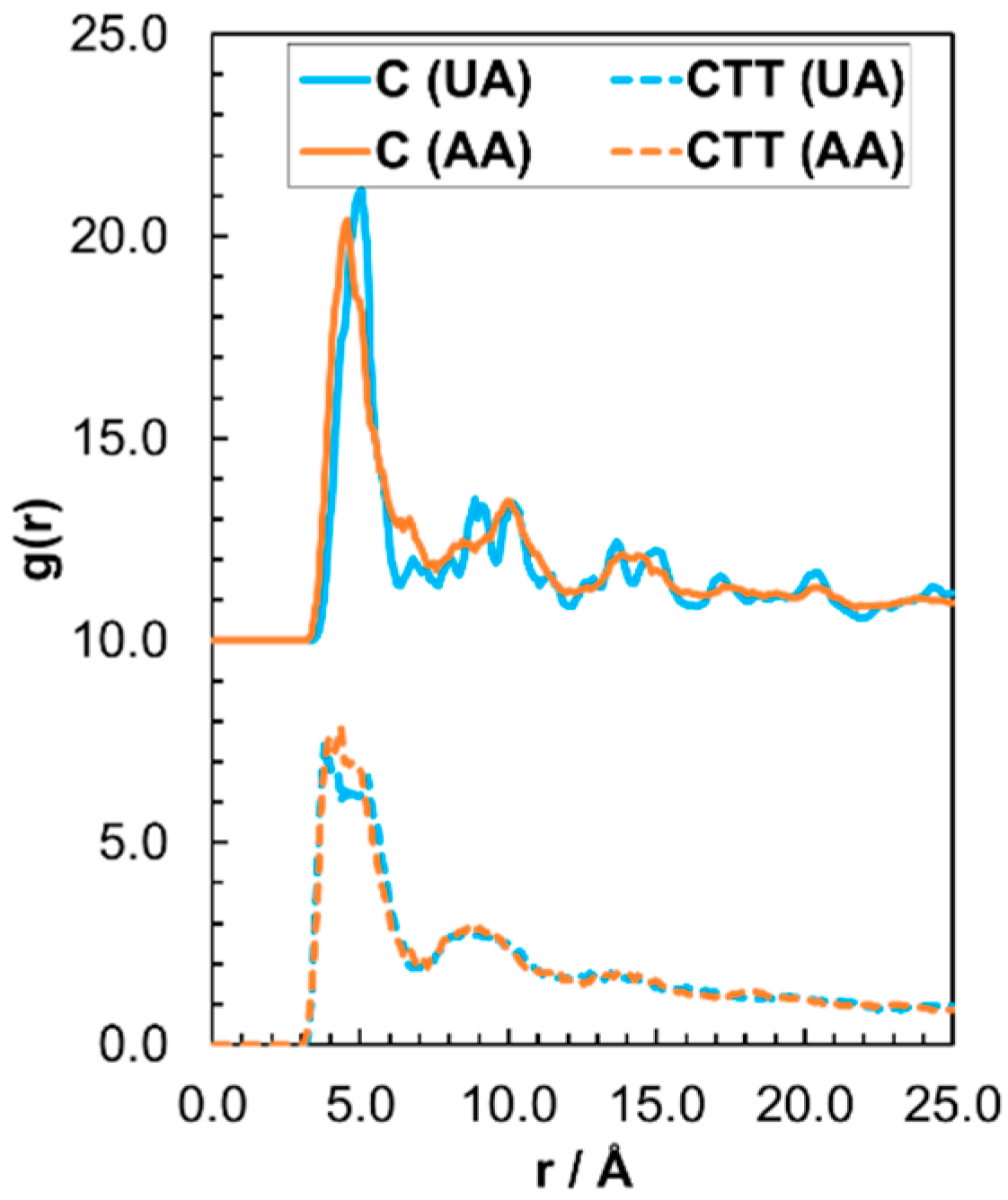
3.2.1. Structure
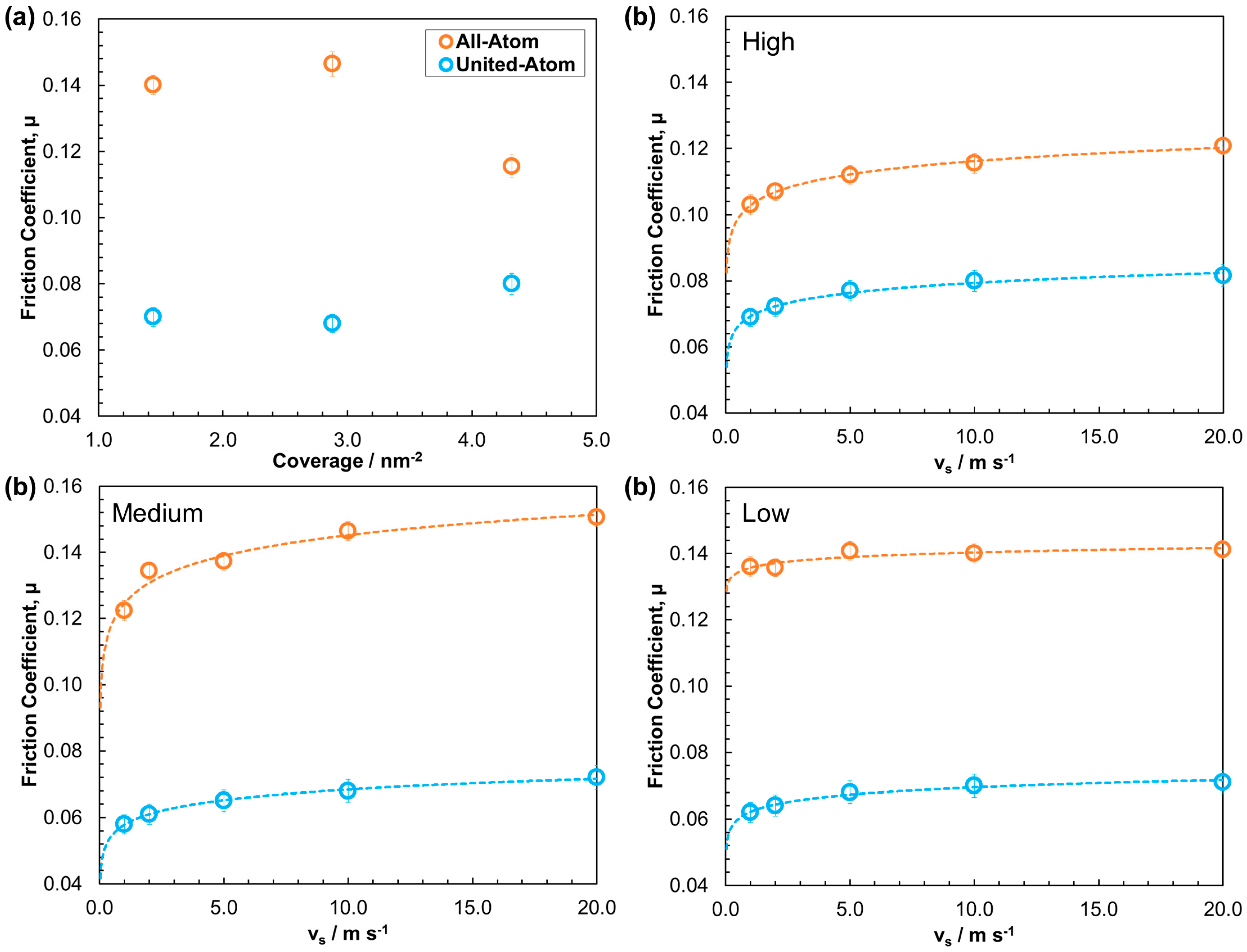
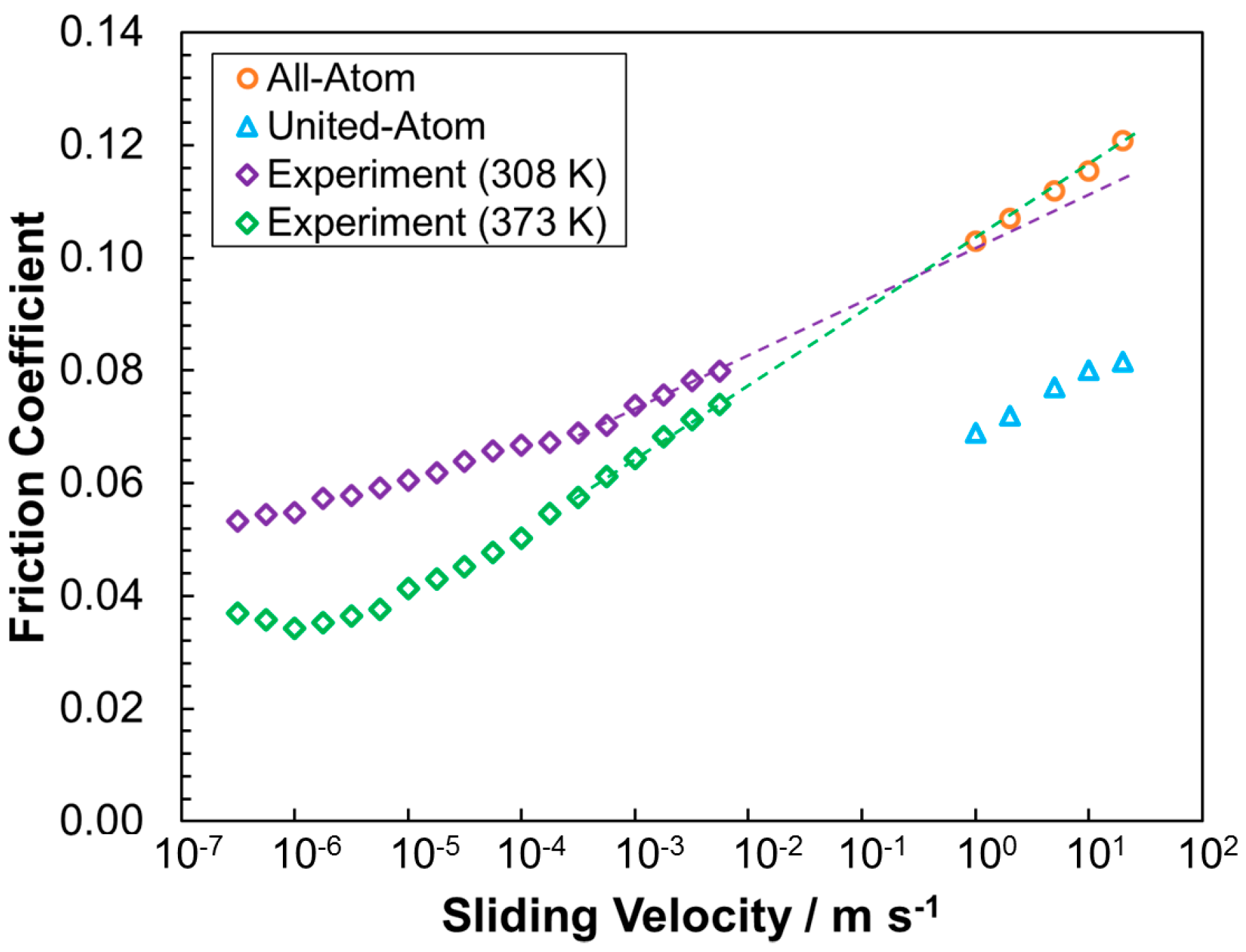
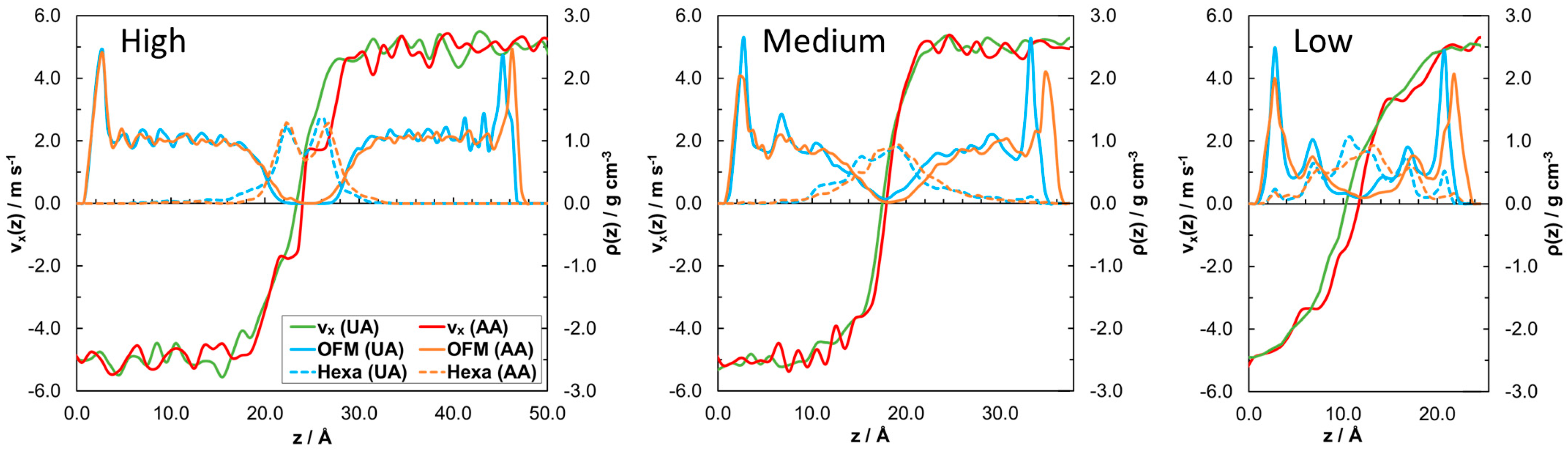
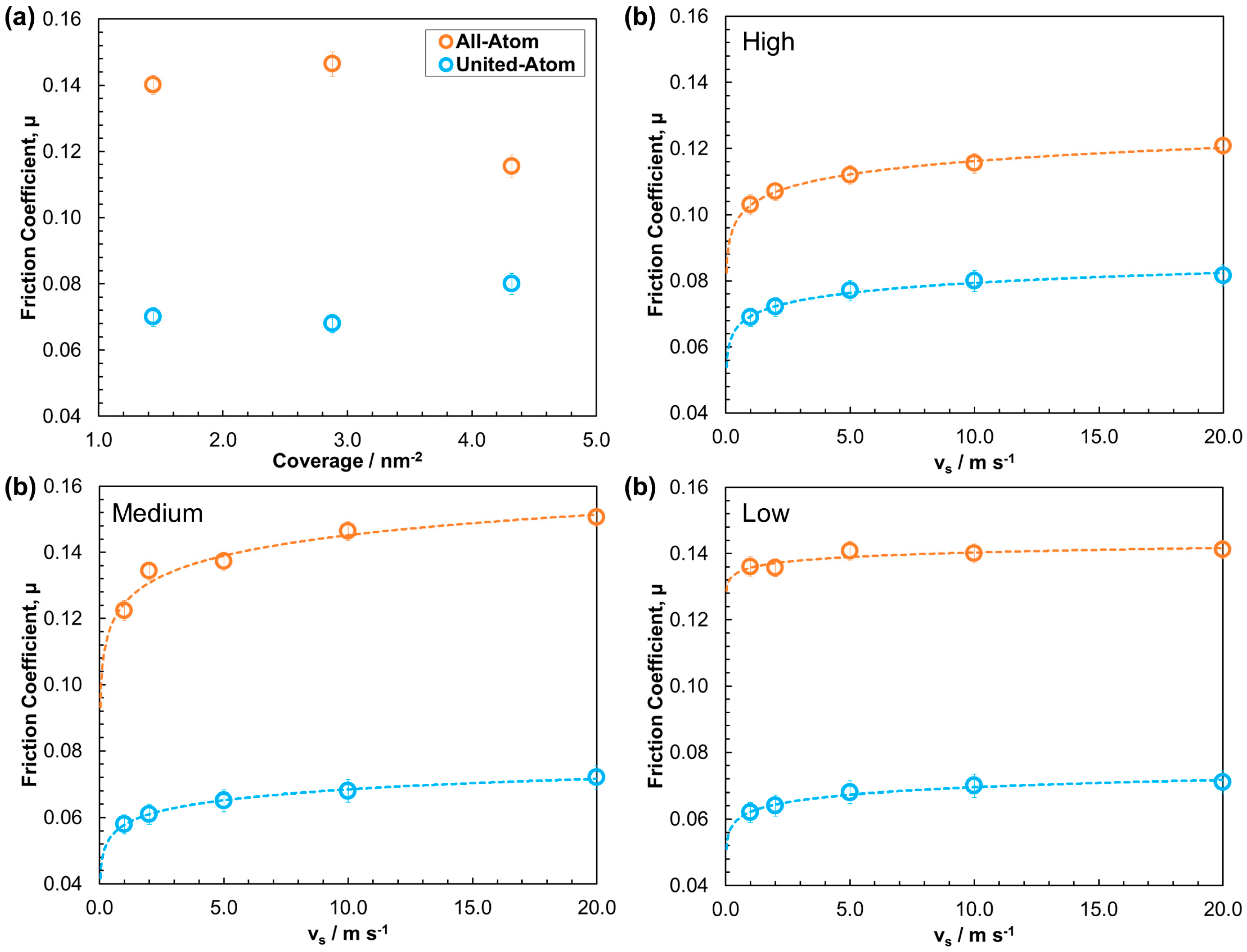
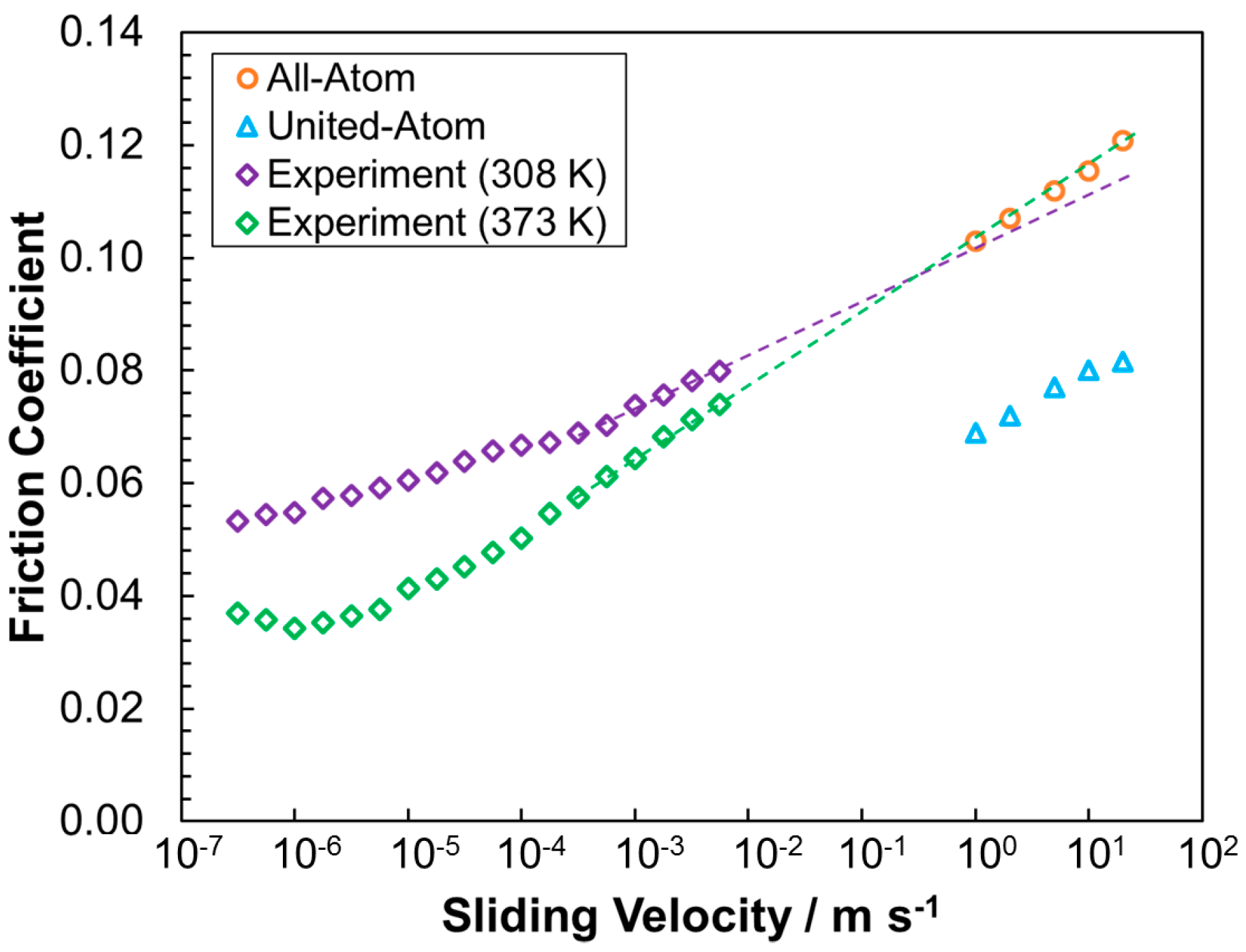
3.2.2. Friction
4. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cui, S.T.; Gupta, S.A.; Cummings, P.T.; Cochran, H.D. Molecular dynamics simulations of the rheology of normal decane, hexadecane, and tetracosane. J. Chem. Phys. 1996, 105, 1214–1220. [Google Scholar] [CrossRef]
- Kioupis, L.I.; Maginn, E.J. Molecular simulation of poly-alpha-olefin synthetic lubricants: Impact of molecular architecture on performance properties. J. Phys. Chem. B 1999, 103, 10781–10790. [Google Scholar] [CrossRef]
- Moore, J.D.; Cui, S.T.; Cochran, H.D.; Cummings, P.T. Rheology of lubricant basestocks: A molecular dynamics study of C-30 isomers. J. Chem. Phys. 2000, 113, 8833–8840. [Google Scholar] [CrossRef]
- Savio, D.; Fillot, N.; Vergne, P. A Molecular Dynamics Study of the Transition from Ultra-Thin Film Lubrication Toward Local Film Breakdown. Tribol. Lett. 2013, 50, 207–220. [Google Scholar] [CrossRef]
- Liu, P.Z.; Yu, H.L.; Ren, N.; Lockwood, F.E.; Wang, Q.J. Pressure-Viscosity Coefficient of Hydrocarbon Base Oil through Molecular Dynamics Simulations. Tribol. Lett. 2015, 60. [Google Scholar] [CrossRef]
- Kong, Y.C.; Tildesley, D.J.; Alejandre, J. The molecular dynamics simulation of boundary-layer lubrication. Mol. Phys. 1997, 92, 7–18. [Google Scholar] [CrossRef]
- Berro, H.; Fillot, N.; Vergne, P. Molecular dynamics simulation of surface energy and ZDDP effects on friction in nano-scale lubricated contacts. Tribol. Int. 2010, 43, 1811–1822. [Google Scholar] [CrossRef]
- Eder, S.J.; Vernes, A.; Betz, G. On the Derjaguin Offset in Boundary-Lubricated Nanotribological Systems. Langmuir 2013, 29, 13760–13772. [Google Scholar] [CrossRef] [PubMed]
- Doig, M.; Warrens, C.P.; Camp, P.J. Structure and Friction of Stearic Acid and Oleic Acid Films Adsorbed on Iron Oxide Surfaces in Squalane. Langmuir 2014, 30, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Ewen, J.P.; Gattinoni, C.; Morgan, N.; Spikes, H.A.; Dini, D. Nonequilibrium Molecular Dynamics Simulations of Organic Friction Modifiers Adsorbed on Iron Oxide Surfaces. Langmuir 2016, 32, 4450–4463. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.G.; Siepmann, J.I. Transferable potentials for phase equilibria. 1. United-atom description of n-alkanes. J. Phys. Chem. B 1998, 102, 2569–2577. [Google Scholar] [CrossRef]
- Braga, C.; Travis, K.P. Computer simulation of the role of torsional flexibility on mass and momentum transport for a series of linear alkanes. J. Chem. Phys. 2012, 137. [Google Scholar] [CrossRef] [PubMed]
- Allen, W.; Rowley, R.L. Predicting the viscosity of alkanes using nonequilibrium molecular dynamics: Evaluation of intermolecular potential models. J. Chem. Phys. 1997, 106, 10273–10281. [Google Scholar] [CrossRef]
- Dysthe, D.K.; Fuchs, A.H.; Rousseau, B. Fluid transport properties by equilibrium molecular dynamics. III. Evaluation of united atom interaction potential models for pure alkanes. J. Chem. Phys. 2000, 112, 7581–7590. [Google Scholar] [CrossRef]
- Payal, R.S.; Balasubramanian, S.; Rudra, I.; Tandon, K.; Mahlke, I.; Doyle, D.; Cracknell, R. Shear viscosity of linear alkanes through molecular simulations: Quantitative tests for n-decane and n-hexadecane. Mol. Simulat. 2012, 38, 1234–1241. [Google Scholar] [CrossRef]
- Murzyn, K.; Bratek, M.; Pasenkiewicz-Gierula, M. Refined OPLS All-Atom Force Field Parameters for n-Pentadecane, Methyl Acetate, and Dimethyl Phosphate. J. Phys. Chem. B 2013, 117, 16388–16396. [Google Scholar] [CrossRef] [PubMed]
- Siu, S.W.I.; Pluhackova, K.; Bockmann, R.A. Optimization of the OPLS-AA Force Field for Long Hydrocarbons. J. Chem. Theory Comput. 2012, 8, 1459–1470. [Google Scholar] [CrossRef] [PubMed]
- Gordon, P.A. Development of intermolecular potentials for predicting transport properties of hydrocarbons. J. Chem. Phys. 2006, 125. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.G.; Thompson, A.P. Industrial property prediction using Towhee and LAMMPS. Fluid Phase Equilibr. 2004, 217, 105–110. [Google Scholar] [CrossRef]
- Ye, X.G.; Cui, S.T.; de Almeida, V.F.; Khomami, B. Effect of varying the 1-4 intramolecular scaling factor in atomistic simulations of long-chain N-alkanes with the OPLS-AA model. J. Mol. Model. 2013, 19, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Jabbarzadeh, A.; Harrowell, P.; Tanner, I. The structural origin of the complex rheology in thin dodecane films: Three routes to low friction. Tribol. Int. 2007, 40, 1574–1586. [Google Scholar] [CrossRef]
- Docherty, H.; Cummings, P.T. Direct evidence for fluid-solid transition of nanoconfined fluids. Soft Matter 2010, 6, 1640–1643. [Google Scholar] [CrossRef]
- Bareman, J.P.; Klein, M.L. Collective tilt behavior in dense, substrate-supported monolayers of long-chain molecules—A molecular dynamics study. J. Phys. Chem. 1990, 94, 5202–5205. [Google Scholar] [CrossRef]
- Moller, M.A.; Tildesley, D.J.; Kim, K.S.; Quirke, N. Molecular-Dynamics Simulation of a Langmuir-Blodgett Film. J. Chem. Phys. 1991, 94, 8390–8401. [Google Scholar] [CrossRef]
- Mar, W.; Klein, M.L. Molecular-dynamics study of the self-assembled monolayer composed of S(CH2)(14)CH3 molecules using an all-atoms model. Langmuir 1994, 10, 188–196. [Google Scholar] [CrossRef]
- Bolton, K.; Bosio, S.B.M.; Hase, W.L.; Schneider, W.F.; Hass, K.C. Comparison of explicit and united atom models for alkane chains physisorbed on alpha-Al2O3 (0001). J. Phys. Chem. B 1999, 103, 3885–3895. [Google Scholar] [CrossRef]
- Kong, L.T.; Denniston, C.; Muser, M.H. The crucial role of chemical detail for slip-boundary conditions: Molecular dynamics simulations of linear oligomers between sliding aluminum surfaces. Model. Simul. Mater. Sci. Eng. 2010, 18. [Google Scholar] [CrossRef]
- Ta, D.T.; Tieu, A.K.; Zhu, H.T.; Kosasih, B. Thin film lubrication of hexadecane confined by iron and iron oxide surfaces: A crucial role of surface structure. J. Chem. Phys. 2015, 143. [Google Scholar] [CrossRef] [PubMed]
- Plimpton, S. Fast parallel algorithms for short-range molecular-dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical-integration of Cartesian equations of motion of a system with constraints—Molecular-dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Green, M.S. Markoff Random Processes and the Statistical Mechanics of Time-Dependent Phenomena. II. Irreversible Processes in Fluids. J. Chem. Phys. 1954, 22, 398–413. [Google Scholar]
- Kubo, R. Statistical-Mechanical Theory of Irreversible Processes. I. General Theory and Simple Applications to Magnetic and Conduction Problems. J. Phys. Soc. Jpn. 1957, 12, 570–586. [Google Scholar] [CrossRef]
- Cui, S.T.; Cummings, P.T.; Cochran, H.D. The calculation of viscosity of liquid n-decane and n-hexadecane by the Green-Kubo method. Mol. Phys. 1998, 93, 117–121. [Google Scholar] [CrossRef]
- Chynoweth, S.; Michopoulos, Y. An improved potential model for n-hexadecane molecular-dynamics simulations under extreme conditions. Mol. Phys. 1994, 81, 133–141. [Google Scholar] [CrossRef]
- Nath, S.K.; Escobedo, F.A.; de Pablo, J.J. On the simulation of vapor-liquid equilibria for alkanes. J. Chem. Phys. 1998, 108, 9905–9911. [Google Scholar] [CrossRef]
- Schuler, L.D.; Daura, X.; Van Gunsteren, W.F. An improved GROMOS96 force field for aliphatic hydrocarbons in the condensed phase. J. Comput. Chem. 2001, 22, 1205–1218. [Google Scholar] [CrossRef]
- Potoff, J.J.; Bernard-Brunel, D.A. Mie Potentials for Phase Equilibria Calculations: Application to Alkanes and Perfluoroalkanes. J. Phys. Chem. B 2009, 113, 14725–14731. [Google Scholar] [CrossRef] [PubMed]
- Mayo, S.L.; Olafson, B.D.; Goddard, W.A. Dreiding—A generic force-field for molecular simulations. J. Phys. Chem. 1990, 94, 8897–8909. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; TiradoRives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Sun, H. COMPASS: An ab initio force-field optimized for condensed-phase applications—Overview with details on alkane and benzene compounds. J. Phys. Chem. B 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Hockney, R.W.; Eastmond, J.W. Computer Simulation Using Particles; CRC Press: Bristol, PA, USA, 1989. [Google Scholar]
- Nose, S. A Molecular-Dynamics Method for Simulations in the Canonical Ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Shinoda, W.; Shiga, M.; Mikami, M. Rapid estimation of elastic constants by molecular dynamics simulation under constant stress. Phys. Rev. B 2004, 69. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Muller-Plathe, F. Reversing the perturbation in nonequilibrium molecular dynamics: An easy way to calculate the shear viscosity of fluids. Phys. Rev. E 1999, 59, 4894–4898. [Google Scholar] [CrossRef]
- Campen, S.; Green, J.; Lamb, G.; Atkinson, D.; Spikes, H. On the Increase in Boundary Friction with Sliding Speed. Tribol. Lett. 2012, 48, 237–248. [Google Scholar] [CrossRef]
- Maslen, E.N.; Streltsov, V.A.; Streltsova, N.R.; Ishizawa, N. Synchrotron X-ray study of the electron-density in alpha-Fe2O3. Acta Crystallogr. Sect. B Struct. Sci. 1994, 50, 435–441. [Google Scholar] [CrossRef]
- Ramachandran, S.; Tsai, B.L.; Blanco, M.; Chen, H.; Tang, Y.C.; Goddard, W.A. Self-assembled monolayer mechanism for corrosion inhibition of iron by imidazolines. Langmuir 1996, 12, 6419–6428. [Google Scholar] [CrossRef]
- Kamath, G.; Cao, F.; Potoff, J.J. An improved force field for the prediction of the vapor-liquid equilibria for carboxylic acids. J. Phys. Chem. B 2004, 108, 14130–14136. [Google Scholar] [CrossRef]
- Yeh, I.C.; Berkowitz, M.L. Ewald summation for systems with slab geometry. J. Chem. Phys. 1999, 111, 3155–3162. [Google Scholar] [CrossRef]
- Schneider, T.; Stoll, E. Molecular-dynamics study of a three-dimensional one-component model for distortive phase-transitions. Phys. Rev. B 1978, 17, 1302–1322. [Google Scholar] [CrossRef]
- Liem, S.Y.; Brown, D.; Clarke, J.H.R. Investigation of the homogeneous-shear nonequilibrium-molecular-dynamics method. Phys. Rev. A 1992, 45, 3706–3713. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, S.; Todd, B.D.; Searles, D.J. Thermostating highly confined fluids. J. Chem. Phys. 2010, 132. [Google Scholar] [CrossRef] [PubMed]
- Toton, D.; Lorenz, C.D.; Rompotis, N.; Martsinovich, N.; Kantorovich, L. Temperature control in molecular dynamic simulations of non-equilibrium processes. J. Phys. Condens. Matter 2010, 22. [Google Scholar] [CrossRef] [PubMed]
- Campen, S.; Green, J.H.; Lamb, G.D.; Spikes, H.A. In Situ Study of Model Organic Friction Modifiers Using Liquid Cell AFM; Saturated and Mono-unsaturated Carboxylic Acids. Tribol. Lett. 2015, 57. [Google Scholar] [CrossRef]
- Lundgren, S.M.; Ruths, M.; Danerlov, K.; Persson, K. Effects of unsaturation on film structure and friction of fatty acids in a model base oil. J. Colloid Interface Sci. 2008, 326, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Savio, D.; Fillot, N.; Vergne, P.; Hetzler, H.; Seemann, W.; Espejel, G.E.M. A Multiscale Study on the Wall Slip Effect in a Ceramic-Steel Contact With Nanometer-Thick Lubricant Film by a Nano-to-Elastohydrodynamic Lubrication Approach. J. Tribol. 2015, 137. [Google Scholar] [CrossRef]
- Yoshizawa, H.; Chen, Y.L.; Israelachvili, J. Fundamental mechanisms of interfacial friction: 1. relation between adhesion and friction. J. Phys. Chem. 1993, 97, 4128–4140. [Google Scholar] [CrossRef]
- Jahanmir, S.; Beltzer, M. An adsorption model for friction in boundary lubrication. ASLE Trans. 1986, 29, 423–430. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ewen, J.P.; Gattinoni, C.; Thakkar, F.M.; Morgan, N.; Spikes, H.A.; Dini, D. A Comparison of Classical Force-Fields for Molecular Dynamics Simulations of Lubricants. Materials 2016, 9, 651. https://doi.org/10.3390/ma9080651
Ewen JP, Gattinoni C, Thakkar FM, Morgan N, Spikes HA, Dini D. A Comparison of Classical Force-Fields for Molecular Dynamics Simulations of Lubricants. Materials. 2016; 9(8):651. https://doi.org/10.3390/ma9080651
Chicago/Turabian StyleEwen, James P., Chiara Gattinoni, Foram M. Thakkar, Neal Morgan, Hugh A. Spikes, and Daniele Dini. 2016. "A Comparison of Classical Force-Fields for Molecular Dynamics Simulations of Lubricants" Materials 9, no. 8: 651. https://doi.org/10.3390/ma9080651
APA StyleEwen, J. P., Gattinoni, C., Thakkar, F. M., Morgan, N., Spikes, H. A., & Dini, D. (2016). A Comparison of Classical Force-Fields for Molecular Dynamics Simulations of Lubricants. Materials, 9(8), 651. https://doi.org/10.3390/ma9080651