
H2 Adsorbed Site-to-Site Electronic Delocalization within IRMOF-1: Understanding Non-Negligible Interactions at High Pressure

Abstract
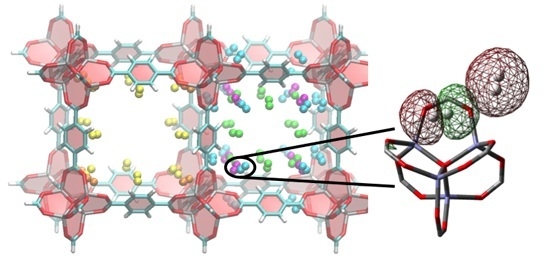
:
1. Introduction
2. Computational Methods
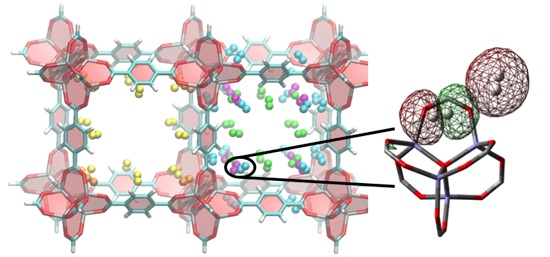
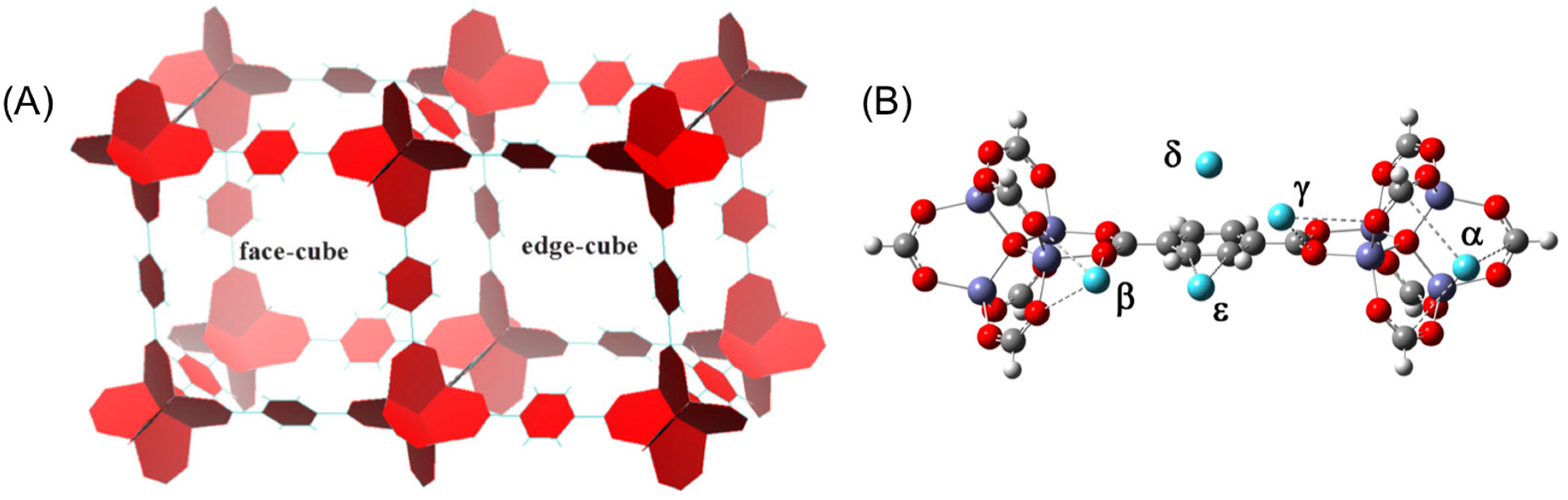
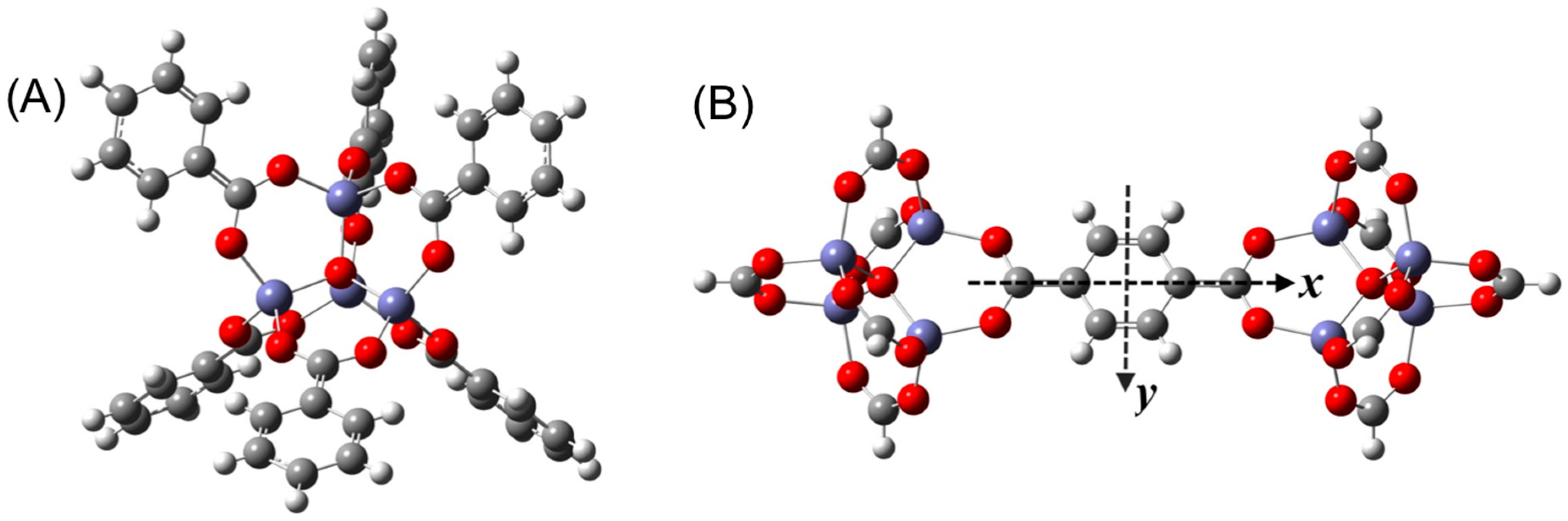
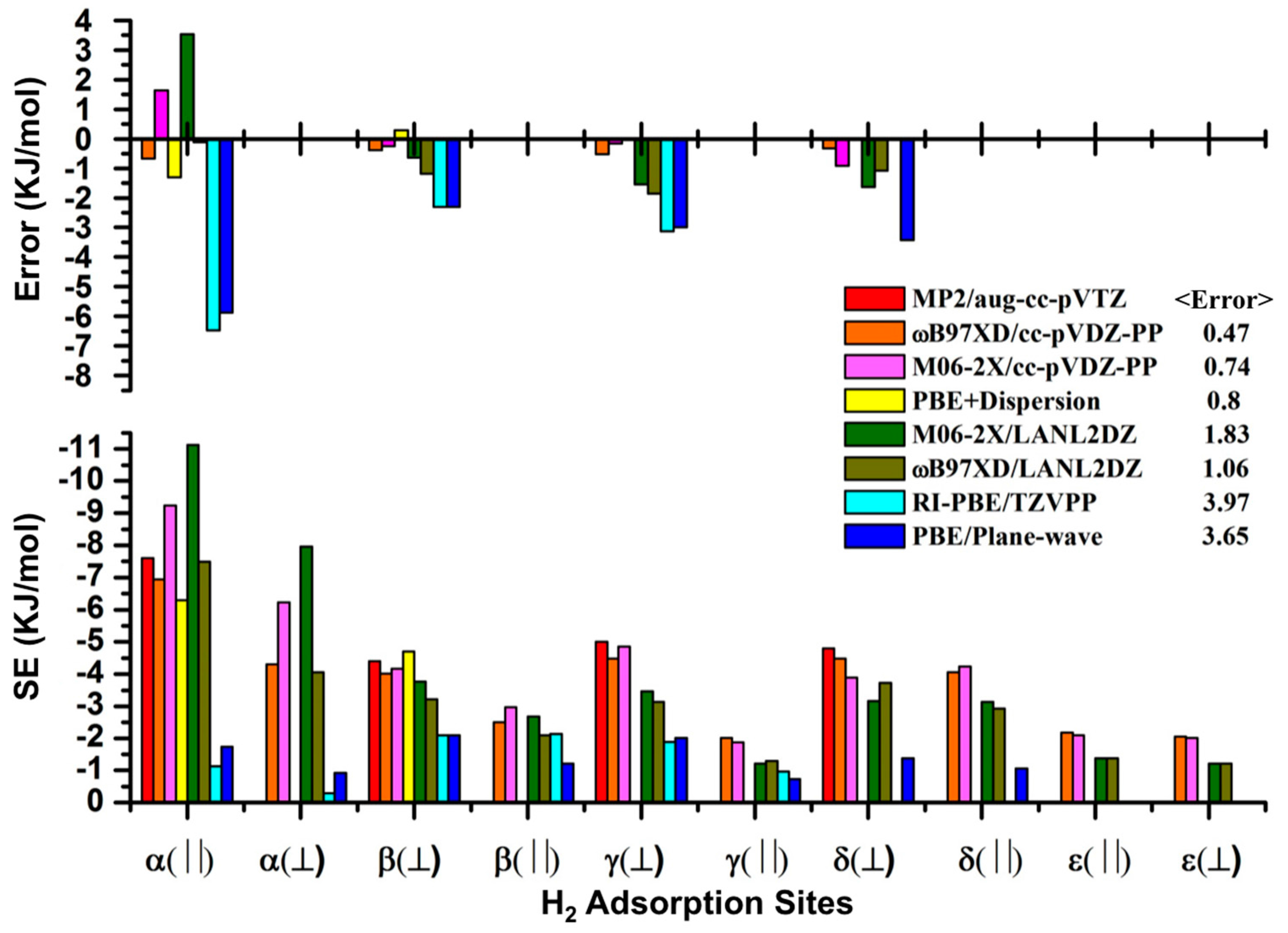
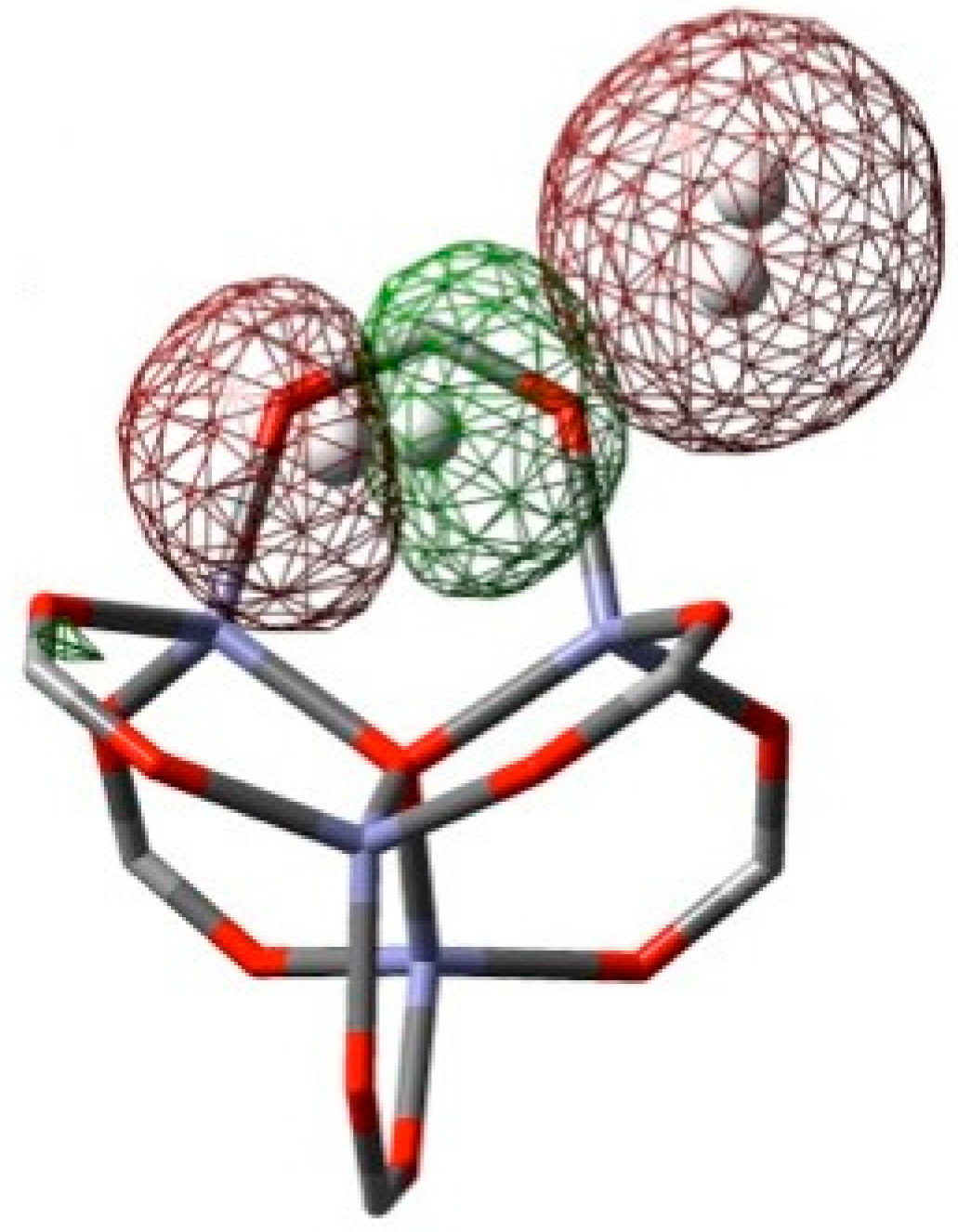
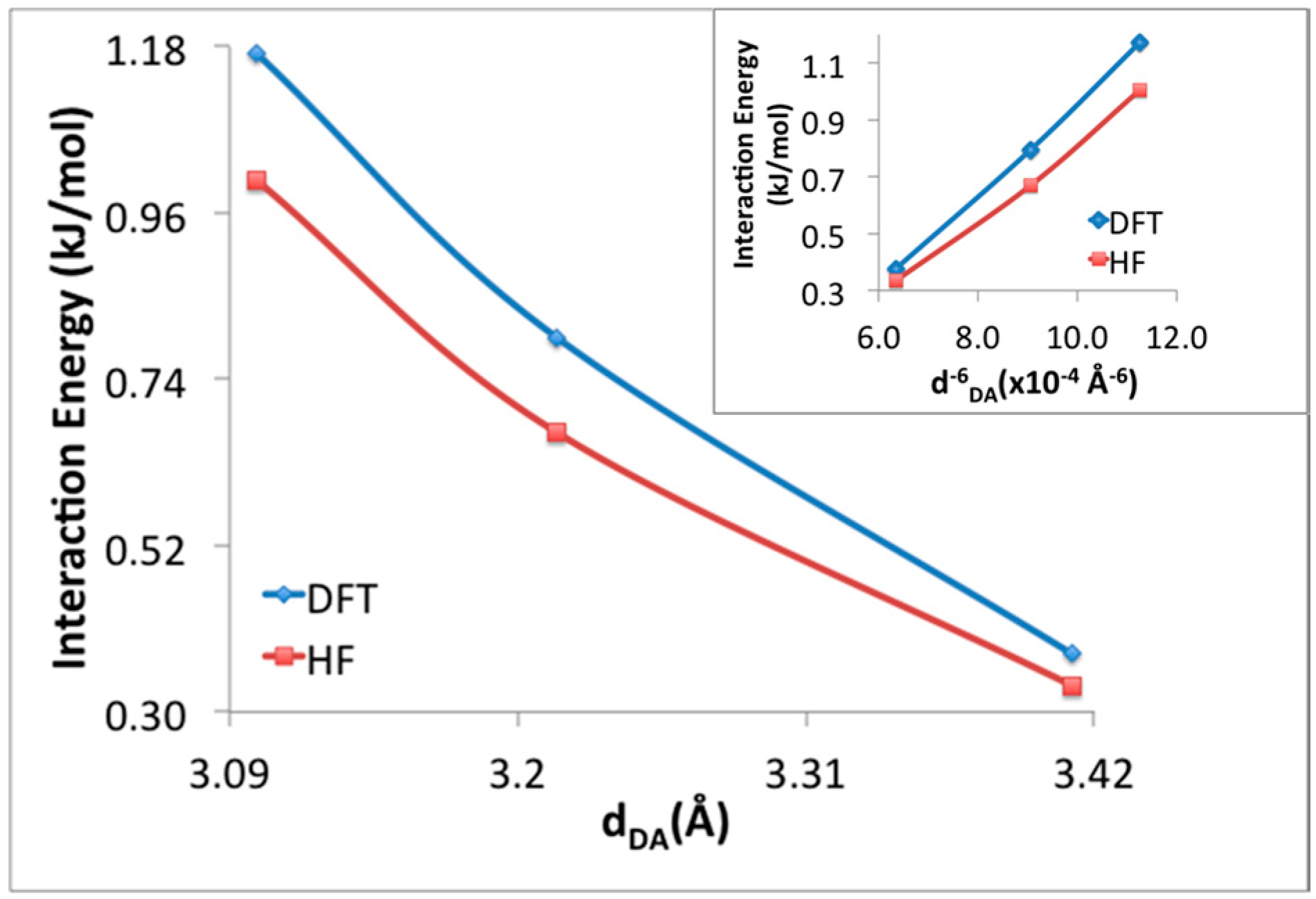
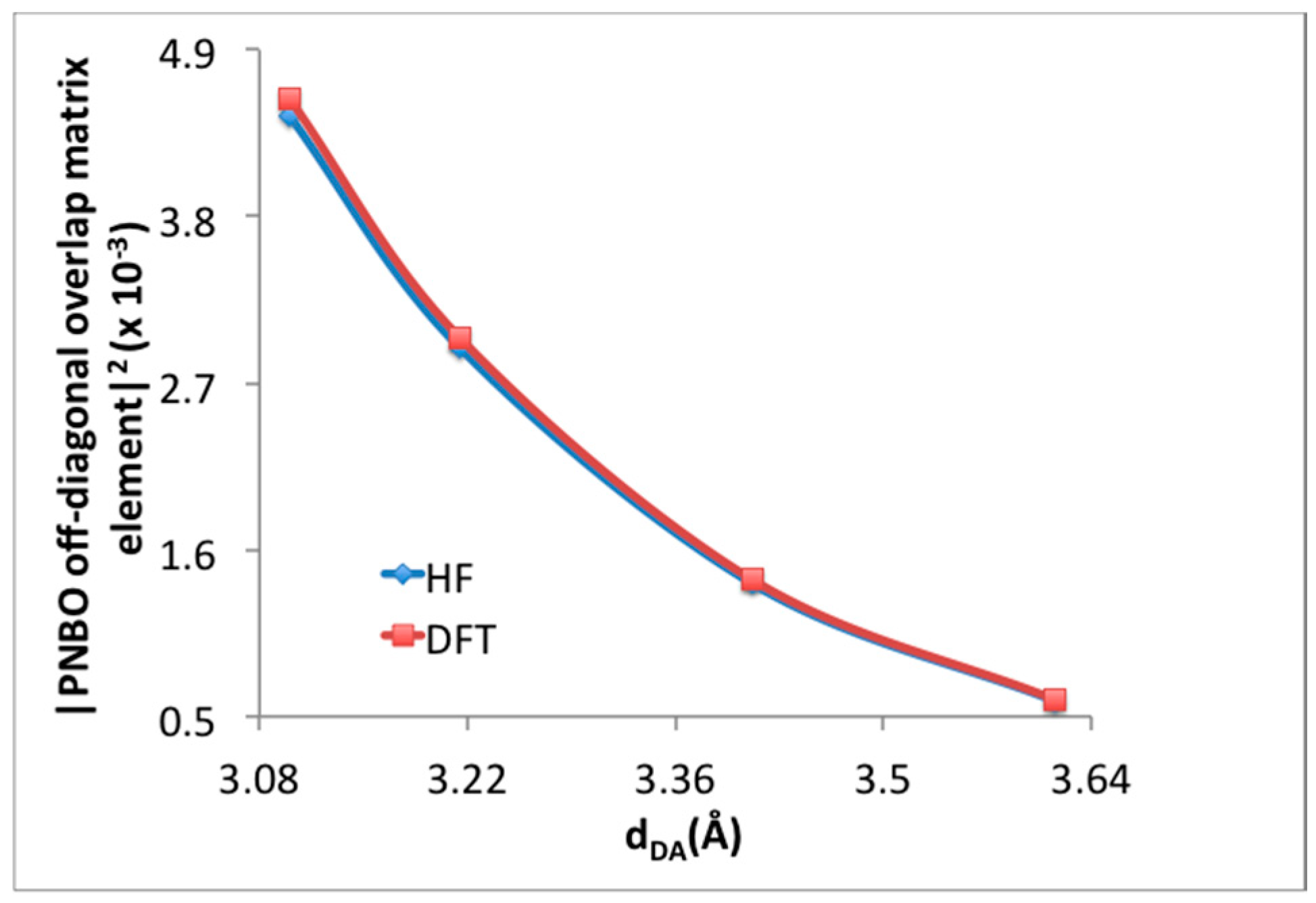
3. Results and Discussion
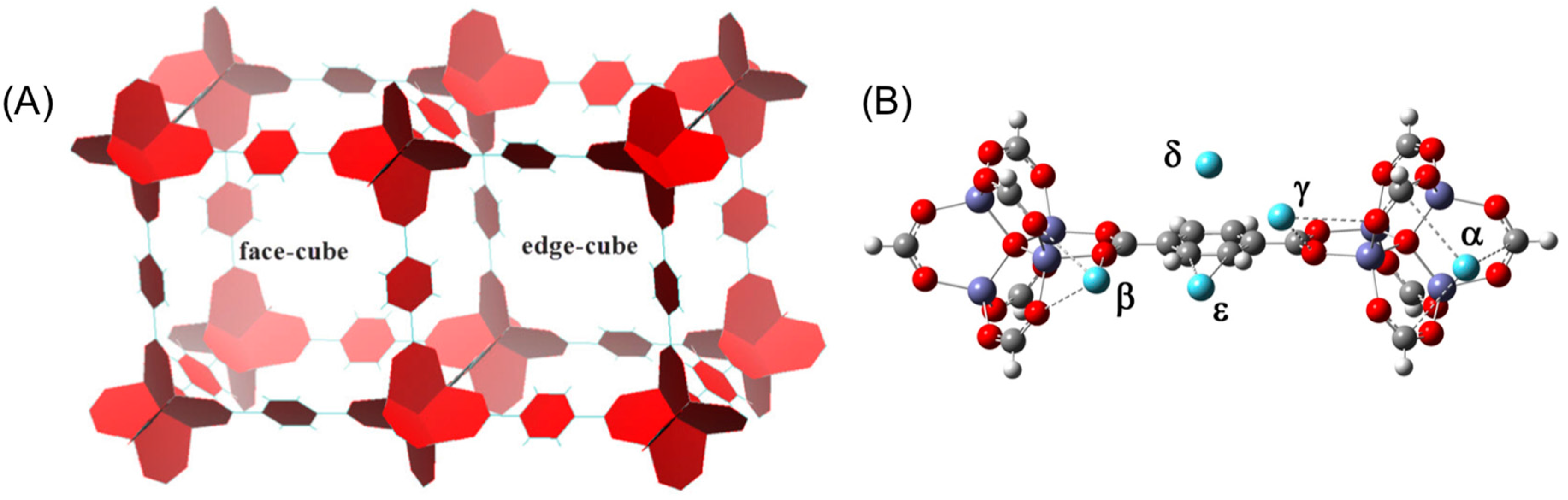
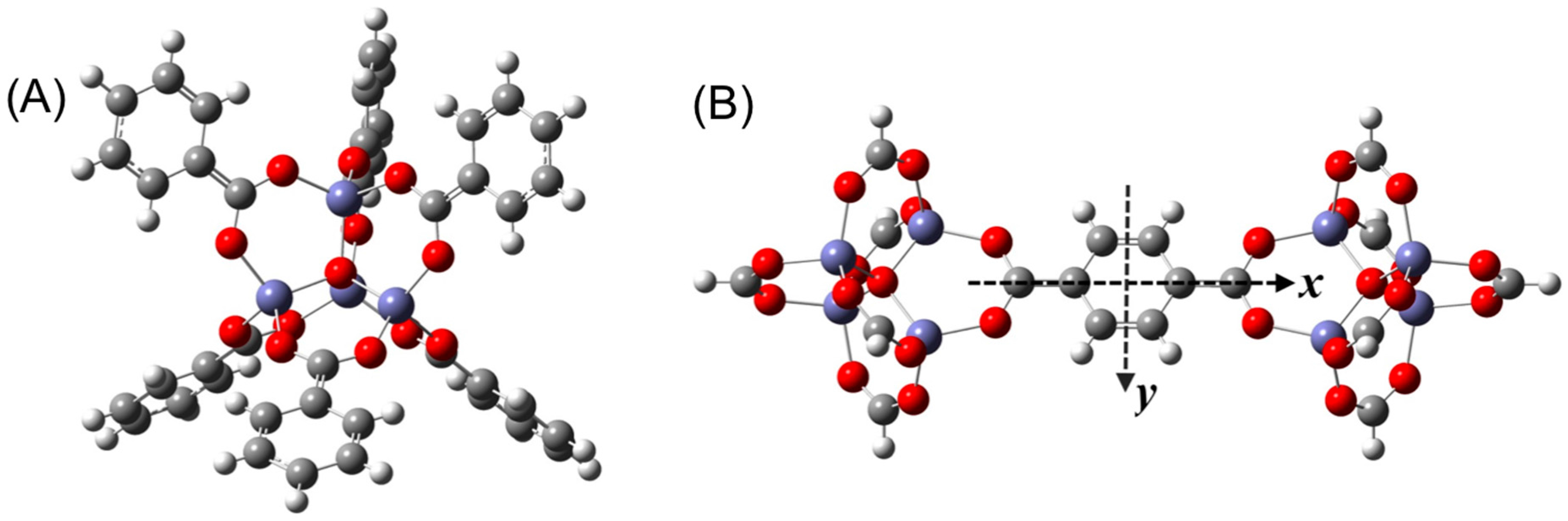
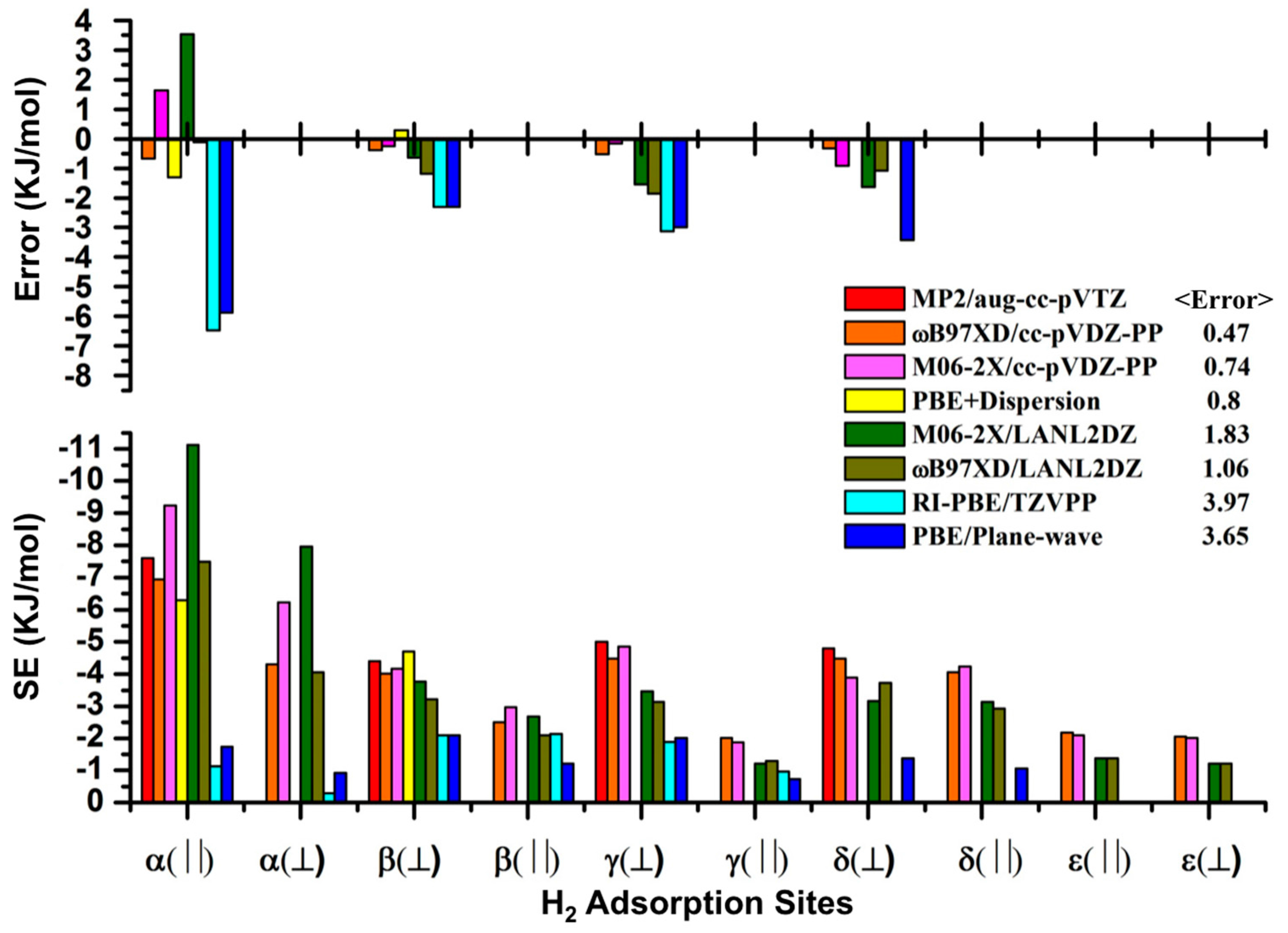
3.1. Benchmarking DFT Methods in Fragment Models
3.2. DFT Optimization of H2 Configurations in the Super-Cell Cluster as a Function of Concentration
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| IRMOFs | Isoreticular metal organic frameworks |
| QM | Quantum Mechanical |
| NBO | Natural Bond Order |
| MP2 | Møller-Plesset 2nd order perturbation theory |
| BDC | 1,4-benzenedicarboxylate |
| BSSE | basis set superposition error |
| DFT | Density Functional Theory |
| EDA | Energy Decomposition Analysis |
| NBO | Natural Bond Order |
| HF | Hartree-Fock |
| CP | counterpoise |
| PES | Potential Energy Surface |
| NBOs | Natural Bond Orbitals |
| SE | Stabilization Energy |
| NLMOs | Natural Localized Molecular Orbitals |
| SAPT | Symmetry-Adapted Perturbation Theory |
References
- Rosi, N.L.; Eckert, J.; Eddaoudi, M.; Vodak, D.; Kim, J.; O’Keeffe, M.; Yaghi, O.M. Hydrogen Storage in Microporous Metal-Organic Frameworks. Science 2003, 300, 1127–1129. [Google Scholar] [CrossRef] [PubMed]
- Eddaoudi, M.; Kim, J.; Rosi, N.L.; Vodak, D.; Wachter, J.; O’Keeffe, M.; Yaghi, O.M. Systematic Design of Pore Size and Functionality in Isoreticular MOFs and Their Application in Methane Storage. Science 2002, 295, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Kaye, S.S.; Dailly, A.; Yaghi, O.M.; Long, J.R. Impact of Preperation and Handling on the Hydrogen Storage Properties of Zn4O(1,4-benzenedicarboxylate)3(MOF-5). J. Am. Chem. Soc. 2007, 129, 14176–14177. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Wu, H.; Hartman, M.R.; Yildirim, T. Hydrogen and Methane Adsorption in Metal−Organic Frameworks: A High-Pressure Volumetric Study. J. Phys. Chem. C 2007, 111, 16131–16137. [Google Scholar] [CrossRef]
- Dailly, A.; Vajo, J.J.; Ahn, C.C. Saturation of Hydrogen Sorption in Zn Benzenedicarboxylate and Zn Napthalenedicarboxylate. J. Phys. Chem. B 2006, 110, 1099–1101. [Google Scholar] [CrossRef] [PubMed]
- Panella, B.; Hirscher, M. Hydrogen Physisorption in Metal-Organic Porous Crystals. Adv. Mater. 2005, 17, 538–541. [Google Scholar] [CrossRef]
- Panella, B.; Hirscher, M.; Pütter, H.; Müller, U. Hydrogen Adsorption in Metal-Organic Frameworks: Cu-MOFs and Zn-MOFs Compared. Adv. Funct. Mater. 2006, 16, 520–524. [Google Scholar] [CrossRef]
- Li, Y.; Yang, R.T. Hydrogen Storage in Metal-Organic Frameworks by Bridged Hydrogen Spillover. J. Am. Chem. Soc. 2006, 128, 8136–8137. [Google Scholar] [CrossRef] [PubMed]
- Sillar, K.; Hofmann, A.; Sauer, J. Ab Initio Study of Hydrogen Adsorption in MOF-5. J. Am. Chem. Soc. 2009, 131, 4143–4150. [Google Scholar] [CrossRef] [PubMed]
- Rowsell, J.L.C.; Eckert, J.; Yaghi, O.M. Characterization of H2 binding Sites in Prototypical Metal-Organic Frameworks by Inelastic Neutron Scattering. J. Am. Chem. Soc. 2005, 127, 14904–14910. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, T.; Hartman, M.R. Direct Observation of Hydrogen Adsorption Sites and Nanocage Formation in Metal-Organic Frameworks. Phys. Rev. Lett. 2005, 95, 215504. [Google Scholar] [CrossRef] [PubMed]
- Sagara, T.; Klassen, J.; Ganz, E. Computational study of hydrogen binding by metal-organic framework-5. J. Chem. Phys. 2004, 121, 12543–12547. [Google Scholar] [CrossRef] [PubMed]
- Klontzas, E.; Mavrandonakis, A.; Froudakis, G.E.; Carissan, Y.; Klopper, W. Molecular Hydrogen Interaction with IRMOF-1: A Multiscale Theoretical Study. J. Phys. Chem. C 2007, 111, 13635–13640. [Google Scholar] [CrossRef]
- Kuc, A.; Heine, T.; Seifert, G.; Duarte, H.A. H2 Adsorption in Metal-Organic Frameworks: Dispersion or Electrostatic Interactions? Chem. Eur. J. 2008, 14, 6597–6600. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.; Ceder, G. A Density Functional Theory Study of Hydrogen Adsorption in MOF-5. J. Phys. Chem. B 2005, 109, 17974–17983. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhong, C. Molecular Simulation of Adsorption and Diffusion of Hydrogen in Metal-Organic Frameworks. J. Phys. Chem. B 2005, 109, 11862–11864. [Google Scholar] [CrossRef] [PubMed]
- Bordiga, S.; Vitillo, J.G.; Ricchiardi, G.; Regli, L.; Cocina, D.; Zecchina, A.; Arstad, B.; Bjorgen, M.; Hafizovic, J.; Lillerud, K.P. Interaction of Hydrogen with MOF-5. J. Phys. Chem. B 2005, 109, 18237–18242. [Google Scholar] [CrossRef] [PubMed]
- Tatsuhiko, S.; Klassen, J.; Ortony, J.; Ganz, E. Binding Energies of Hydrogen Molecules to Isoreticular Metal-Organic Framework Materials. J. Chem. Phys. 2005, 123, 014701–014704. [Google Scholar]
- Tsivion, E.; Long, J.R.; Head-Gordon, M. Hydrogen Physisorption on Metal–Organic Framework Linkers and Metalated Linkers: A Computational Study of the Factors That Control Binding Strength. J. Am. Chem. Soc. 2014, 136, 17827–17835. [Google Scholar] [CrossRef] [PubMed]
- Mulder, F.M.; Dingemans, T.J.; Wagemaker, M.; Kearley, G.J. Modeling of Hydrogen Adsorption in the Metal Organic Framework MOF5. Chem. Phys. 2005, 317, 113–118. [Google Scholar] [CrossRef]
- Samanta, A.; Furuta, T.; Li, J. Theoretical Assessment of the Elastic Constants and Hydrogen Storage Capacity of Some Metal-Organic Framework Materials. J. Chem. Phys. 2006, 125, 084714–084718. [Google Scholar] [CrossRef] [PubMed]
- Grosch, J.S.; Paesani, F. Molecular-Level Characterization of the Breathing Behavior of the Jungle-Gym-type DMOF-1 Metal−Organic Framework. J. Am. Chem. Soc. 2012, 134, 4207–4215. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Figgen, D.; Rauhut, G.; Dolg, M.; Stoll, H. Energy-consistent pseudopotentials for group 11 and 12 atoms: Adjustment to multi-configuration Dirac–Hartree–Fock data. Chem. Phys. 2005, 311, 227–244. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. Calculation of Small Molecular Interactions by Differences of Separate Total Energies—Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Simon, S.; Duran, M.; Dannenberg, J.J. How does basis set superposition error change the potential surfaces for hydrogen bonded dimers? J. Chem. Phys. 1996, 105, 11024–11031. [Google Scholar] [CrossRef]
- Li, H.; Eddaoudi, M.; O’Keeffe, M.; Yaghi, O.M. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 1999, 402, 276–279. [Google Scholar]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Montgomery, J.A. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, 1st ed.; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Mulder, F.M.; Dingemans, T.J.; Schimmel, H.G.; Ramirez-Cuesta, A.J.; Kearley, G. Hydrogen adsorption strength and sites in the metal organic framework MOF5: Comparing experiment and model calculations. Chem. Phys. 2008, 351, 72–76. [Google Scholar] [CrossRef]
- Dincă, M.; Han, W.S.; Liu, Y.; Dailly, A.; Brown, C.M.; Long, J.R. Observation of Cu2+–H2 Interactions in a Fully Desolvated Sodalite-Type Metal–Organic Framework. Angew. Chem. Int. Ed. 2007, 46, 1419–1422. [Google Scholar] [CrossRef] [PubMed]
- Hamel, S.; Côté, M. First-principles study of the rotational transitions of physisorbed over benzene. J. Chem. Phys. 2004, 121, 12618–12625. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, S.; Sedlak, R.; Hobza, P. On the Nautre of Stabilization: In Weak, Medium and Strong Charge-Transfer Complexes: CCSD(T)/CBS and SAPT Calculations. J. Phys. Chem. A 2011, 115, 9422–9428. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
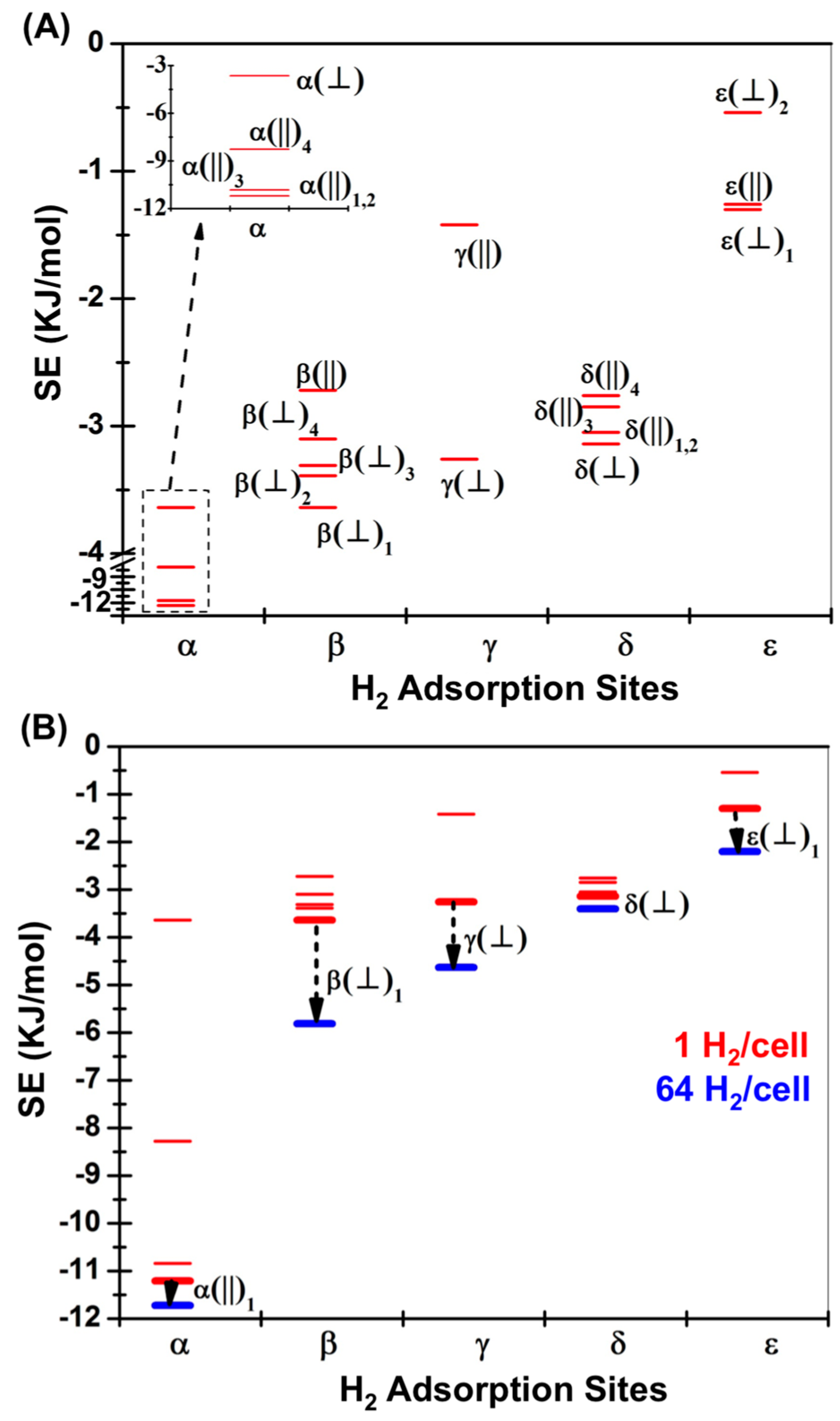
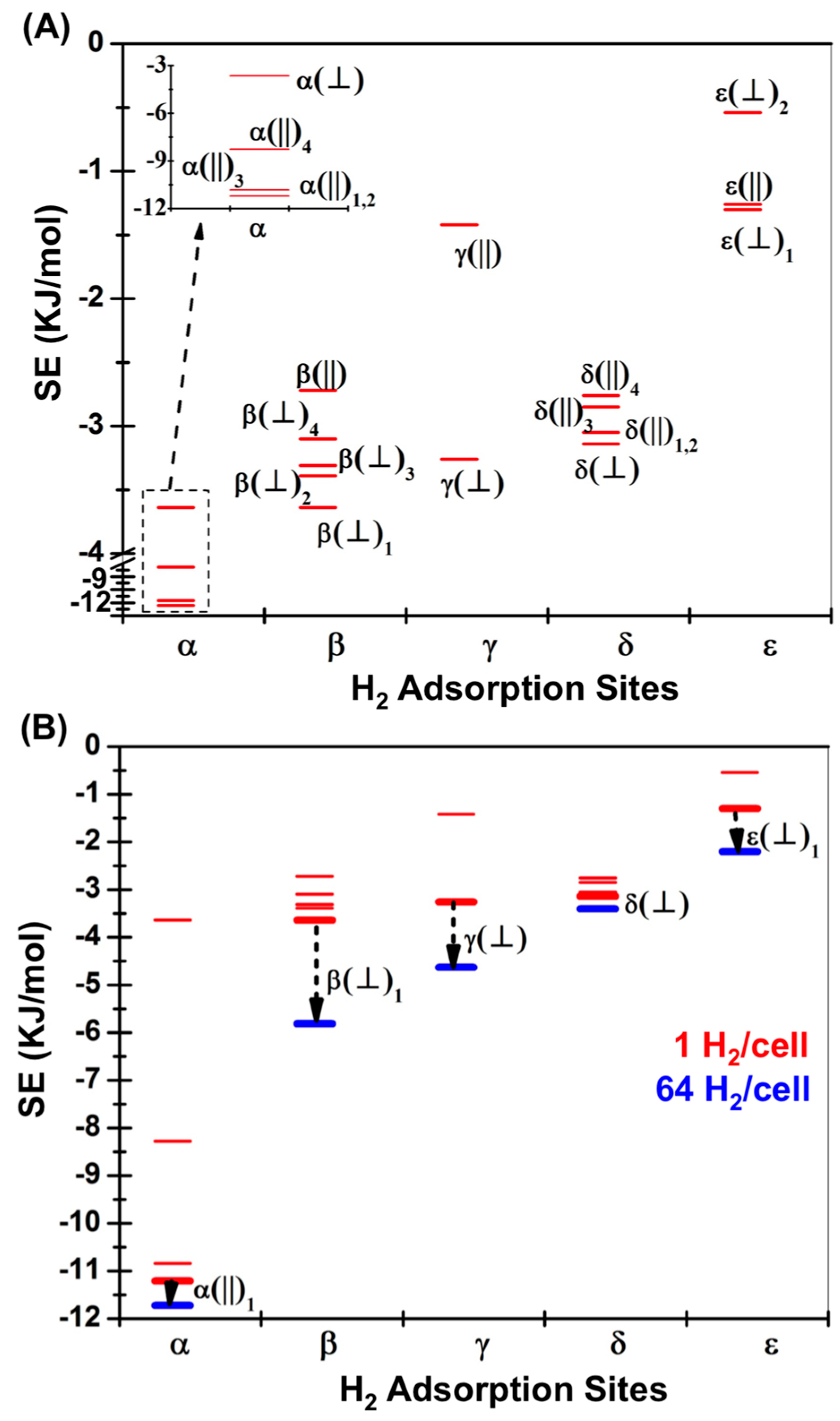
| H2 Position | PBE in VASP [14] | M06-2X (1 H2/Cell) | M06-2X (64 H2/Cell) | ΩB97XD (1 H2/Cell) | |
|---|---|---|---|---|---|
| α(||)1 | II(B) | −1.31 | −11.21 (−11.13) | −11.72 | −9.37 (−9.24) |
| α(||)2 | II(C) | 1.73 | −11.21 | – | −9.41 |
| α(||)3 | II(D) | −1.28 | −10.84 | – | −9.41 |
| α(||)4 | II(E) | −1.73 | −3.64 | – | −9.41 |
| α(⏊) | II(A) | −0.92 | −8.28 (−7.96) | – | −6.61 (−6.23) |
| β(⏊)1 | I(A) | −2.09 | −3.64 (−3.77) | −5.81 | −4.05 (−4.16) |
| β(⏊)2 | I(B) | −1.92 | −3.39 | – | −4.01 |
| β(⏊)3 | I(D) | −1.95 | −3.31 | – | −3.81 |
| β(⏊)4 | I(E) | −1.43 | −3.10 | – | −4.02 |
| β(||) | I(C) | −1.21 | −2.72 (−2.68) | – | −3.14 (−2.97) |
| γ(⏊) | III(A) | −2.01 | −3.26 (−3.46) | −4.63 | −4.85 (−4.85) |
| γ(||) | III(C) | −1.15 | −1.42 (−1.21) | – | −2.22 (−1.87) |
| γ | III(B) | −0.73 | – | – | – |
| δ(⏊) | IV(A) | −1.38 | −3.14 (−3.17) | −3.40 | −4.06 (−3.89) |
| δ(||)1 | IV(B) | −1.06 | −3.05 (−3.14) | – | −4.35 (−4.23) |
| δ(||)2 | IV(C) | −1.02 | −3.05 | – | −4.27 |
| δ(||)3 | IV(D) | −0.96 | −2.85 | – | −2.97 |
| δ(||)4 | IV(E) | −0.92 | −2.76 | – | −4.06 |
| ε(⏊)1 | V(A) | −1.22 | −1.30 (−1.21) | −2.20 | −2.30 (−2.01) |
| ε(||) | V(B) | −0.98 | −1.26 (−1.38) | – | −2.22 (−2.09) |
| ε(⏊)2 | V(C) | −0.50 | −0.54 | – | −1.21 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Kucukkal, M.U.; Clark, A.E. H2 Adsorbed Site-to-Site Electronic Delocalization within IRMOF-1: Understanding Non-Negligible Interactions at High Pressure. Materials 2016, 9, 578. https://doi.org/10.3390/ma9070578
Wu J, Kucukkal MU, Clark AE. H2 Adsorbed Site-to-Site Electronic Delocalization within IRMOF-1: Understanding Non-Negligible Interactions at High Pressure. Materials. 2016; 9(7):578. https://doi.org/10.3390/ma9070578
Chicago/Turabian StyleWu, Jian, Mustafa U. Kucukkal, and Aurora E. Clark. 2016. "H2 Adsorbed Site-to-Site Electronic Delocalization within IRMOF-1: Understanding Non-Negligible Interactions at High Pressure" Materials 9, no. 7: 578. https://doi.org/10.3390/ma9070578
APA StyleWu, J., Kucukkal, M. U., & Clark, A. E. (2016). H2 Adsorbed Site-to-Site Electronic Delocalization within IRMOF-1: Understanding Non-Negligible Interactions at High Pressure. Materials, 9(7), 578. https://doi.org/10.3390/ma9070578