
Effect of Na3PO4 on the Hydration Process of Alkali-Activated Blast Furnace Slag

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Sample Preparation
2.2. Physical‑Mechanical Tests
2.3. Heat Evolution Rates Measurement
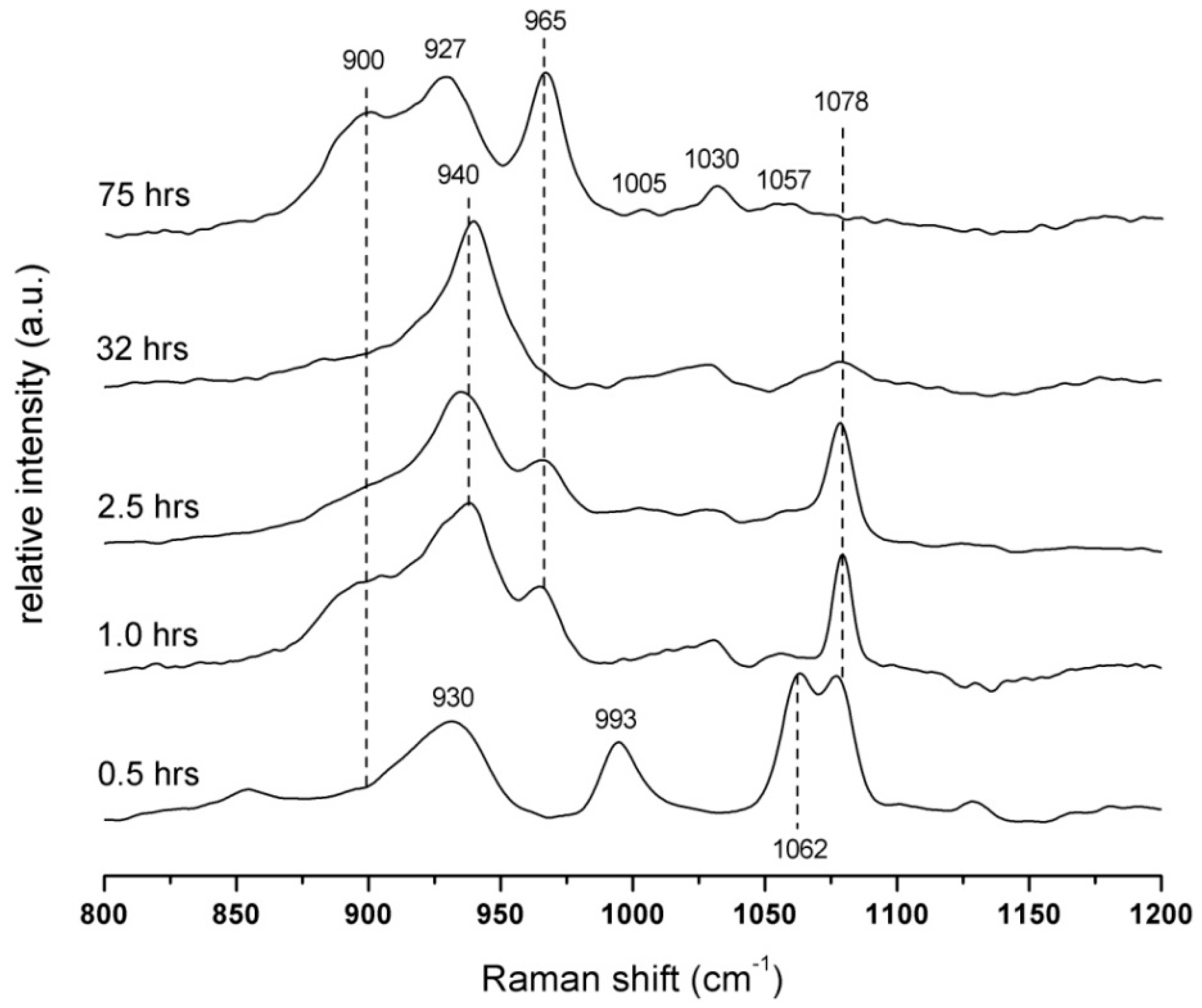
2.4. Raman Spectroscopy
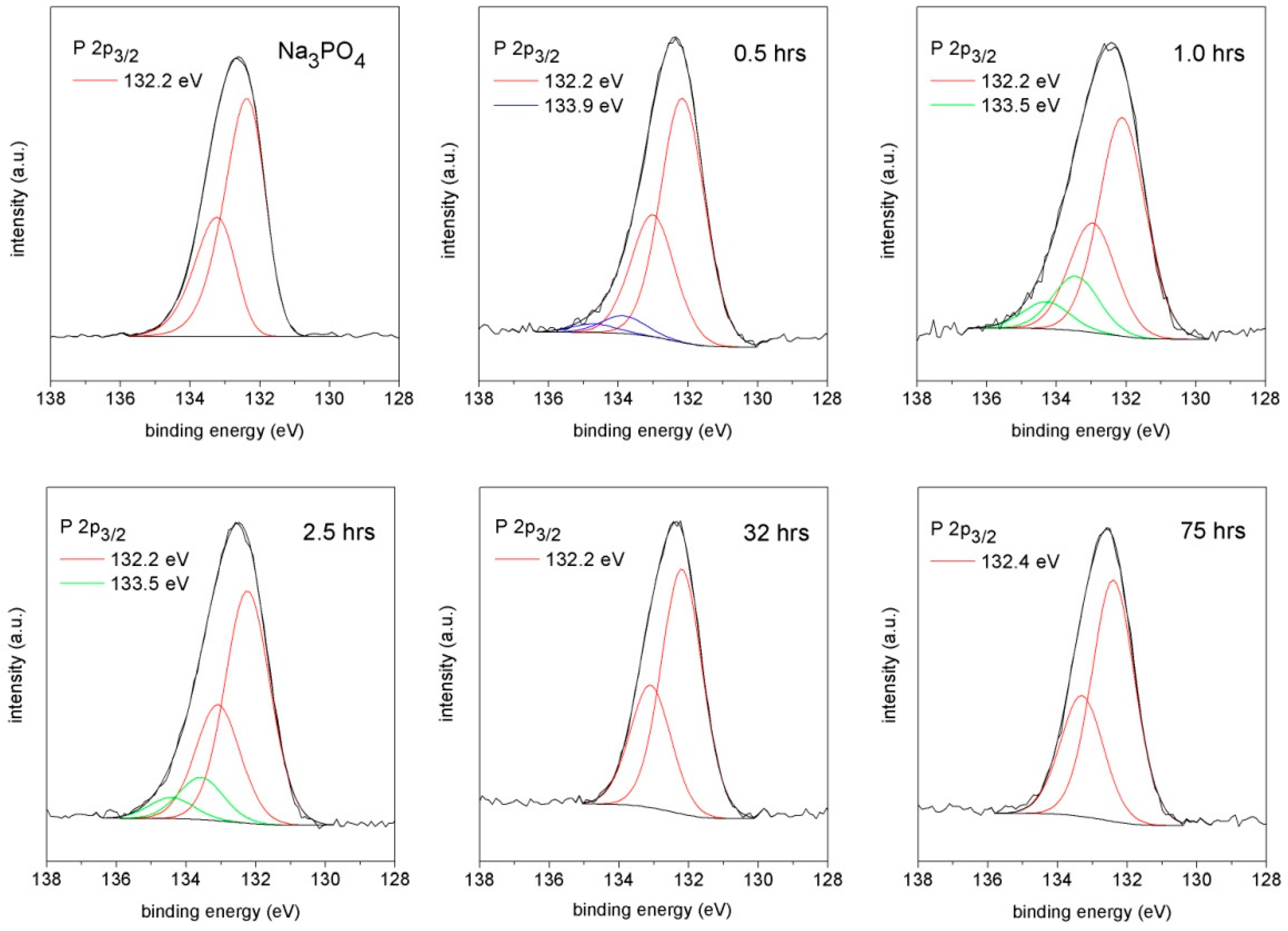
2.5. X-ray Photoelectron Spectroscopy
3. Results and Discussion
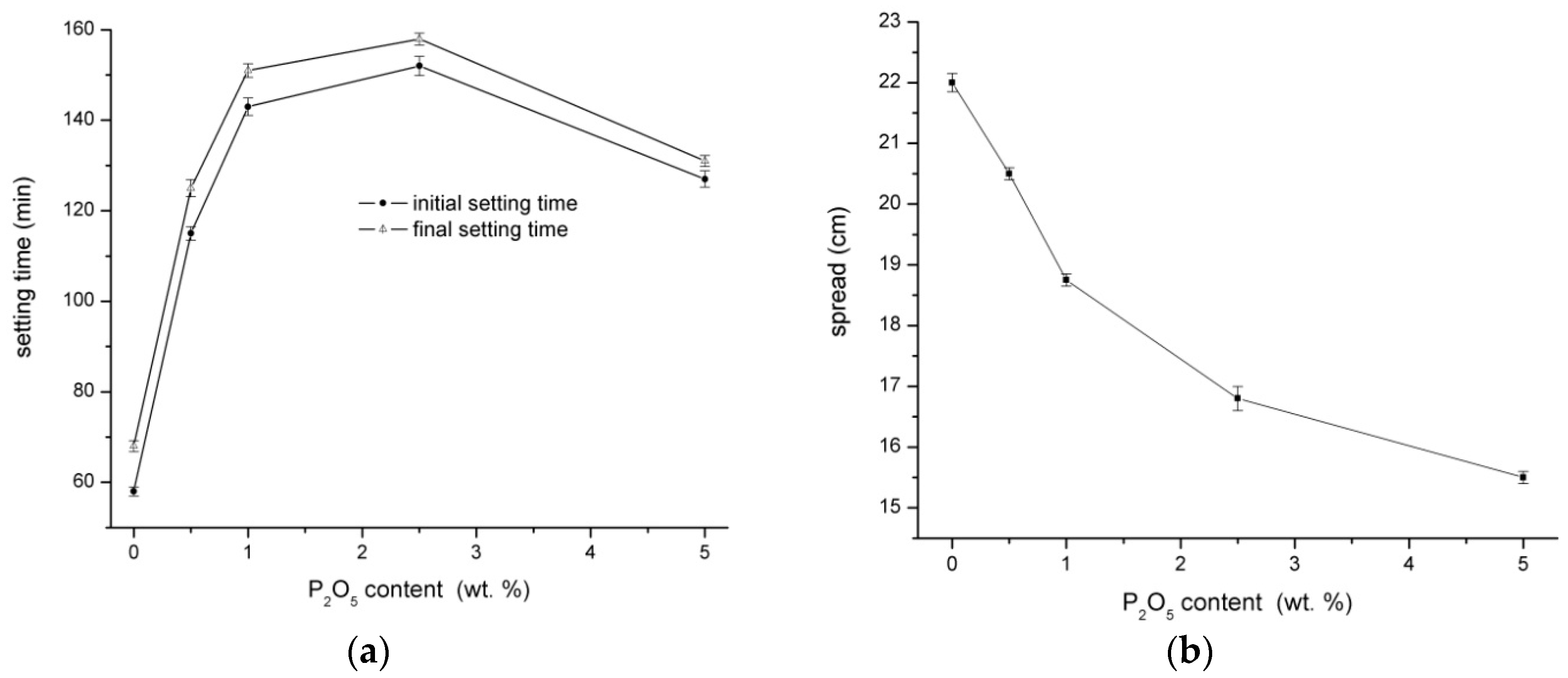
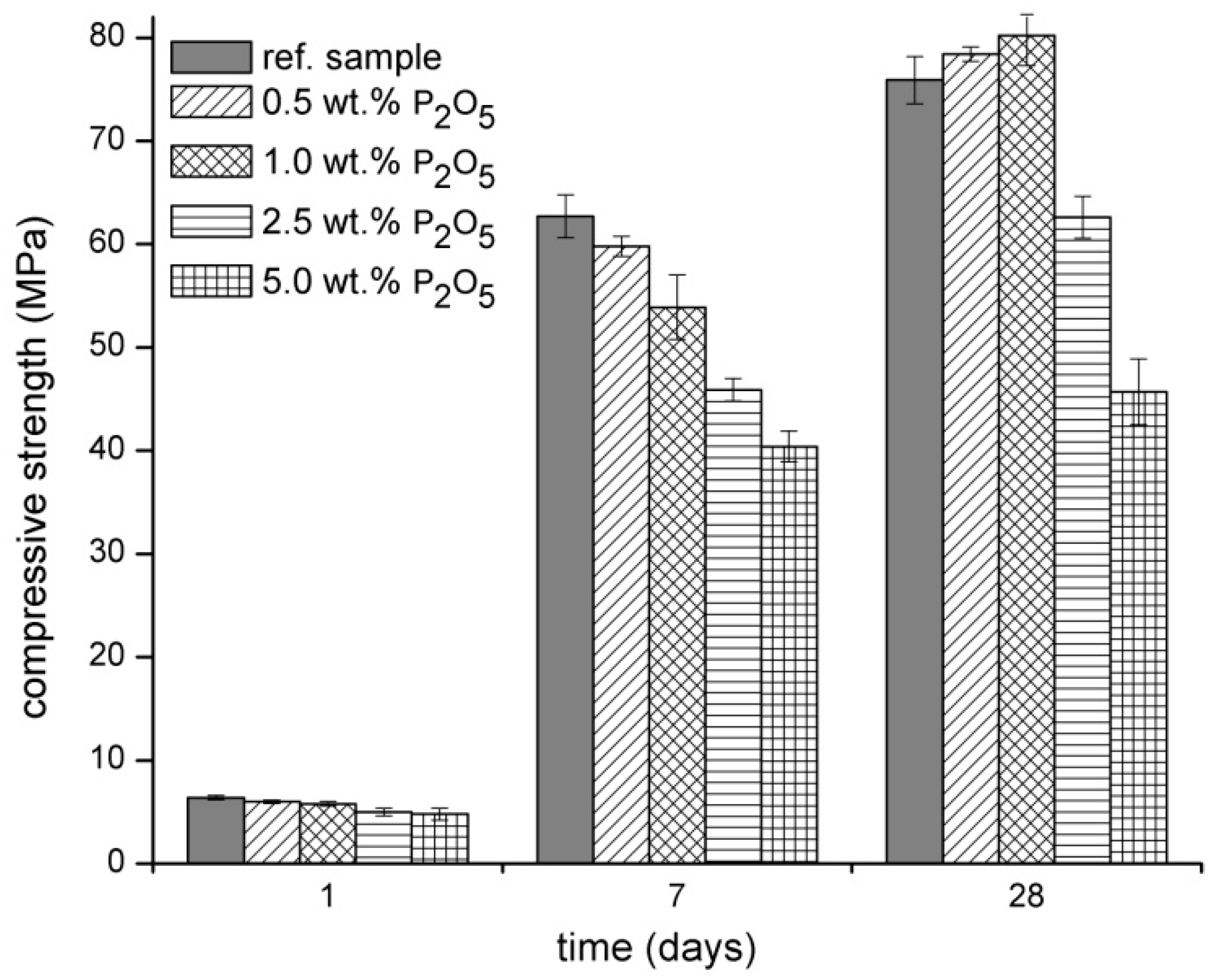
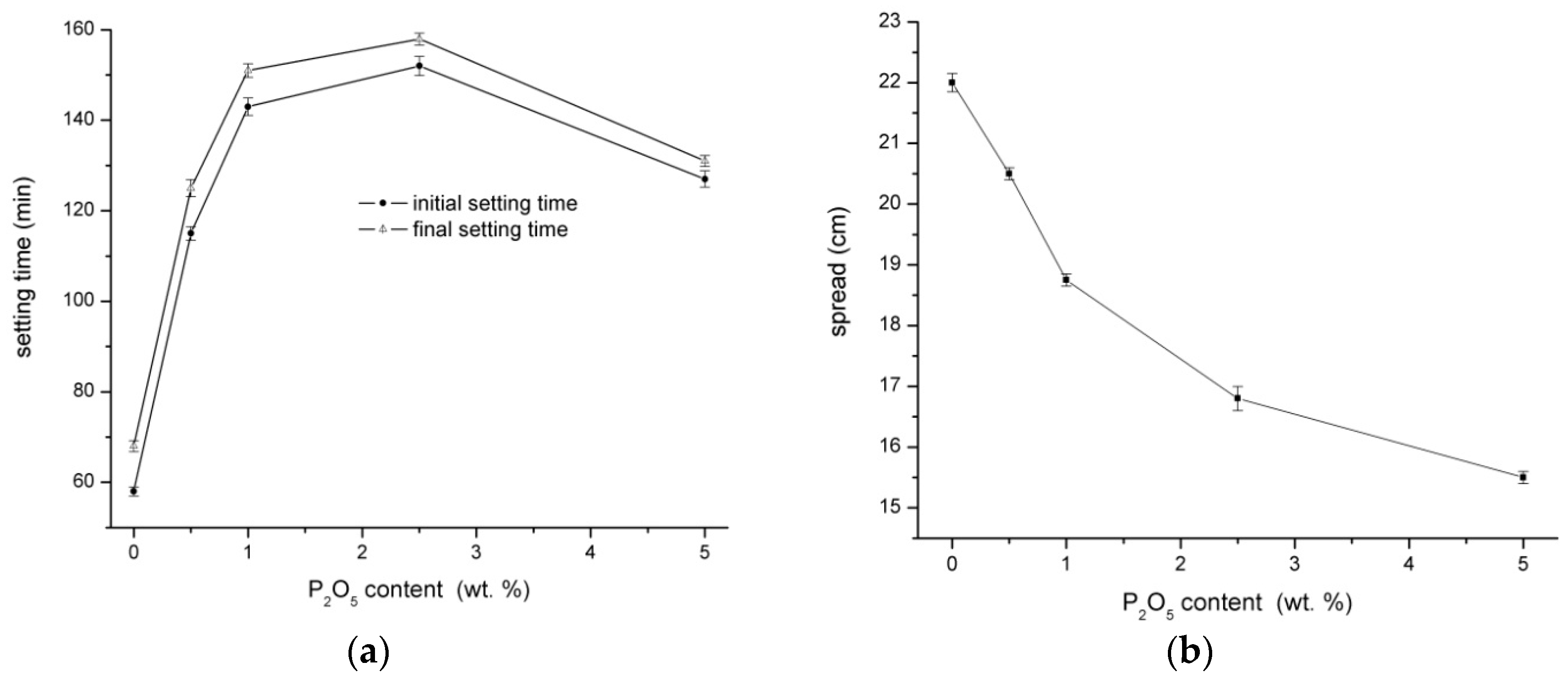
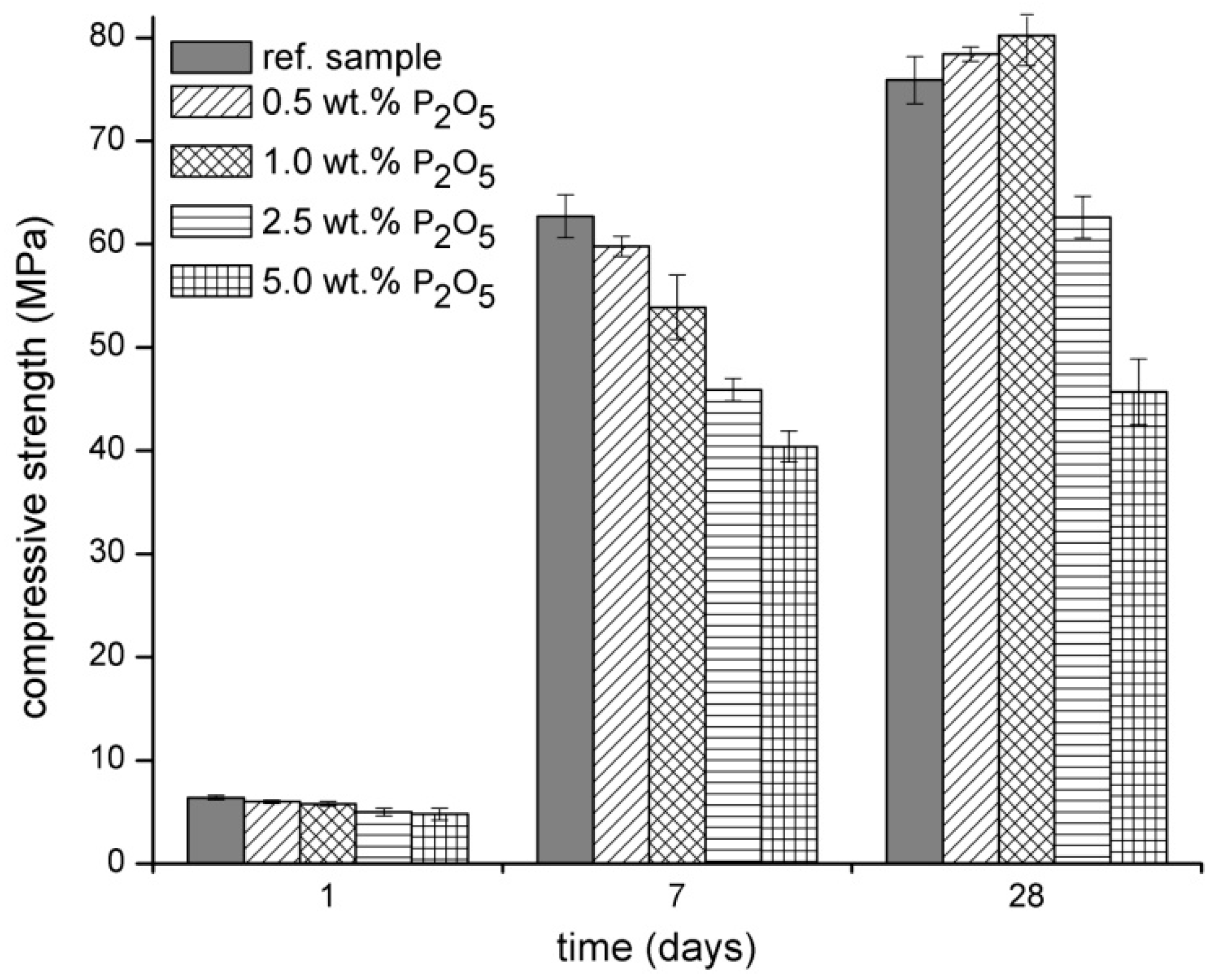
3.1. Physical‑Mechanical Properties
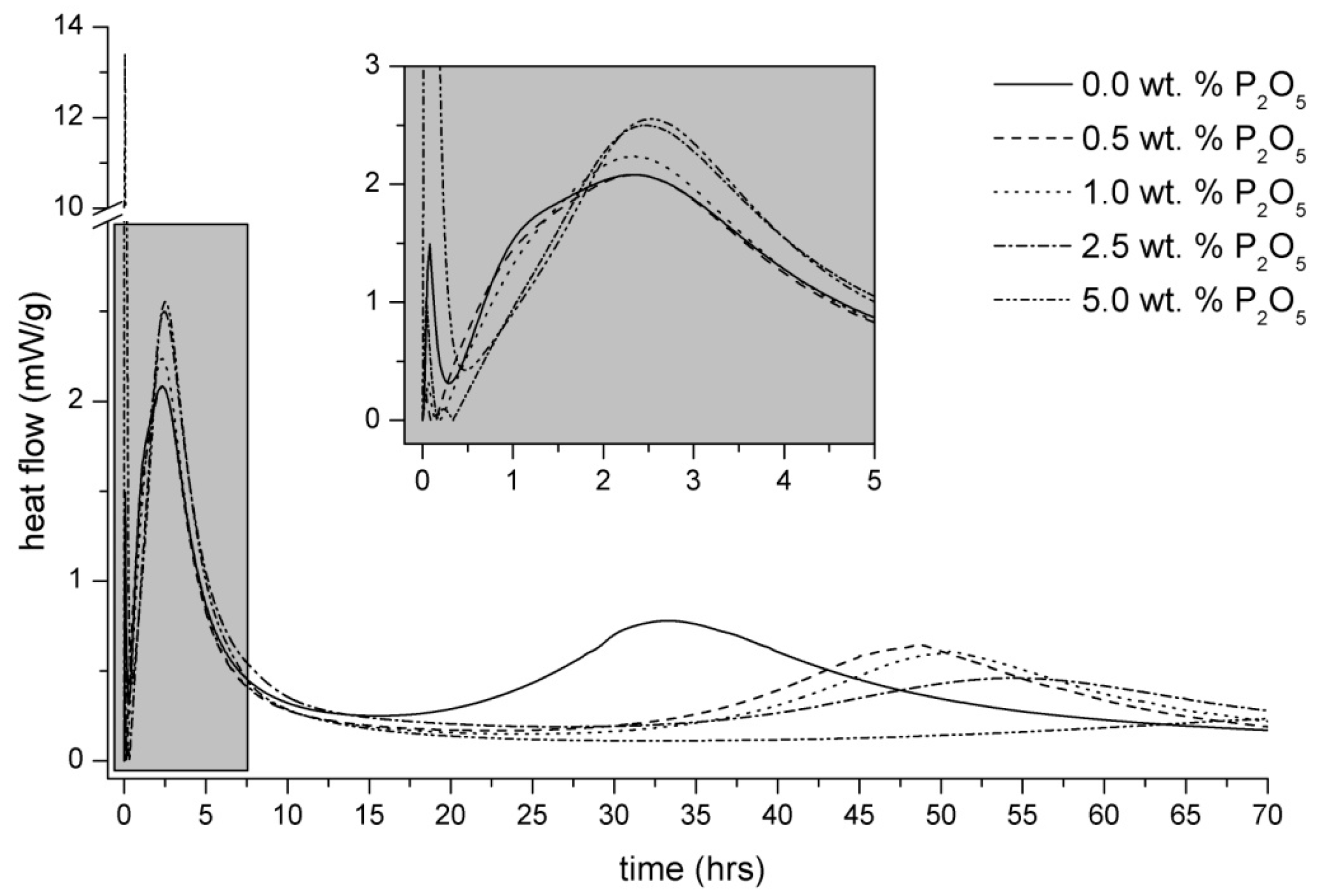
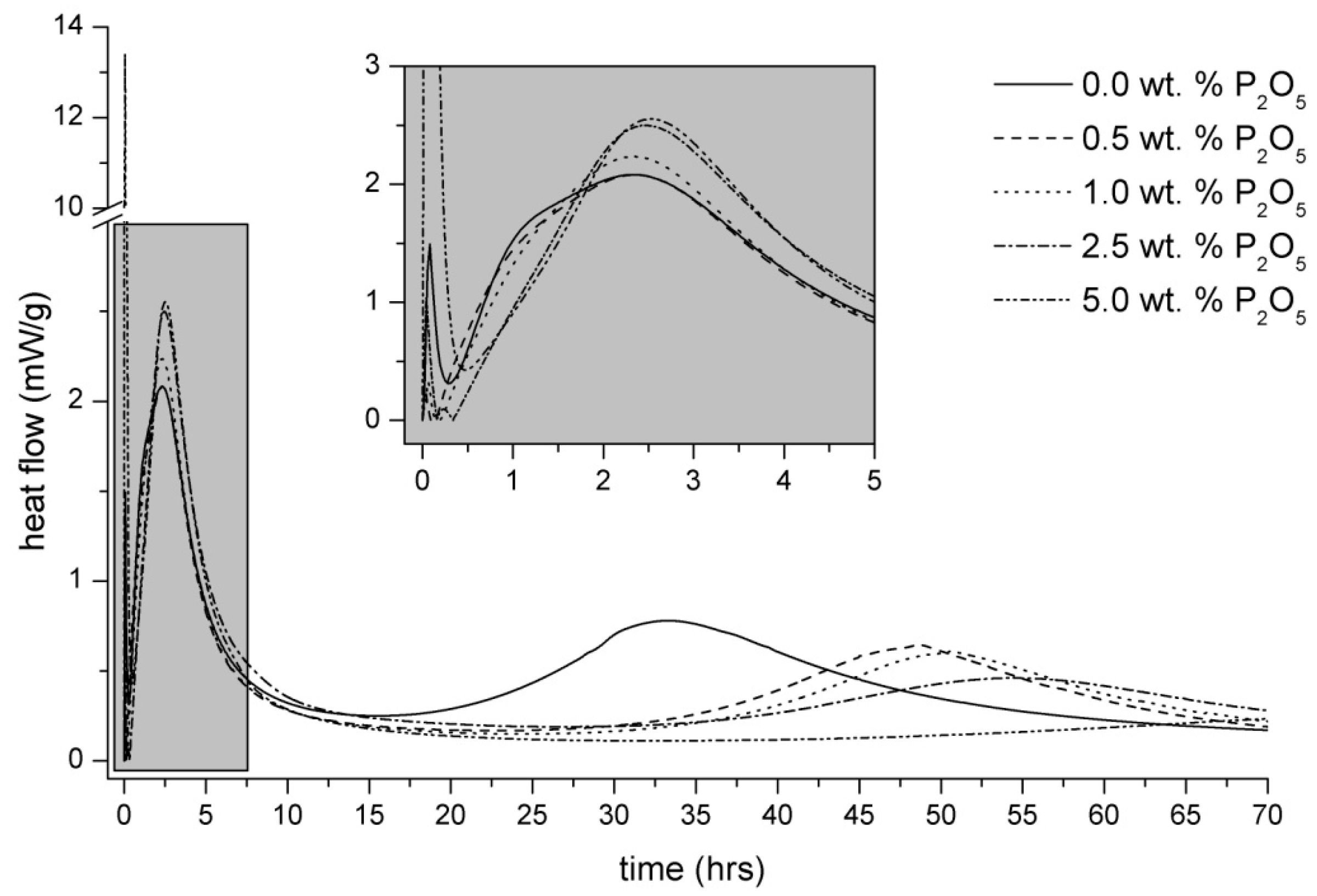
3.2. Heat Evolution
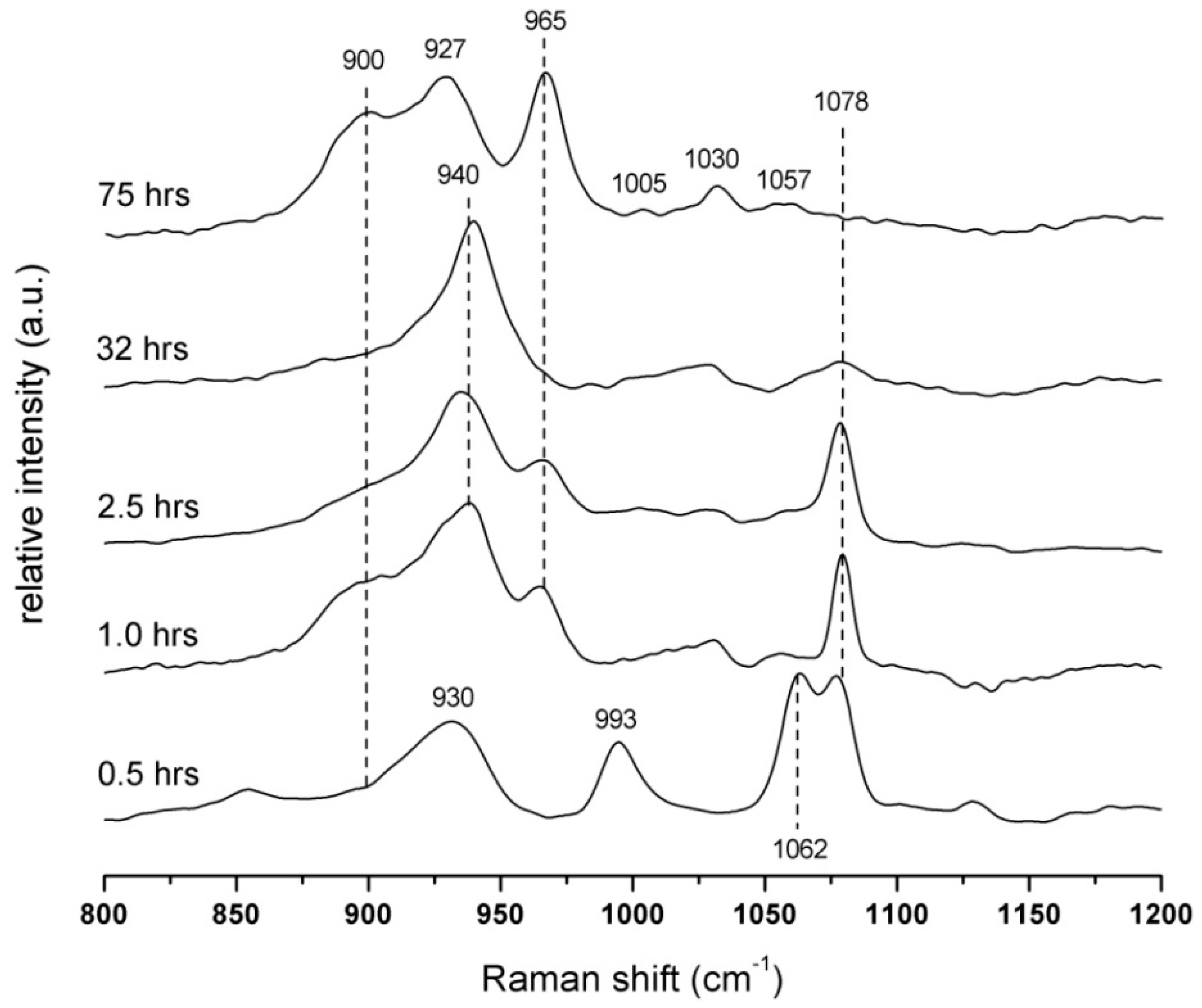
3.3. Raman Spectroscopy
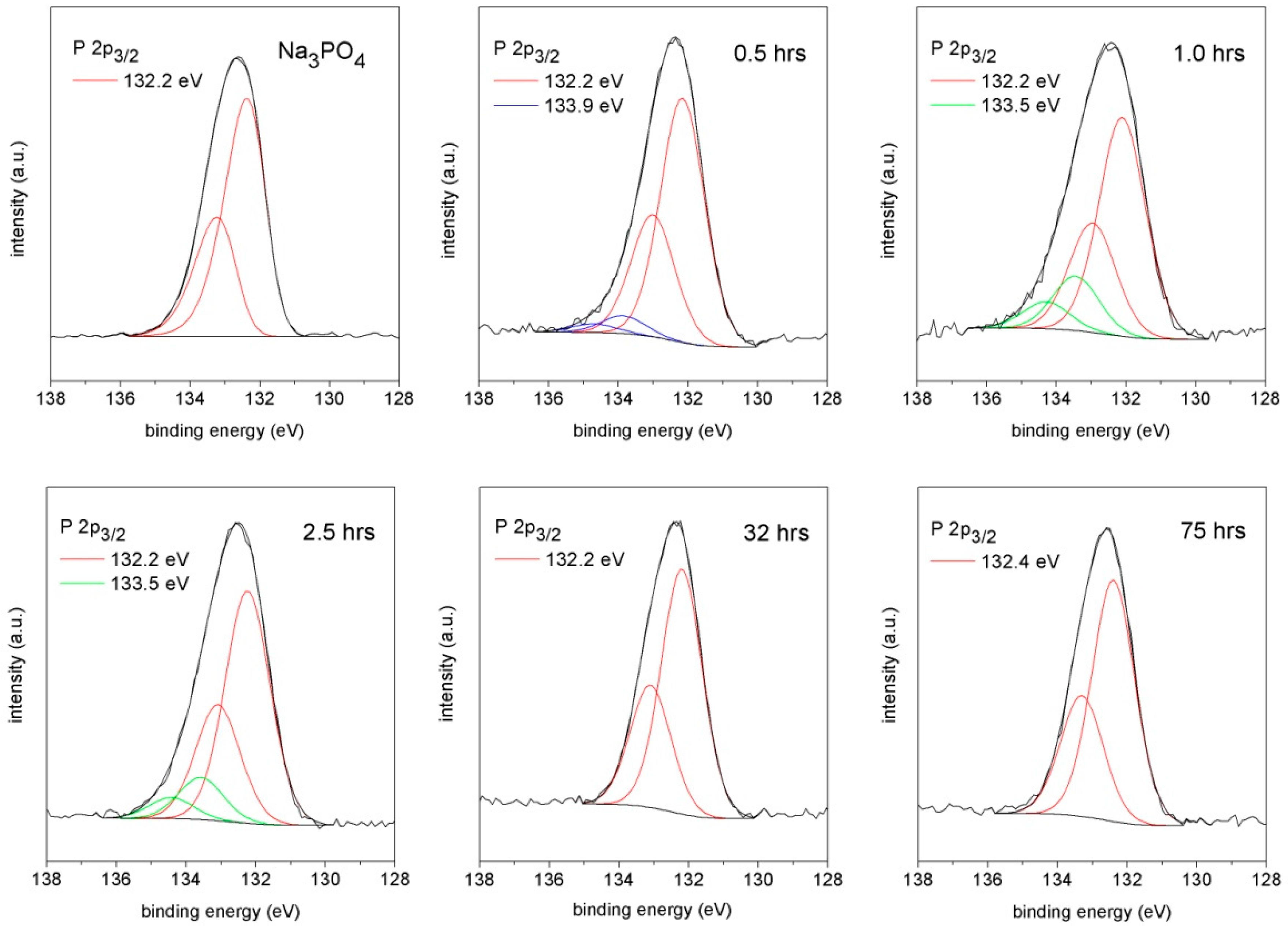
3.4. X-ray Photoelectron Spectroscopy
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Ksp | solubility product constant |
| BFS | blast furnace slag |
| XRF | X-ray fluorescence |
| XRD | X-ray diffraction |
| β-C2S | β-dicalcium silicate |
| SP | sodium phosphate |
| XPS | X-ray photoelectron spectroscopy |
| CSH gel | calcium-silicate-hydrate gel |
| FWHM | full width at half maximum |
| BE | binding energy |
| υ1(A1) | symmetric stretching vibration |
| υ3(F2) | asymmetric stretching vibration |
References
- Provis, J.L.; van Deventer, J.S.J. Alkali Activated Materials; Springer: Houten, The Netherlands, 2014; p. 388. [Google Scholar]
- Wu, C.; Zhang, Y.; Hu, Z. Properties and application of alkali-slag cement. J. Chin. Ceram. Soc. 1993, 21, 175–181. [Google Scholar]
- Nicholson, C.L.; Murray, B.J.; Fletcher, R.A.; Brew, D.; Mackenzie, K.J.; Schmücker, M. Novel Geopolymer Materials Containing Borate Structural Units. In Proceedings of the World Congress Geopolymer, Perth, Australia, September 2005; pp. 31–33.
- Brough, A.; Holloway, M.; Sykes, J.; Atkinson, A. Sodium silicate-based alkali-activated slag mortars: Part II. The retarding effect of additions of sodium chloride or malic acid. Cem. Concr. Res. 2000, 30, 1375–1379. [Google Scholar] [CrossRef]
- Chang, J.J.; Yeih, W.; Hung, C.C. Effects of gypsum and phosphoric acid on the properties of sodium silicate-based alkali-activated slag pastes. Cem. Concr. Compos. 2005, 27, 85–91. [Google Scholar] [CrossRef]
- Gong, C.; Yang, N. Effect of phosphate on the hydration of alkali-activated red mud–slag cementitious material. Cem. Concr. Res. 2000, 30, 1013–1016. [Google Scholar] [CrossRef]
- Shi, C.; Li, Y. Investigation on some factors affecting the characteristics of alkali-phosphorus slag cement. Cem. Concr. Res. 1989, 19, 527–533. [Google Scholar]
- Lee, W.; van Deventer, J.J. Effects of anions on the formation of aluminosilicate gel in geopolymers. Ind. Eng. Chem. Res. 2002, 41, 4550–4558. [Google Scholar] [CrossRef]
- Shi, C.; Day, R.L. A calorimetric study of early hydration of alkali-slag cements. Cem. Concr. Res. 1995, 25, 1333–1346. [Google Scholar] [CrossRef]
- Ghule, A.; Baskaran, N.; Murugan, R.; Chang, H. Phase transformation studies of Na3PO4 by thermo-raman and conductivity measurements. Solid State Ion. 2003, 161, 291–299. [Google Scholar] [CrossRef]
- McAfee, L. Infrared and raman spectra of inorganic and coordination compounds. Part A: Theory and applications in inorganic chemistry; part B: Application in coordination, organometallic, and bioinorganic chemistry, (Nakamoto, Kazuo). J. Chem. Educ. 2000, 77, 1122. [Google Scholar] [CrossRef]
- Ghule, A.; Murugan, R.; Chang, H. Thermo-raman studies on dehydration of Na3PO4·12H2O. Thermochim. Acta 2001, 371, 127–135. [Google Scholar] [CrossRef]
- Frost, R.L.; López, A.; Xi, Y.; Scholz, R. Vibrational spectroscopic characterization of the phosphate mineral althausite Mg2(PO4)(OH,F,O)—Implications for the molecular structure. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 120, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Taddei, P.; Tinti, A.; Gandolfi, M.G.; Rossi, P.L.; Prati, C. Ageing of calcium silicate cements for endodontic use in simulated body fluids: A micro-raman study. J. Raman Spectrosc. 2009, 40, 1858–1866. [Google Scholar] [CrossRef]
- Koutsopoulos, S. Synthesis and characterization of hydroxyapatite crystals: A review study on the analytical methods. J. Biomed. Mater. Res. 2002, 62, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Moulder, J.F.; Chastain, J. Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data; Physical Electronics Division, Perkin-Elmer Corporation: Eden Prairie, MN, USA, 1995; pp. 84–85. [Google Scholar]
- Textor, M.; Ruiz, L.; Hofer, R.; Rossi, A.; Feldman, K.; Hähner, G.; Spencer, N.D. Structural chemistry of self-assembled monolayers of octadecylphosphoric acid on tantalum oxide surfaces. Langmuir 2000, 16, 3257–3271. [Google Scholar] [CrossRef]
- Berner, U.R. Modelling of incongruent dissolution of hydrated cement minerals. Radiochim. Acta 1988, 44/45, 387–393. [Google Scholar] [CrossRef]
- Moreno, E.C.; Gregory, T.M.; Brown, W.E. Preparation and solubility of hydroxyapatite. Phys. Chem. 1968, 72A, 773–782. [Google Scholar] [CrossRef]
- Shin, H.-Y.; Jung, J.-Y.; Kim, S.-W.; Lee, W.-K. XPS analysis on chemical properties of calcium phosphate thin films and osteoblastic hos cell responses. J. Ind. Eng. Chem. 2006, 12, 476–483. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Raw Material | Chemical Composition wt % | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| BFS | SiO2 | Al2O3 | CaO | Na2O | K2O | MgO | SO3 | Fe2O3 | TiO2 | MnO |
| 34.7 | 9.1 | 41.1 | 0.4 | 0.9 | 10.5 | 1.4 | 0.3 | 1.0 | 0.6 | |
| Component | Binding Energy (FWHM)/eV | ||
|---|---|---|---|
| Na3PO4 | 0.5 h | 1.0 h | |
| P 2p3/2 | 132.2 (1.4) | 132.2 (1.4) | 132.2 (1.4) |
| 133.9 (1.4) | 133.5 (1.4) | ||
| 2.5 h | 32 h | 75 h | |
| 132.2 (1.4) | 132.2 (1.4) | 132.4 (1.4) | |
| 133.5 (1.4) | |||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalina, L.; Bílek, V.; Novotný, R.; Mončeková, M.; Másilko, J.; Koplík, J. Effect of Na3PO4 on the Hydration Process of Alkali-Activated Blast Furnace Slag. Materials 2016, 9, 395. https://doi.org/10.3390/ma9050395
Kalina L, Bílek V, Novotný R, Mončeková M, Másilko J, Koplík J. Effect of Na3PO4 on the Hydration Process of Alkali-Activated Blast Furnace Slag. Materials. 2016; 9(5):395. https://doi.org/10.3390/ma9050395
Chicago/Turabian StyleKalina, Lukáš, Vlastimil Bílek, Radoslav Novotný, Miroslava Mončeková, Jiří Másilko, and Jan Koplík. 2016. "Effect of Na3PO4 on the Hydration Process of Alkali-Activated Blast Furnace Slag" Materials 9, no. 5: 395. https://doi.org/10.3390/ma9050395
APA StyleKalina, L., Bílek, V., Novotný, R., Mončeková, M., Másilko, J., & Koplík, J. (2016). Effect of Na3PO4 on the Hydration Process of Alkali-Activated Blast Furnace Slag. Materials, 9(5), 395. https://doi.org/10.3390/ma9050395