
Fc-Binding Ligands of Immunoglobulin G: An Overview of High Affinity Proteins and Peptides

Abstract
:1. Introduction
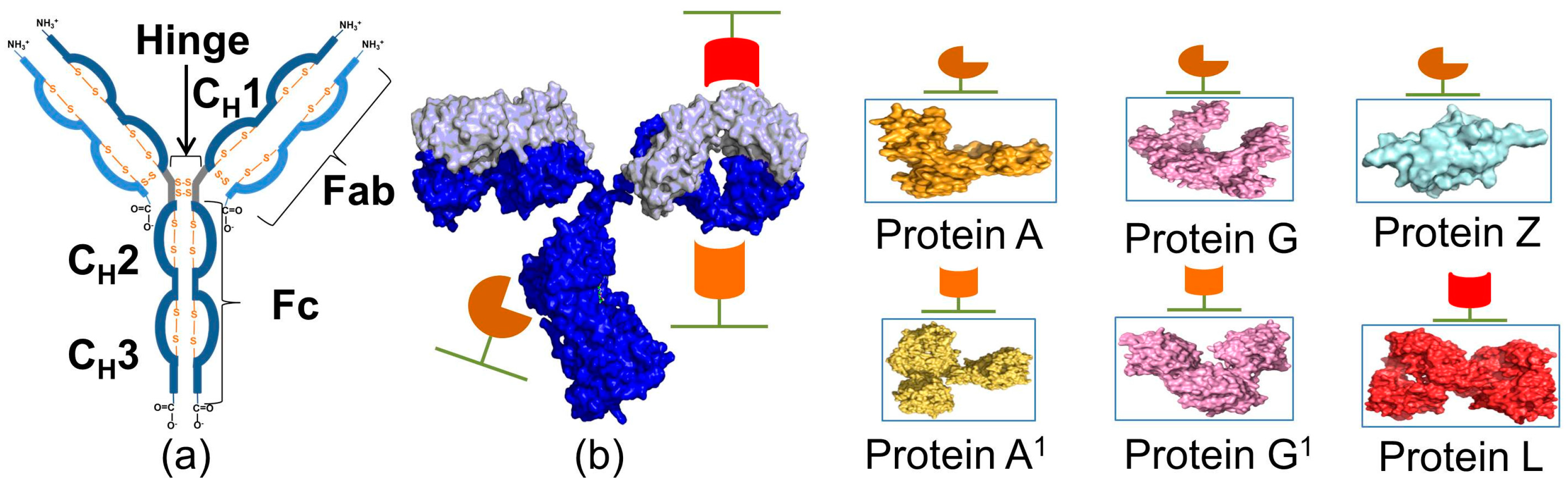
2. Immunoglobulin Binding Proteins
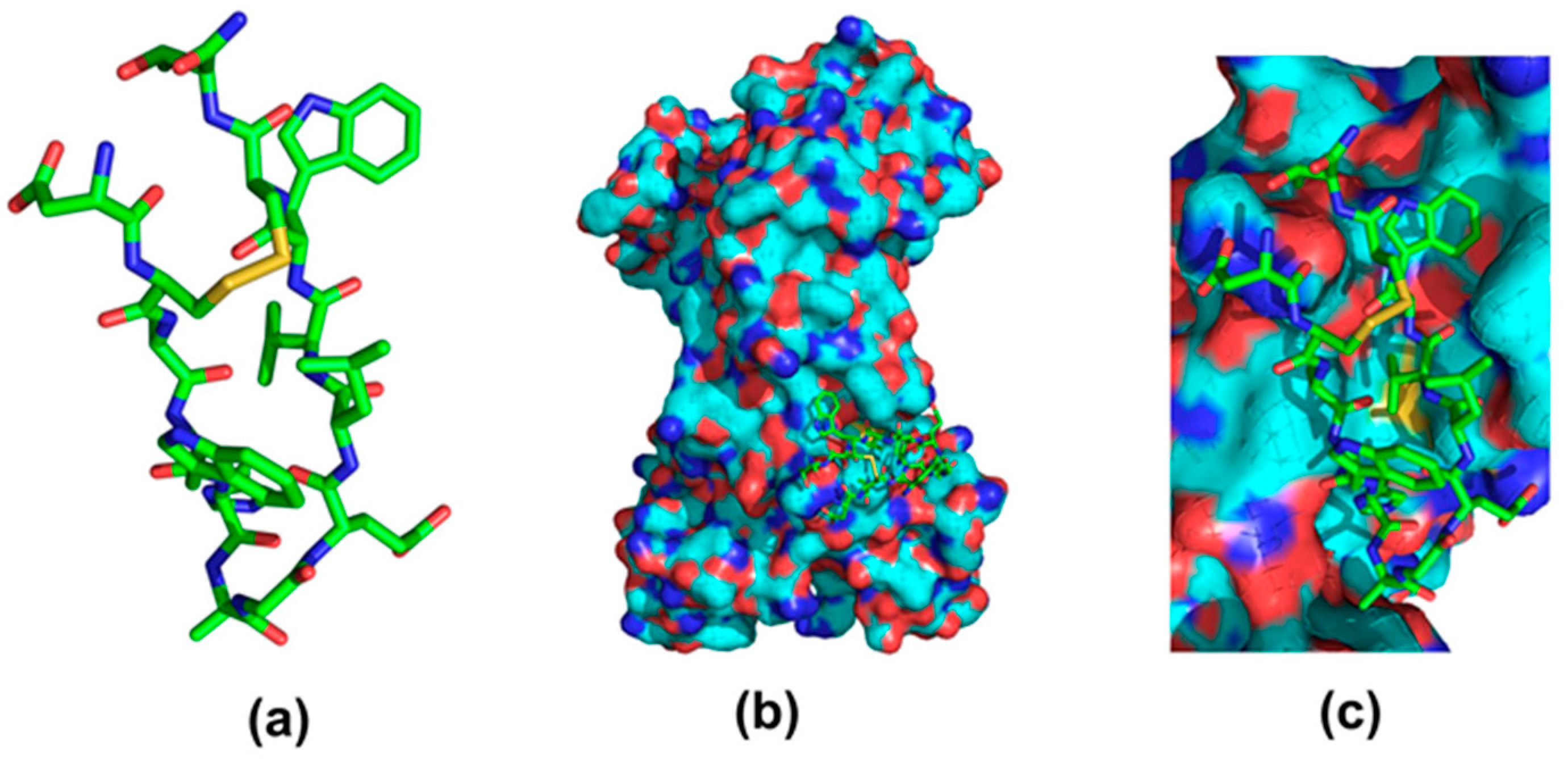
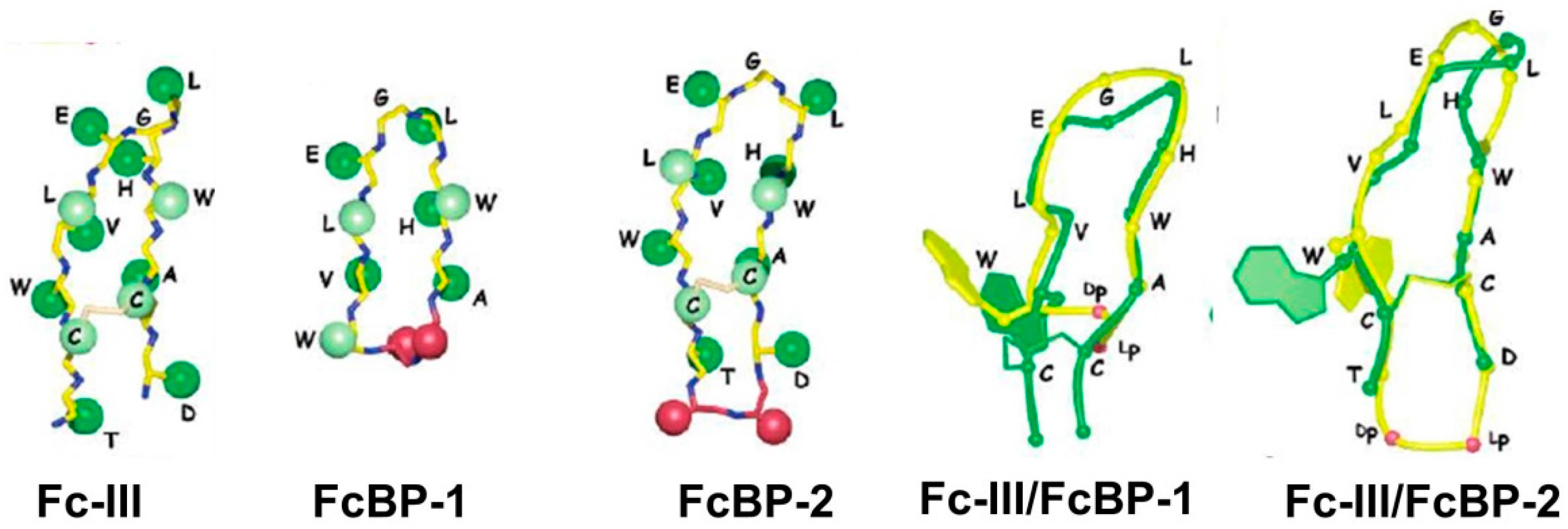
3. Immunoglobulin Binding Peptides and Peptidomimetics
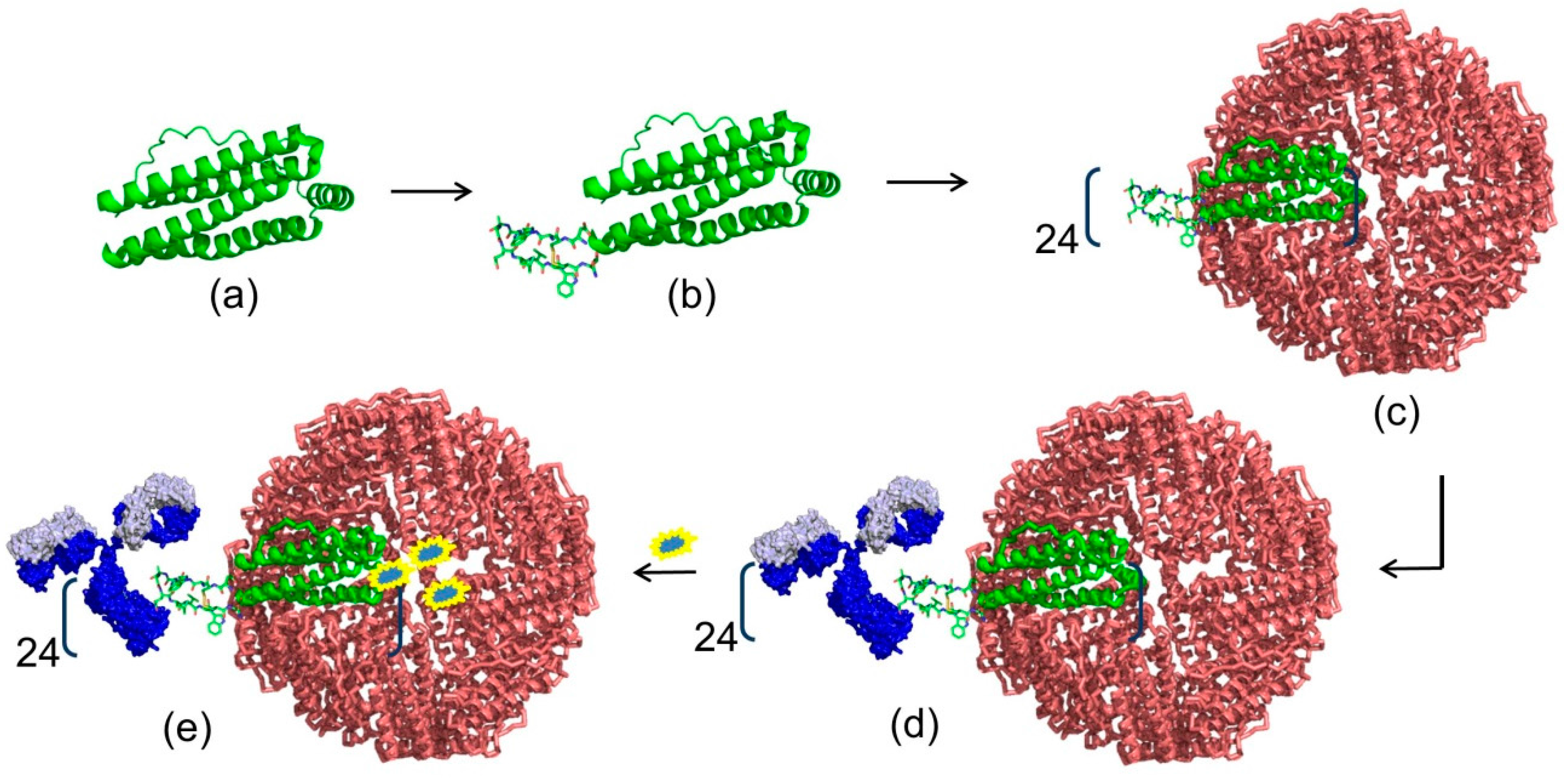
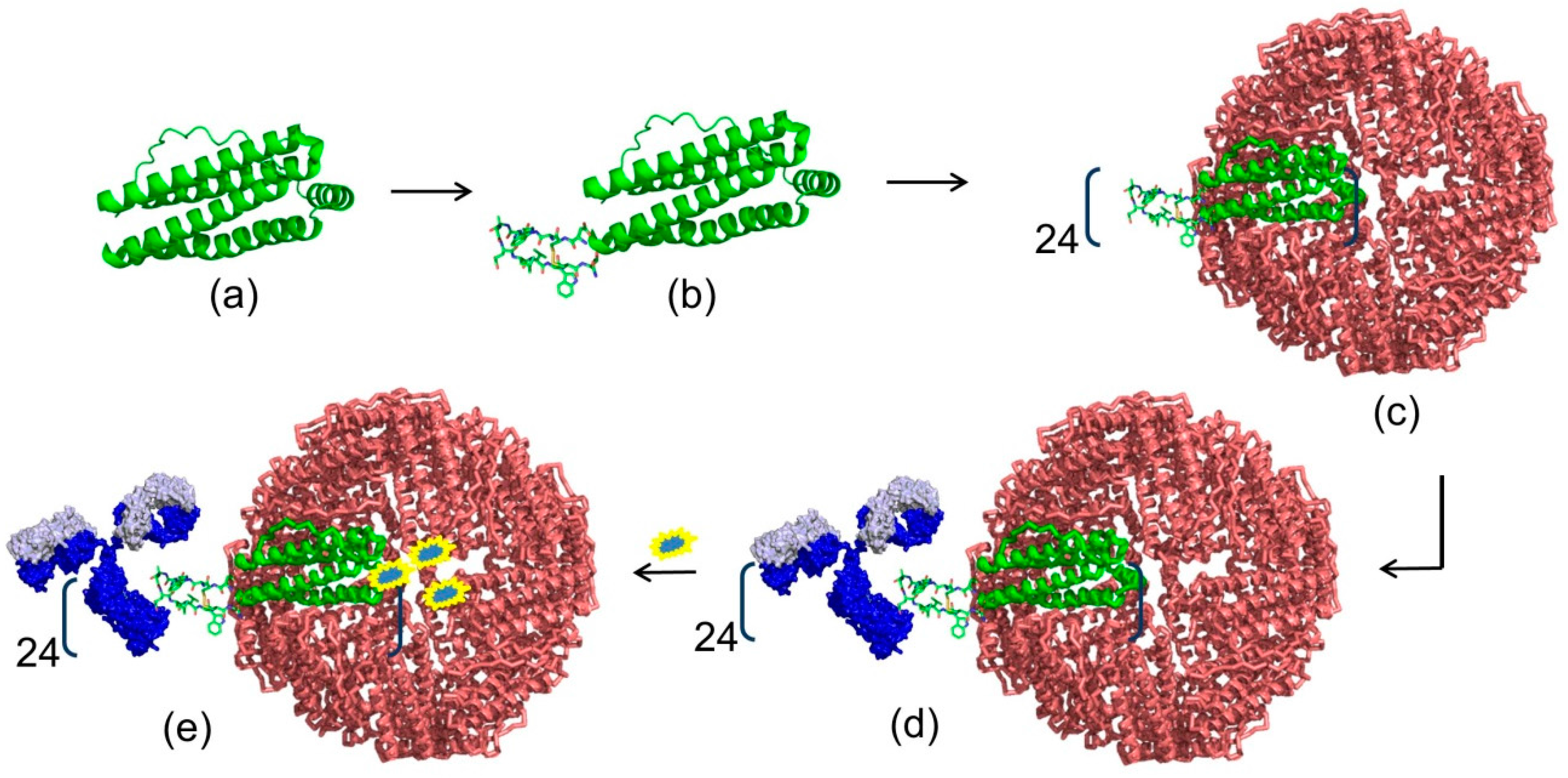
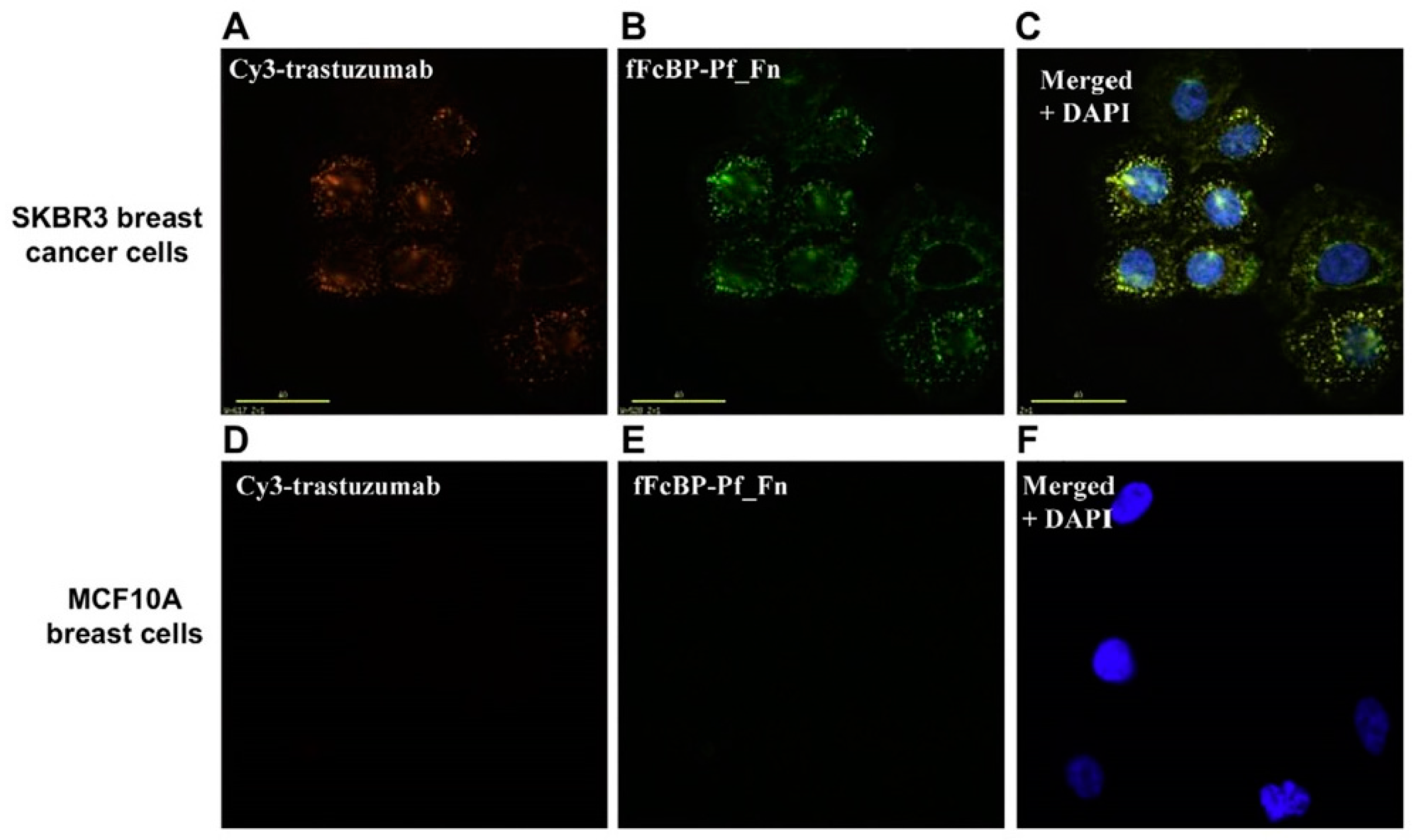
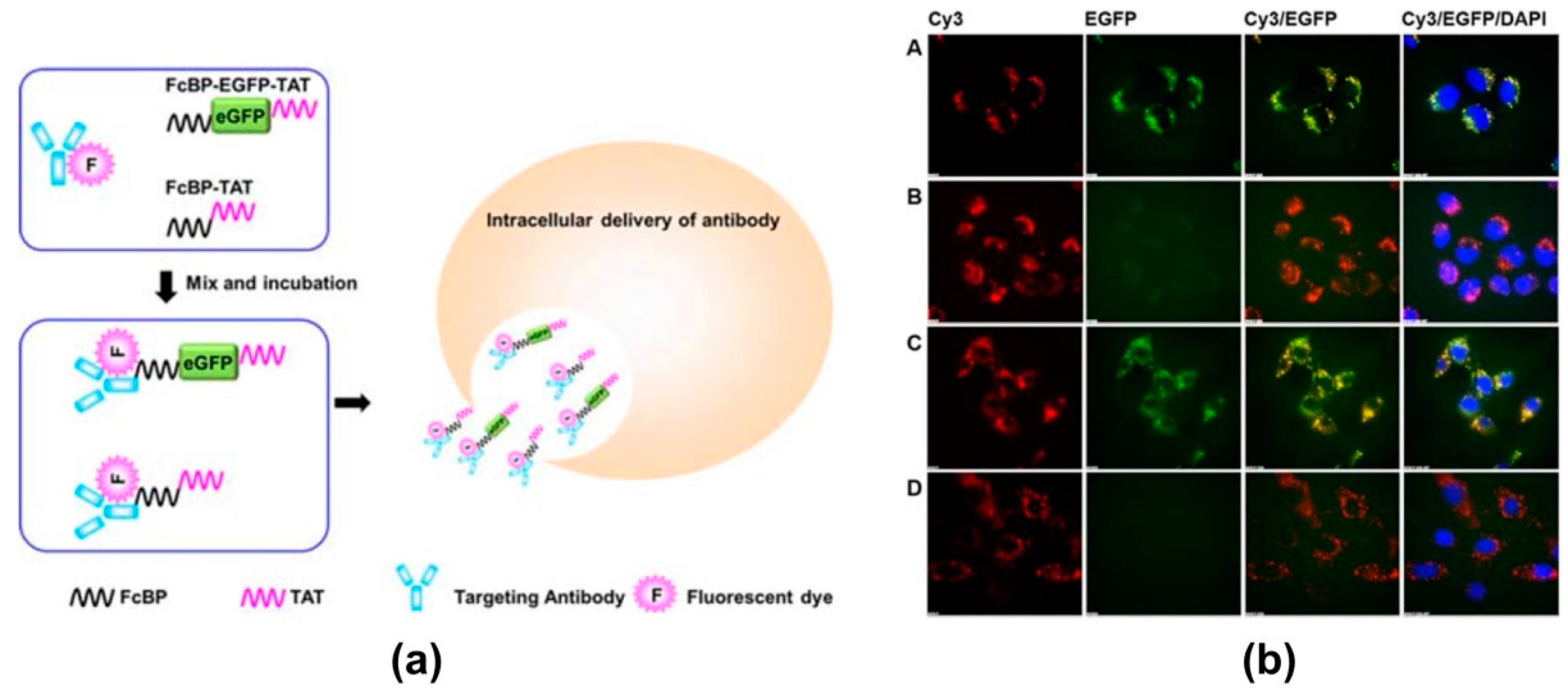
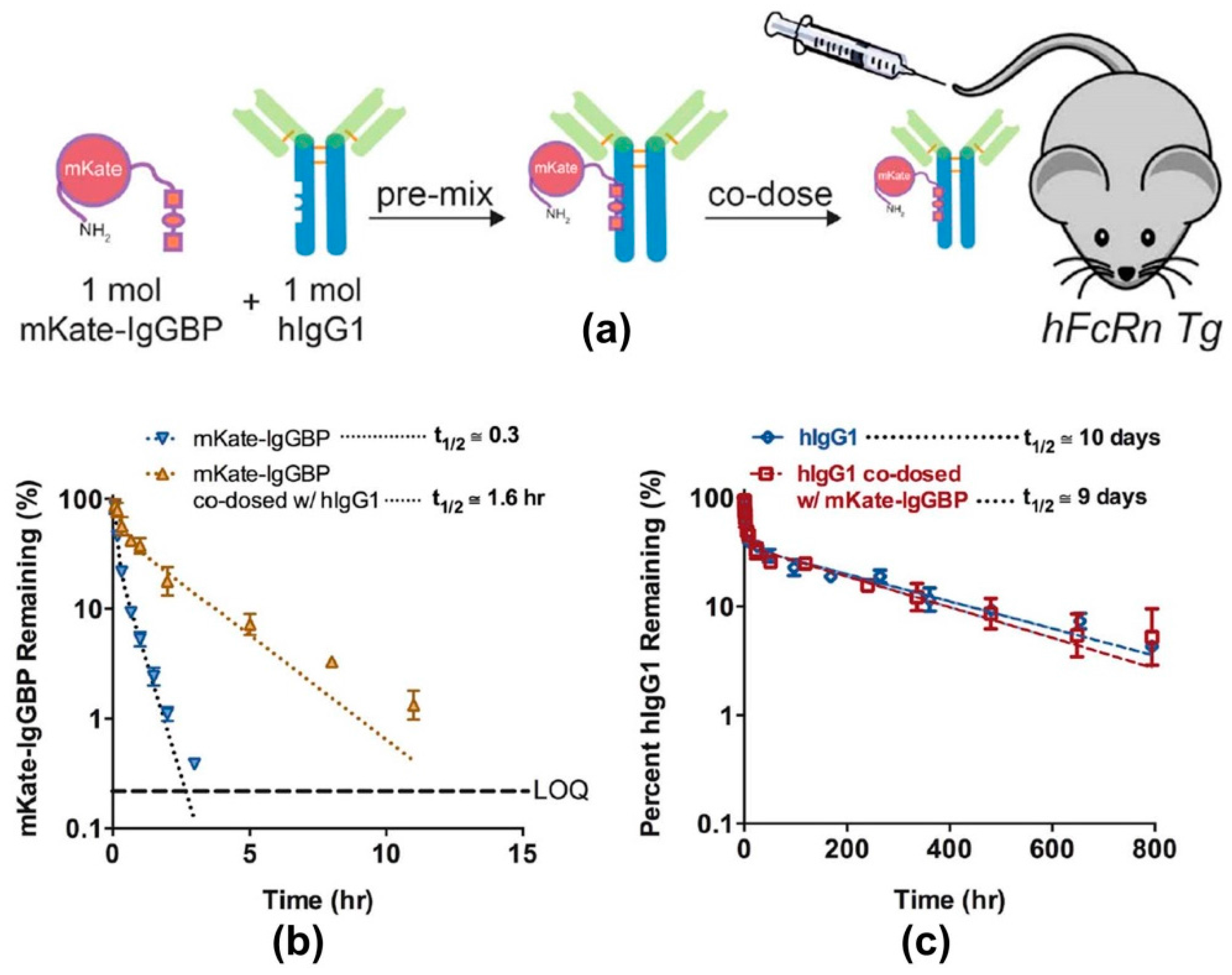
4. Modern Applications of Immunoglobulin Binding Ligands
5. Conclusions and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Steinitz, M. Three decades of human monoclonal antibodies: Past, present and future developments. Hum. Antibodies 2009, 18, 1–10. [Google Scholar] [PubMed]
- Leavy, O. Therapeutic antibodies: Past, present and future. Nat. Rev. Immunol. 2010, 10, 297. [Google Scholar] [CrossRef] [PubMed]
- Matera, M.G.; Page, C.; Rogliani, P.; Calzetta, L.; Cazzola, M. Therapeutic monoclonal antibodies for the treatment of chronic obstructive pulmonary disease. Drugs 2016, 76, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Mould, D.R.; Meibohm, B. Drug development of therapeutic monoclonal antibodies. BioDrugs 2016, 30, 275–293. [Google Scholar] [CrossRef] [PubMed]
- Lambour, J.; Naranjo-Gomez, M.; Piechaczyk, M.; Pelegrin, M. Converting monoclonal antibody-based immunotherapies from passive to active: Bringing immune complexes into play. Emerg. Microbes Infect. 2016, 5, e92. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.K. The history of monoclonal antibody development—Progress, remaining challenges and future innovations. Ann. Med. Surg. (Lond.) 2014, 3, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Hamblett, K.J.; Le, T.; Rock, B.M.; Rock, D.A.; Siu, S.; Huard, J.N.; Conner, K.P.; Milburn, R.R.; O’Neill, J.W.; Tometsko, M.E.; et al. Altering antibody-drug conjugate binding to the neonatal Fc receptor impacts efficacy and tolerability. Mol. Pharm. 2016, 13, 2387–2396. [Google Scholar] [CrossRef] [PubMed]
- Jendeberg, L.; Nilsson, P.; Larsson, A.; Denker, P.; Uhlen, M.; Nilsson, B.; Nygren, P.-A. Engineering of Fc1 and Fc3 from human immunoglobulin G to analyse subclass specificity for staphylococcal protein A. J. Immunol. Methods 1997, 201, 25–34. [Google Scholar] [CrossRef]
- Deisenhofer, J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from staphylococcus aureus at 2.9- and 2.8-Å resolution. Biochemistry 1981, 20, 2361–2370. [Google Scholar] [CrossRef] [PubMed]
- Arora, I. Chromatographic methods for the purification of monoclonal antibodies and their alternatives: A review. IJETAE 2013, 3, 475–481. [Google Scholar]
- Gaughan, C.L. The present state of the art in expression, production and characterization of monoclonal antibodies. Mol. Divers. 2016, 20, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Ey, P.L.; Prowse, S.J.; Jenkin, C.R. Isolation of pure IgG1, IgG2a and IgG2b immunoglobulins from mouse serum using protein A-sepharose. Immunochemistry 1978, 15, 429–436. [Google Scholar] [CrossRef]
- Huse, K.; Bohme, H.J.; Scholz, G.H. Purification of antibodies by affinity chromatography. J. Biochem. Biophys. Methods 2002, 51, 217–231. [Google Scholar] [CrossRef]
- Billakanti, J.M.; Fee, C.J.; Naik, A.D.; Carbonell, R.G. Application of peptide chromatography for the isolation of antibodies from bovine skim milk, acid whey and colostrum. Food Bioprod. Process. 2014, 92, 199–207. [Google Scholar] [CrossRef]
- Hober, S.; Nord, K.; Linhult, M. Protein A chromatography for antibody purification. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 848, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Gronemeyer, P.; Ditz, R.; Strube, J. Trends in upstream and downstream process development for antibody manufacturing. Bioengineering 2014, 1, 188–212. [Google Scholar] [CrossRef]
- Roque, A.C.; Silva, C.S.; Taipa, M.A. Affinity-based methodologies and ligands for antibody purification: Advances and perspectives. J. Chromatogr. A 2007, 1160, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Fassina, G.; Ruvo, M.; Palombo, G.; Verdoliva, A.; Marino, M. Novel ligands for the affinity-chromatographic purification of antibodies. J. Biochem. Biophys. Methods 2001, 49, 481–490. [Google Scholar] [CrossRef]
- Kabir, S. Immunoglobulin purification by affinity chromatography using protein A mimetic ligands prepared by combinatorial chemical synthesis. Immunol. Investig. 2002, 31, 263–278. [Google Scholar] [CrossRef]
- Thapa, P.; Espiritu, M.J.; Cabalteja, C.; Bingham, J.P. The emergence of cyclic peptides: The potential of bioengineered peptide drugs. Med. Chem. 2014, 4, 451–468. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the viron surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Bazan, J.; Calkosinski, I.; Gamian, A. Phage display—A powerful technique for immunotherapy: 1. Introduction and potential of therapeutic applications. Hum. Vaccin Immunother. 2012, 8, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, G.; Gruvegard, M.; Van Alstine, J.M. Antibody fragments and their purification by protein L affinity chromatography. Antibodies 2015, 4, 259–277. [Google Scholar] [CrossRef]
- Konrad, A.; Karlstrom, A.E.; Hober, S. Covalent immunoglobulin labeling through a photoactivable synthetic Z domain. Bioconjug. Chem. 2011, 22, 2395–2403. [Google Scholar] [CrossRef] [PubMed]
- Kihlberg, B.M.; Sjoholm, A.G.; Bjorck, L.; Sjobring, U. Characterization of the binding properties of protein LG, an immunoglobulin-binding hybrid protein. Eur. J. Biochem. 1996, 240, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, B.; Moks, T.; Jansson, B.; Abrahmsén, L.; Elmblad, A.; Holmgren, E.; Henrichson, C.; Jones, T.A.; Uhlén, M. A synthetic IgG-binding domain based on staphylococcal protein A. Protein Eng. Des. Sel. 1987, 1, 107–113. [Google Scholar] [CrossRef]
- Ghitescu, L.; Galis, Z.; Bendayan, M. Protein AG-gold complex: An alternative probe in immunocytochemistry. J. Histochem. Cytochem. 1991, 39, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Akerstrom, B.; Bjorck, L. A physicochemical study of protein G, a moleculle with unique immunoglobulin G-binding properties. J. Biol. Chem. 1986, 261, 10240–10247. [Google Scholar] [PubMed]
- Svensson, H.; Hoogenboom, H.R.; Sjöbring, U. Protein LA, a novel hybrid protein with unique single-chain Fv antibody- and Fab-binding properties. Eur. J. Biochem. 1998, 258, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Moks, T.; Abrahmsen, L.; Nilsson, B.; Hellman, B.; Sjöquist, J.; Uhlen, M. Staphylococcal protein A consist of five IgG-binding domains. Eur. J. Biochem. 1986, 156, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Graille, M.; Stura, E.A.; Corper, A.L.; Sutton, B.J.; Taussig, M.J.; Charbonnier, J.B.; Silverman, G.J. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: Structural basis for recognition of B-cell receptors and superantigen activity. Proc. Natl. Acad. Sci. USA 2000, 97, 5399–5404. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Bender, F.; Gizeli, E. Comparative study of IgG binding to proteins G and A: Nonequilibrium kinetic and binding constant determination with the acoustic waveguide device. Anal. Chem. 2003, 75, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Pabst, T.M.; Palmgren, R.; Forss, A.; Vasic, J.; Fonseca, M.; Thompson, C.; Wang, W.K.; Wang, X.; Hunter, A.K. Engineering of novel staphylococcal protein A ligands to enable milder elution pH and high dynamic binding capacity. J. Chromatogr. A 2014, 1362, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Tashira, M.; Tejero, R.; Zimmerman, D.E.; Celda, B.; Nilsson, B.; Montelione, G.T. High-resolution solution NMR structure of the Z domain of staphylococcal protein A. J. Mol. Biol. 1997, 272, 573–590. [Google Scholar] [CrossRef] [PubMed]
- Braisted, A.; Wells, J.A. Minimizing a binding domain from protein. Proc. Natl. Acad. Sci. USA 1996, 93, 5688–5692. [Google Scholar] [CrossRef] [PubMed]
- Mouratou, B.; Behar, G.; Pecorari, F. Artificial affinity proteins as ligands of immunoglobulins. Biomolecules 2015, 5, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Matsumaru, H.; Ooishi, A.; Feng, Y.; Odahara, T.; Suto, K.; Honda, S. Optimizing pH response of affinity between protein G and IgG Fc: How electrostatic modulations affect protein-protein interactions. J. Biol. Chem. 2009, 284, 12373–12383. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, M.; Watanabe, H.; Ooishi, A.; Honda, H. Engineerd protein A ligands, derived from a histidine-scanning library, facilitate the affinity purification of IgG under mild acidic conditions. J. Biol. Eng. 2014, 8, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Gulich, S.; Uhlen, M.; Hober, S. Protein engineering of an IgG-binding domain allows milder elution conditions during affinity chromatography. J. Biotechnol. 2000, 76, 233–243. [Google Scholar] [CrossRef]
- Strauch, E.M.; Fleishman, S.J.; Baker, D. Computational design of a pH-sensitive IgG binding protein. Proc. Natl. Acad. Sci. USA 2014, 111, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Gera, N.; Hill, A.B.; White, D.P.; Carbonell, R.G.; Rao, B.M. Design of pH sensitive binding proteins from the hyperthermophilic Sso7d scaffold. PLoS ONE 2012, 7, e48928. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L.; Ultsch, M.H.; de Vos, A.M.; Wells, J.A. Convergent solutions to binding at a protein-protein interface. Science 2000, 287, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
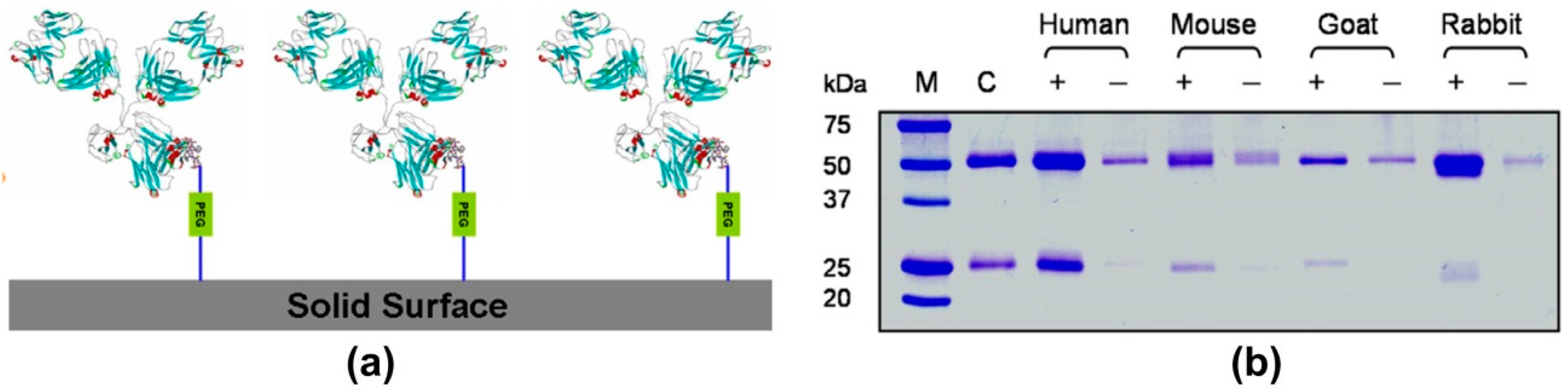
- Makaraviciute, A.; Ramanaviciene, A. Site-directed antibody immobilization techniques for immunosensors. Biosens. Bioelectron. 2013, 50, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Lee, J.M.; Kim, J.; Yoon, J.; Cho, H.; Chung, B.H. Photoactivatable antibody binding protein: Site-selective and covalent coupling of antibody. Anal. Chem. 2009, 81, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Fassina, G.; Verdoliva, A.; Odierna, M.R.; Ruvo, M.; Cassini, G. Protein A mimetic peptide ligan for affinity purification of antibodi. J. Mol. Recognit 1996, 9, 564–569. [Google Scholar] [CrossRef]
- Verdoliva, A.; Pannone, F.; Rossi, M.; Catello, S.; Manfredi, V. Affinity purification of polyclonal antibodies using a new all-D synthetic peptide ligand: Comparison with protein A and protein G. J. Immunol. Methods 2002, 271, 77–88. [Google Scholar] [CrossRef]
- Dinon, F.; Salvalaglio, M.; Gallotta, A.; Beneduce, L.; Pengo, P.; Cavallotti, C.; Fassina, G. Structural refinement of protein A mimetic peptide. J. Mol. Recognit 2011, 24, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Krook, M.; Mosbach, K.; Ramstrom, O. Novel peptides binding to the Fc-portion of immunoglobulins obtained from a combinatorial phage display peptide library. J. Immunol. Methods 1998, 221, 151–157. [Google Scholar] [CrossRef]
- Kang, H.J.; Choe, W.; Min, J.K.; Lee, Y.M.; Kim, B.M.; Chung, S.J. Cyclic peptide ligand with high binding capacity for affinity purification of immunoglobulin G. J. Chromatogr. A 2016, 1466, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Dias, R.L.; Fasan, R.; Moehle, K.; Renard, A.; Obrecht, D.; Robinson, J.A. Protein ligand design: From phage display to synthetic protein epitope mimetics in human antibody Fc-binding peptidomimetics. J. Am. Chem. Soc. 2006, 128, 2726–2732. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Zhang, L.; Li, J.; Feng, S.; Deng, H. Development of the double cyclic peptide ligand for antibody purification and protein detection. Bioconjug. Chem. 2016, 27, 1569–1573. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, G.K.; Bailon, P. Identification of model peptides as affinity ligands for the purification of humanized monoclonal antibodies by means of phage disaplay. J. Biochem. Biophys. Method 2001, 49, 443–454. [Google Scholar] [CrossRef]
- Camperi, S.A.; Iannucci, N.B.; Albanesi, G.J.; Eberhardt, M.O.; Etcheverrigaray, M.; Messequer, A.; Albericio, F.; Cascone, O. Monoclonal antibody purification by affinity chromatography with ligands derived from the screening of peptide combinatory libraries. Biotechnol. Lett. 2003, 25, 1545–1548. [Google Scholar] [CrossRef] [PubMed]
- Verdoliva, A.; Marasco, D.; De Capua, A.; Saporito, A.; Bellofiore, P.; Manfredi, V.; Fattorusso, R.; Pedone, C.; Ruvo, M. A new ligand for immunoglobulin g subdomains by screening of a synthetic peptide library. ChemBioChem 2005, 6, 1242–1253. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Gurgel, P.V.; Carbonell, R.G. Hexamer peptide affinity resin that bind the Fc region of human immunoglobulin G. J. Peptide Res. 2006, 66, 110–137. [Google Scholar] [CrossRef]
- Yang, H.; Gurgel, P.V.; Carbonell, R.G. Purification of human immunoglobulin G via Fc-specific small peptide ligand affinity chromatography. J. Chromatogr. A 2009, 1216, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Menegatti, S.; Bobay, B.G.; Ward, K.L.; Islam, T.; Kish, W.S.; Naik, A.D.; Carbonell, R.G. Design of protease-resistant peptide ligands for the purification of antibodies from human plasma. J. Chromatogr. A 2016, 1445, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Lund, L.N.; Gustavsson, P.E.; Michael, R.; Lindgren, J.; Norskov-Lauritsen, L.; Lund, M.; Houen, G.; Staby, A.; St Hilaire, P.M. Novel peptide ligand with high binding capacity for antibody purification. J. Chromatogr. A 2012, 1225, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Menegatti, S.; Ward, K.L.; Naik, A.D.; Kish, W.S.; Blackburn, R.K.; Carbonell, R.G. Reversible cyclic peptide libraries for the discovery of affinity ligands. Anal. Chem. 2013, 85, 9229–9237. [Google Scholar] [CrossRef] [PubMed]
- Menegatti, S.; Hussain, A.; Naik, A.D.; Carbonell, R.G.; Rao, B.M. mRNA display selection and solid-phase synthesis of Fc-binding cyclic peptide affinity ligands. Biotechnol. Bioeng. 2013, 110, 857–870. [Google Scholar] [CrossRef] [PubMed]
- Sugita, T.; Katayama, M.; Okochi, M.; Kato, R.; Ichihara, T.; Honda, H. Screening of peptide ligands that bind to the Fc region of IgG using peptide array and its application to affinity purification of antibody. Biochem. Eng. J. 2013, 79, 33–40. [Google Scholar] [CrossRef]
- Zhao, W.-W.; Liu, F.-F.; Shi, Q.-H.; Dong, X.-Y.; Sun, Y. Biomimetic design of affinity peptide ligands for human IgG based on protein A-IgG complex. Biochem. Eng. J. 2014, 88, 1–11. [Google Scholar] [CrossRef]
- Zhao, W.W.; Shi, Q.H.; Sun, Y. FYWHCLDE-based affinity chromatography of IgG: Effect of ligand density and purifications of human IgG and monoclonal antibody. J. Chromatogr. A 2014, 1355, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.W.; Liu, F.F.; Shi, Q.H.; Sun, Y. Octapeptide-based affinity chromatography of human immunoglobulin G: Comparisons of three different ligands. J. Chromatogr. A 2014, 1359, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.W.; Shi, Q.H.; Sun, Y. Dual-ligand affinity systems with octapeptide ligands for affinity chromatography of hIgG and monoclonal antibody. J. Chromatogr. A 2014, 1369, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.W.; Jheng, S.L.; Chen, W.Y.; Ruaan, R.C. Strategy of Fc-recognizable Peptide ligand design for oriented immobilization of antibody. Anal. Chem. 2014, 86, 2931–2938. [Google Scholar] [CrossRef] [PubMed]
- Yoo, R.-J.; Choi, S.-J. Identification of a peptide ligand for antibody immobilization on biosensor surfaces. BioChip J. 2015, 10, 88–94. [Google Scholar] [CrossRef]
- Nair, M.; Vijayan, M.; Venkatachalapath, Y.V.; Balaram, P. X-ray Crystal Structure of Pivaloyl-[D]-Pro-L-Pro-L-Ala-N-methylamide; Observation of a Consecutive β-Turn Conformation. J. Chem. Soc. Chem. Commun. 1979, 1183–1184. [Google Scholar] [CrossRef]
- Yang, H.; Gurgel, P.V.; Williams, D.K., Jr.; Bobay, B.G.; Cavanagh, J.; Muddiman, D.C.; Carbonell, R.G. Binding site on human immunoglobulin G for the affinity ligand HWRGWV. J. Mol. Recognit 2010, 23, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Kang, H.J.; Lee, J.M.; Jung, S.O.; Yun, W.S.; Chung, S.J.; Chung, B.H. Controlled antibody immobilization onto immunoanalytical platforms by synthetic peptide. Anal. Biochem. 2008, 374, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Kang, Y.J.; Lee, Y.M.; Shin, H.H.; Chung, S.J.; Kang, S. Developing an antibody-binding protein cage as a molecular recognition drug modular nanoplatform. Biomaterials 2012, 33, 5423–5430. [Google Scholar] [CrossRef] [PubMed]
- Ghisaidoobe, A.B.T.; Chung, S.J. Functionalized protein nanocages as a platform of targeted therapy and immunodetection. Nanomedicine 2015, 10, 3579–3595. [Google Scholar] [CrossRef] [PubMed]
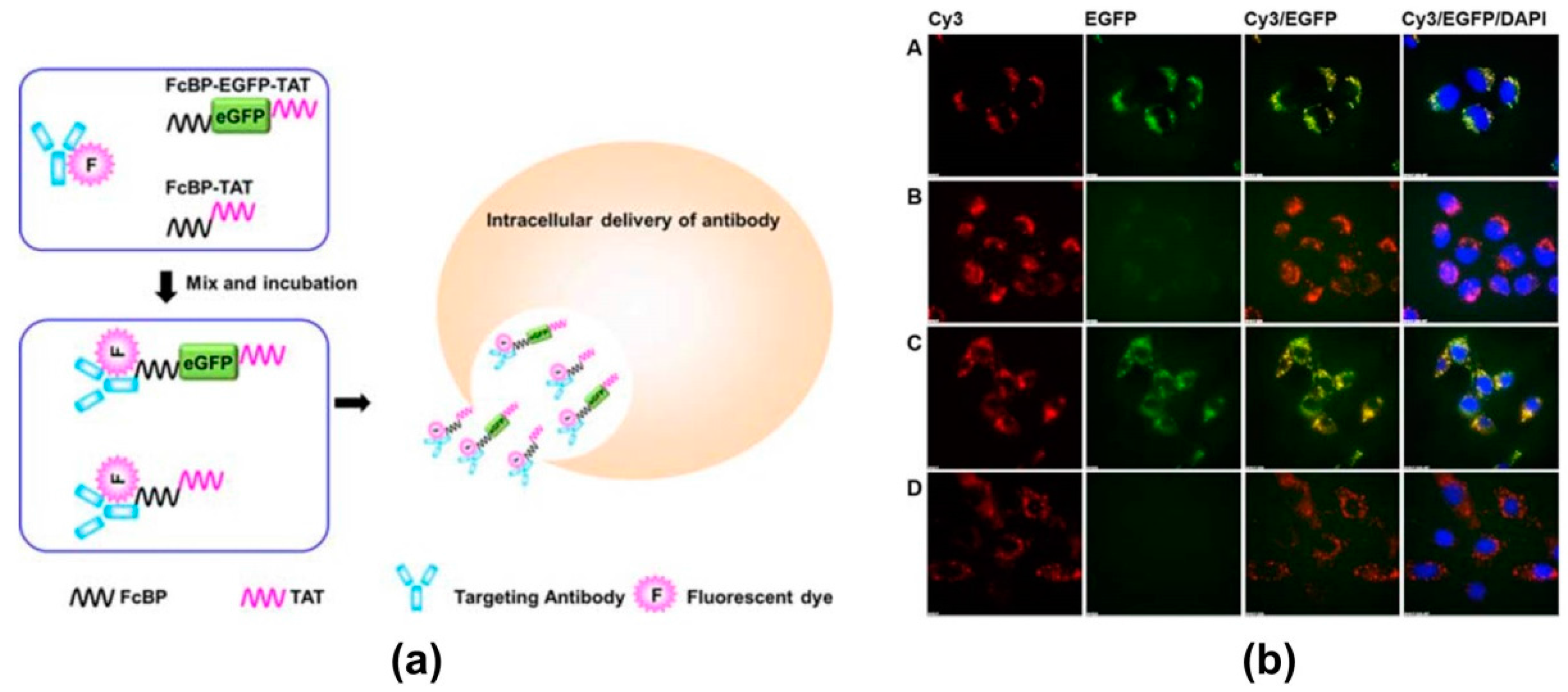
- Kang, H.J.; Choe, W.; Kim, B.M.; Chung, S.J. IgG Fc-binding peptide (FcBP)-tat conjugate as a smart antibody carrier into live cells. Macromol. Res. 2015, 23, 876–881. [Google Scholar] [CrossRef]
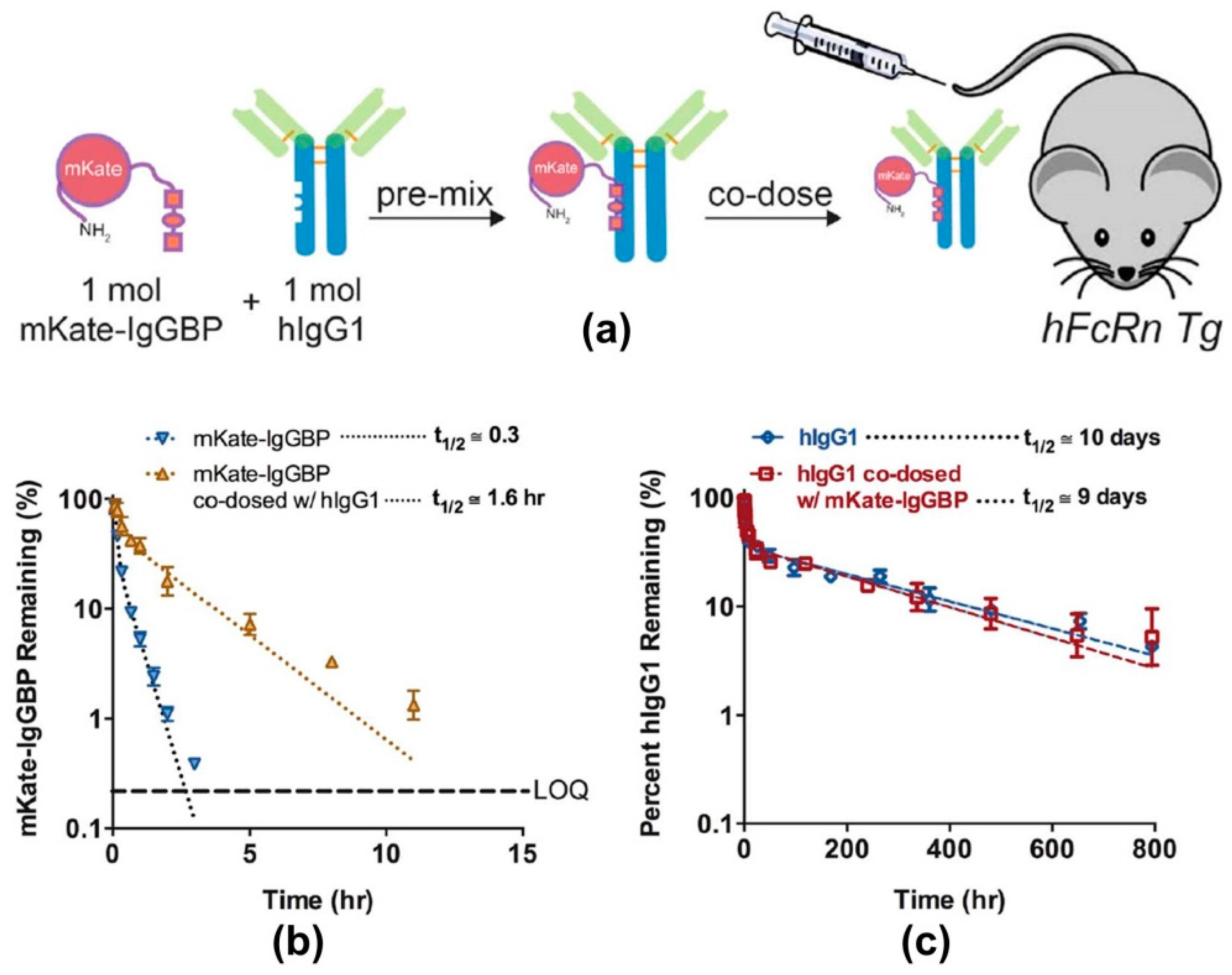
- Sockolosky, J.; Kivimae, S.; Szoka, F. Fusion of a short protein that binds to immunoglobilin G to a recombinat protein substantially increases its plasma half-life in mice. PLoS ONE 2014, 9, e102566. [Google Scholar] [CrossRef] [PubMed]
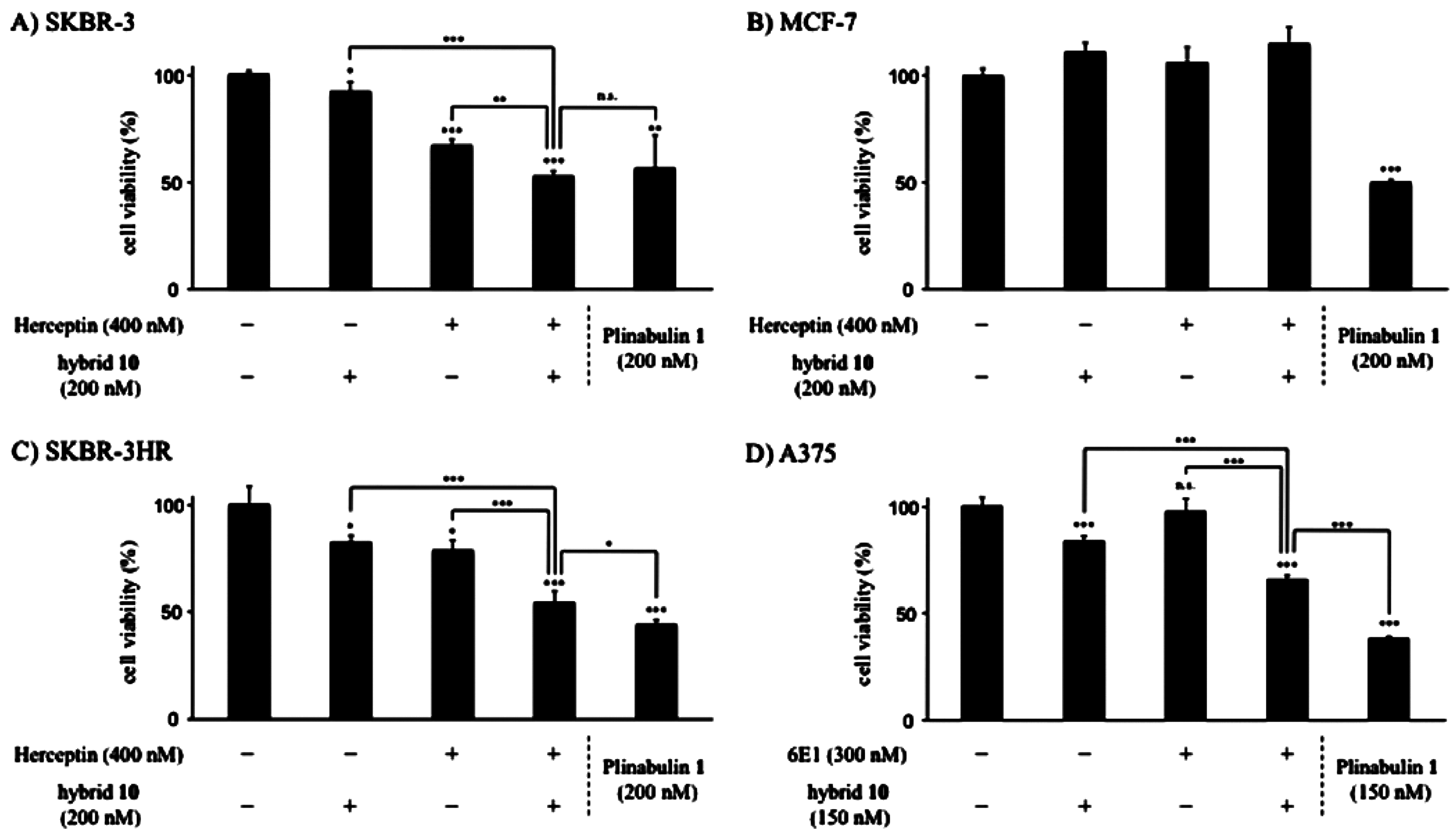
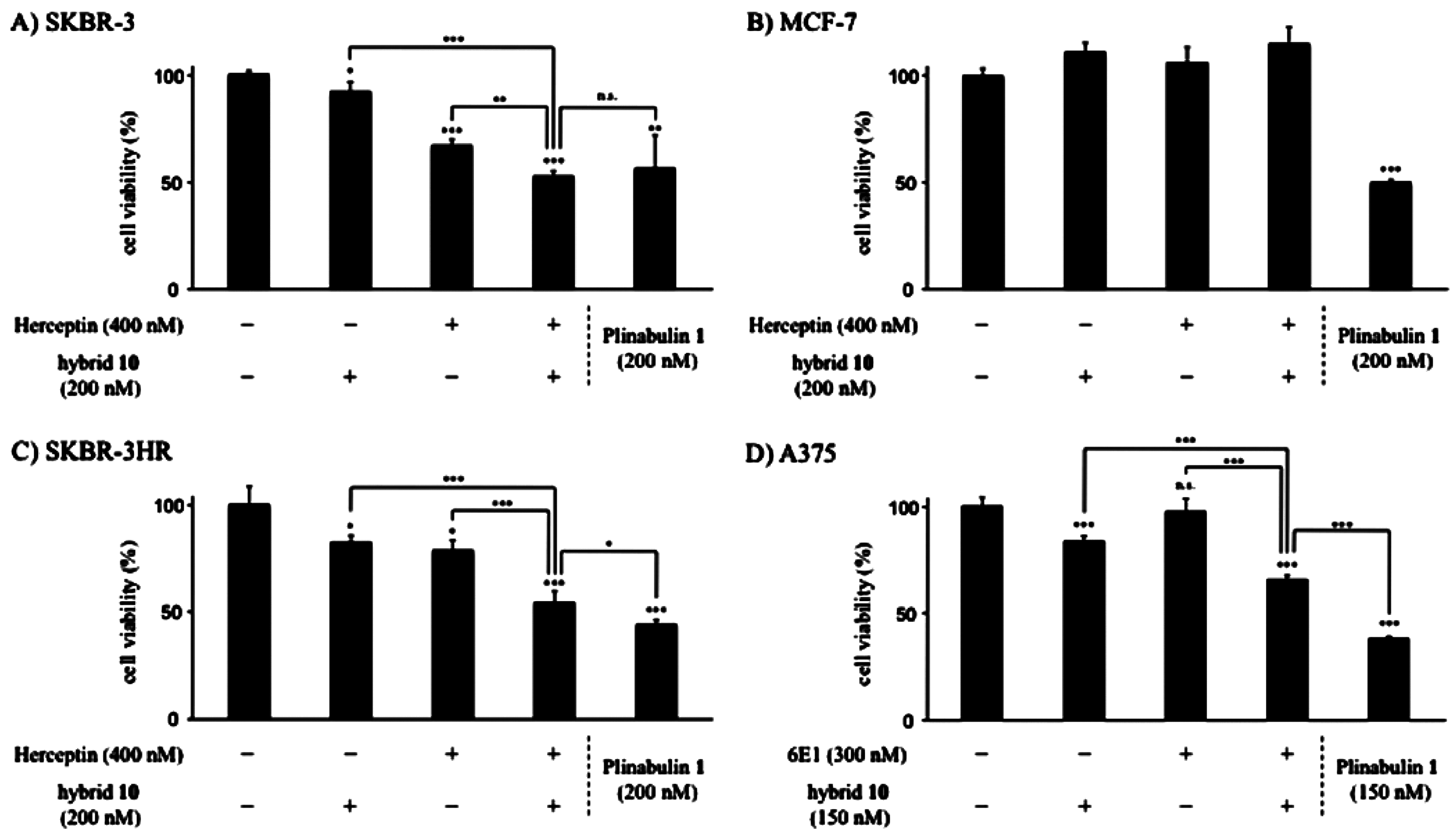
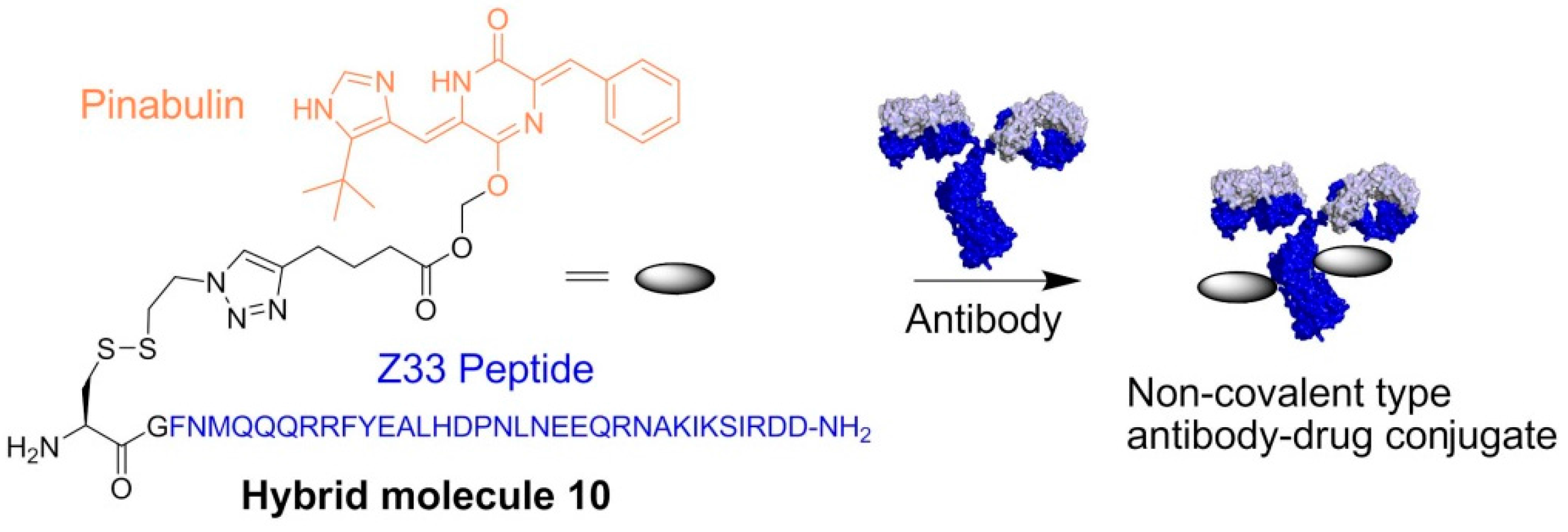
- Muguruma, K.; Yakushiji, F.; Kawamata, R.; Akiyama, D.; Arima, R.; Shirasaka, T.; Kikkawa, Y.; Taguchi, A.; Takayama, K.; Fukuhara, T.; et al. Novel hybrid compound of a plinabulin prodrug with an IgG binding peptide for generating a tumor selective noncovalent-type antibody-drug conjugate. Bioconjug. Chem. 2016, 27, 1606–1613. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
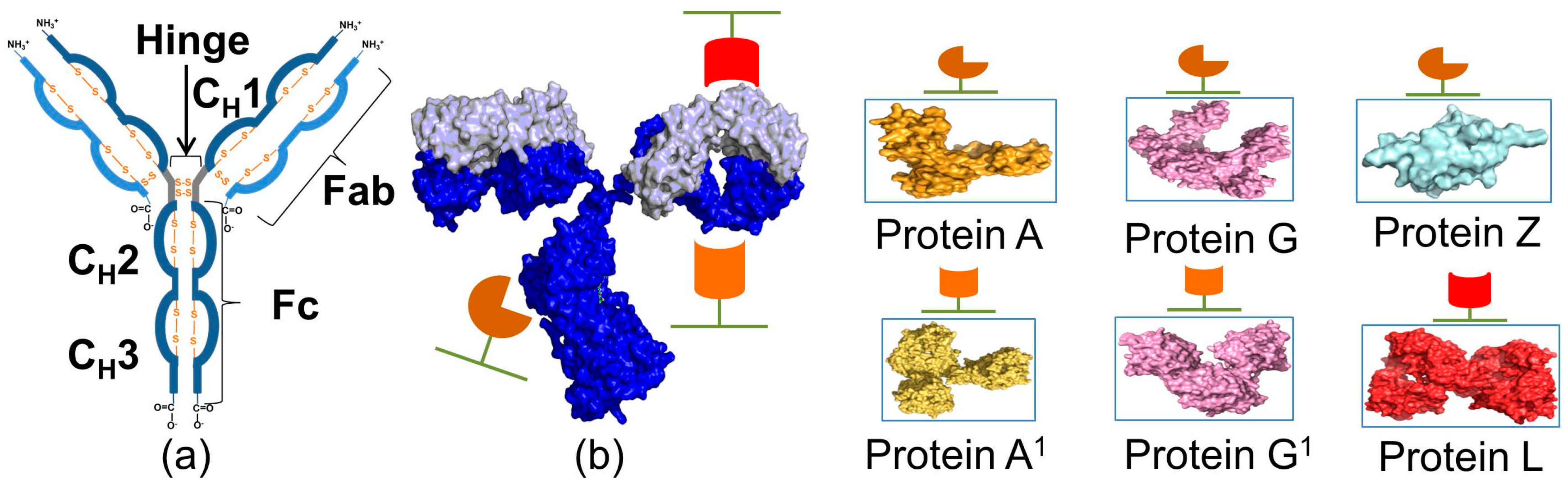
| Protein | Ka * | Size | Target | Elution pH | References |
|---|---|---|---|---|---|
| Protein A | 1.4 × 108 M−1 | 42 kDa | Fc, Fab | 3.5 | [12,29] |
| Protein G | 6.7 × 109 M−1 | 30 kDa | Fc, Fab, ScFv, Dab | 3.2 | [28] |
| Protein L | 5.69 × 107 M−1 | 76 to 106 kDa | Fab, ScFv, Dab, and κ-light chain | 2 | [23] |
| Protein Z | 5.0 × 108 M−1 | 6.7 kDa | Fc | 3.6 | [24] |
| Protein LG | 5.9 × 109 M−1 | 50 kDa | Fc, κ-light chain | 2 | [25] |
| Protein LA | 2.93 × 108 M−1 | 60 kDa | Fc, κ-light chain | 2 | [26] |
| Protein AG | n.a. | 47.3 kDa | Fc | 3 | [27] |
| Peptide | Binding Constant | Binding Capacity * | Elution pH | Remarks | Year (References) |
|---|---|---|---|---|---|
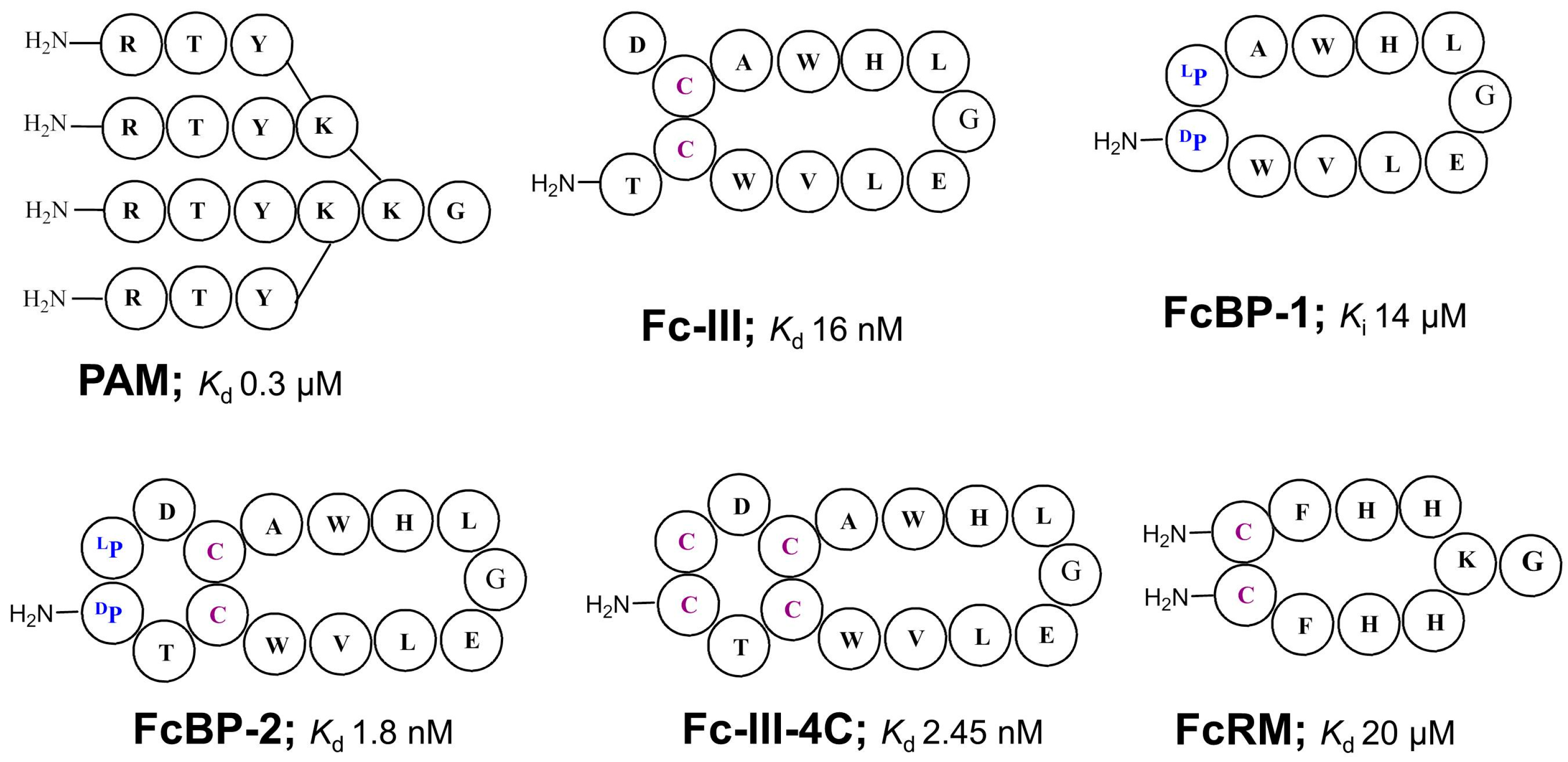
| PAM | Kd = 0.3 μM | n.a. | 3 or 9 | Dendrimer comb. library SpA mimic | 1996 [45] |
| d-PAM | n.a. | 50 | 3.5 | 2002 [46] | |
| d-PAM-Ф | n.a. | 10 | 4 | 2011 [47] | |
| TWKTSRISIF | n.a. | n.a. | n.a. | Phage display library SpA mimic | 1998 [48] |
| FGRLVSSIRY | n.a. | n.a. | n.a. | ||
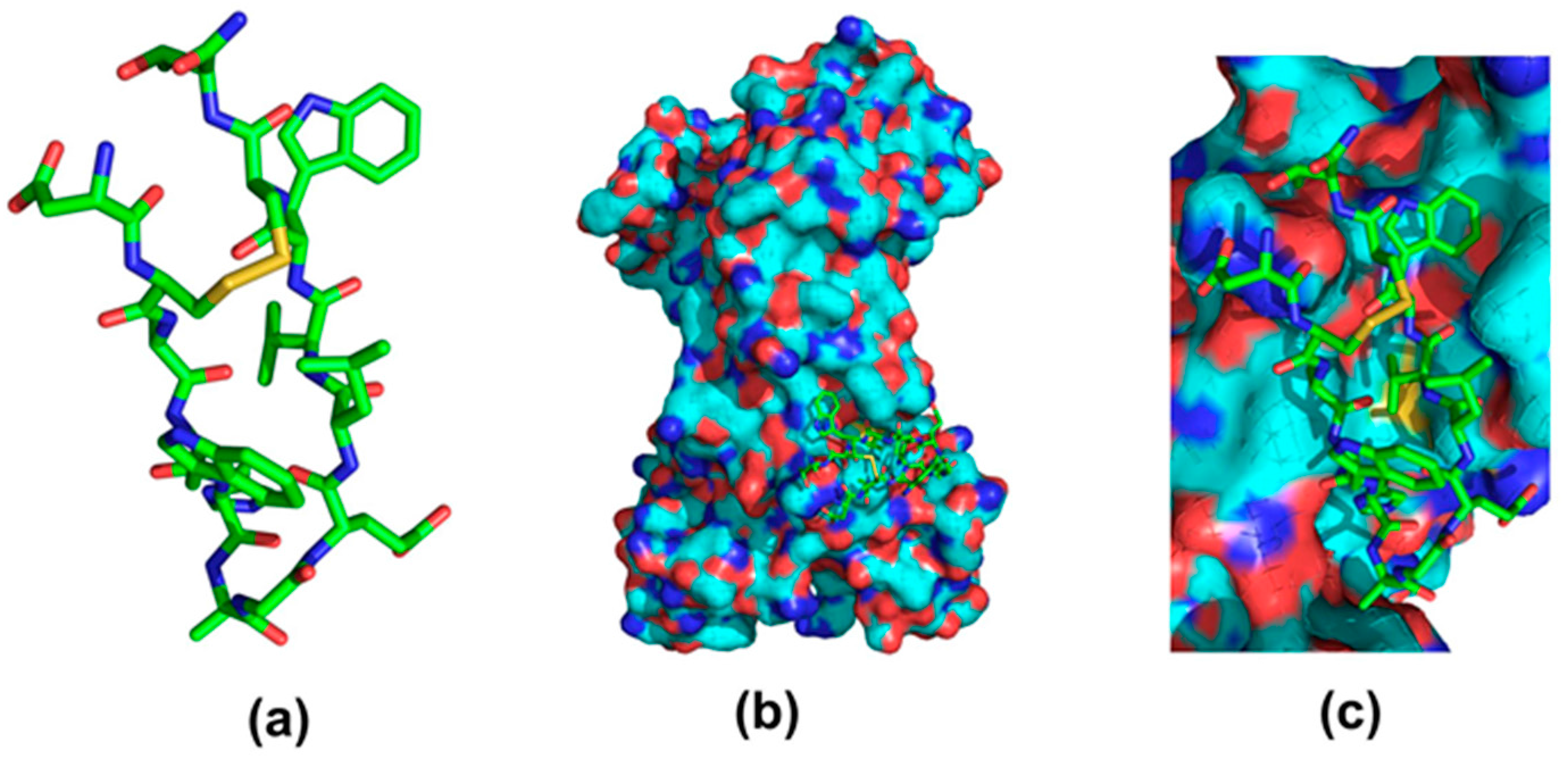
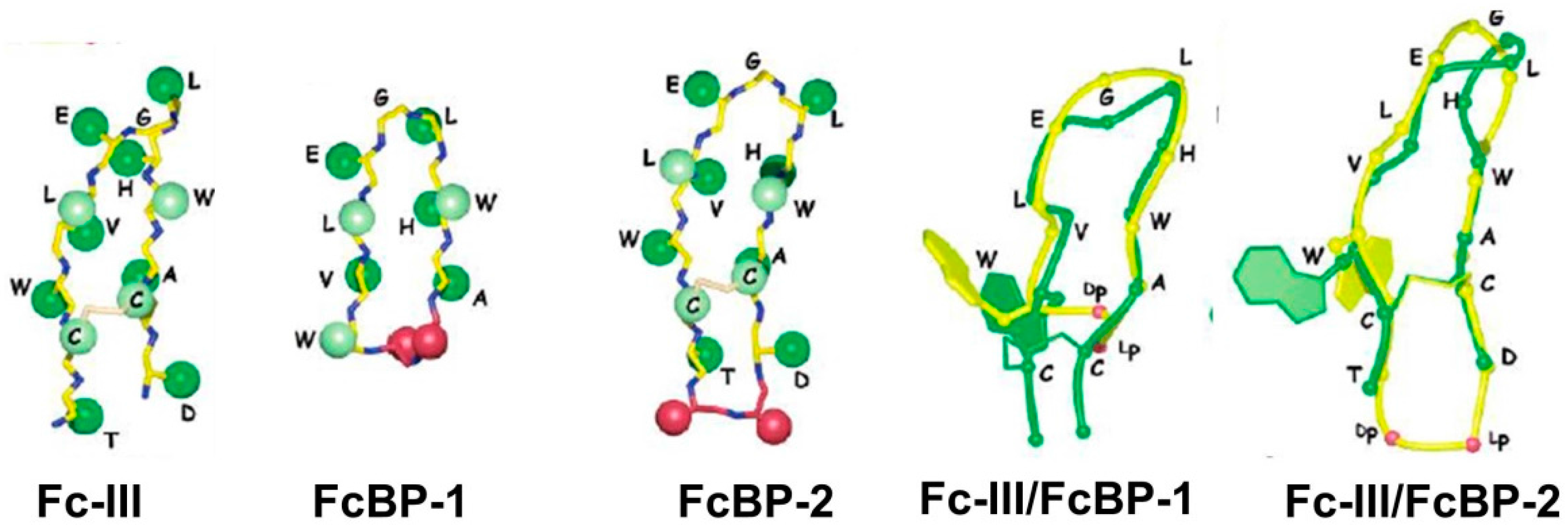
| Fc-III (DeLano et al.) | Kd = 16 nM | Phage display cyclic peptide library SpA mimic | 2000 [42] | ||
| Fc-III-(Sepharose) | 26.6 mg/μmol | 3.5 | 2016 [49] | ||
| FcBP-2 | Kd = 1.8 nM | n.a | n.a. | Bicyclic peptide | 2006 [50] |
| Fc-III-4C | Kd = 2.5 nM | n.a. | 3.5 | Bicyclic peptide | 2016[51] |
| EPIHRSTLTALL | n.a. | 320 μg/g | n.a. | Phage display library SpA mimic | 2001 [52] |
| APAR | Kd = 94 nM | 9.1 | n.a. | Comb. tetrapeptide library | 2003 [53] |
| FcRM | Kd = 20 μM | n.a. | 2.7 | Synthetic cyclic peptide library, Fcγ-receptor mimic | 2005 [54] |
| HWRGWV | Kd = 10 μM | 28.4 | 4 | Comb. library (one-bead-one-peptide) SpA mimic | 2006 [55] |
| HYFKFD | Kd = 11 μM | 27.0 | n.a. | 2009 [56] | |
| HFRRHL | Kd = 26 μM | 33.6 | n.a. | 2016 [57] | |
| HWCitGWV | Kd = 108 μM | 72 | n.a. | ||
| D2AAG | Ka = 7.9 × 105 M−1 | 36.2 | 3.6 | Comb. library SpA mimic | 2012 [58] |
| DAAG | Ka = 2.6 × 106 M−1 | 49.6 | 3.6 | ||
| cyclo[(Nα-Ac)S(A)-RWHYFK-Lact-E] | n.a. | n.a. | 3.5 | Cyclic peptide | 2013 [59] |
| cyclo[(Nα-Ac)–Dap(A)-RWHYFK-Lact-E] | n.a. | n.a. | 3.5 | ||
| cyclo[Link-M-WFRHYK] | Kd = 7.6 μM | 19.7 | 4.0 | mRNA display library SpA mimic | 2013 [60] |
| NKFRGKYK | Ka = 8.6 × 106 M−1 | DBC-4.9 | 4.0 | Spot peptide array Fcγ mimic | 2013 [61] |
| NARKFYKG | Ka = 6.5 × 106 M−1 | DBC-5.0 | 4.0 | ||
| FYWHCLDE(1) | Kd = 1.5 μM | 104.2 | 6.0 | Biomimetic design strategy | 2014 [62] |
| FYCHWALE(2) | Kd = 6.1 μM | 87.6 | 6.0 | Fc-binder (SpA mimic) | 2014 [63] |
| FYCHTIDE(3) | Kd = 5.7 μM | 63.7 | 6.0 | 2014 [64] | |
| Dual 1/3 (2:1) | Kd = 0.69 μM | 137.9 | 6.0 | Dual affinity ligand | 2014 [65] |
| RRGW | Kd = 0.5 nM | n.a. | n.a. | Computer design strategy Fc-binder | 2014 [66] |
| KHRFNKD | Kd = 20 nM | n.a. | n.a. | Phage-display library SpA mimic | 2015 [67] |
| Ligand | IC50 (nM) | Ki (nM) |
|---|---|---|
| Fc-III, pH = 7.4 | 460 ± 74 | 215 |
| Fc-III, pH = 6.0 | 280 ± 41 | 41 |
| Fc-III, reduced | >106 | n.d. |
| FcBP-1, pH = 7.4 | 64,800 ± 4400 | 14,200 |
| FcBP-1, pH = 6.0 | 395,000 ± 7100 | 71,000 |
| FcBP-2, pH = 7.4 | 32 ± 2 | 0.4 |
| FcBP-2, pH = 6.0 | 35 ± 2 | 2.0 |
| FcBP-2, reduced, pH = 7.4 | 550 ± 50 | n.d. |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choe, W.; Durgannavar, T.A.; Chung, S.J. Fc-Binding Ligands of Immunoglobulin G: An Overview of High Affinity Proteins and Peptides. Materials 2016, 9, 994. https://doi.org/10.3390/ma9120994
Choe W, Durgannavar TA, Chung SJ. Fc-Binding Ligands of Immunoglobulin G: An Overview of High Affinity Proteins and Peptides. Materials. 2016; 9(12):994. https://doi.org/10.3390/ma9120994
Chicago/Turabian StyleChoe, Weonu, Trishaladevi A. Durgannavar, and Sang J. Chung. 2016. "Fc-Binding Ligands of Immunoglobulin G: An Overview of High Affinity Proteins and Peptides" Materials 9, no. 12: 994. https://doi.org/10.3390/ma9120994
APA StyleChoe, W., Durgannavar, T. A., & Chung, S. J. (2016). Fc-Binding Ligands of Immunoglobulin G: An Overview of High Affinity Proteins and Peptides. Materials, 9(12), 994. https://doi.org/10.3390/ma9120994