Polymer-Derived Boron Nitride: A Review on the Chemistry, Shaping and Ceramic Conversion of Borazine Derivatives


Abstract
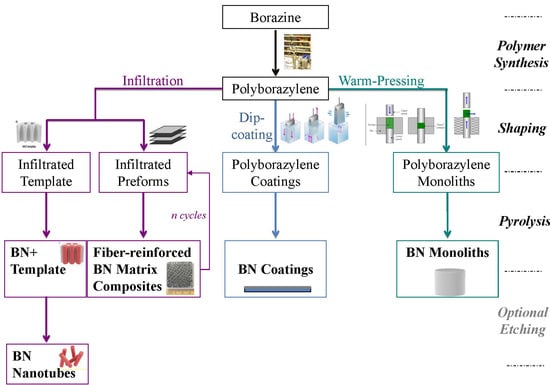
:
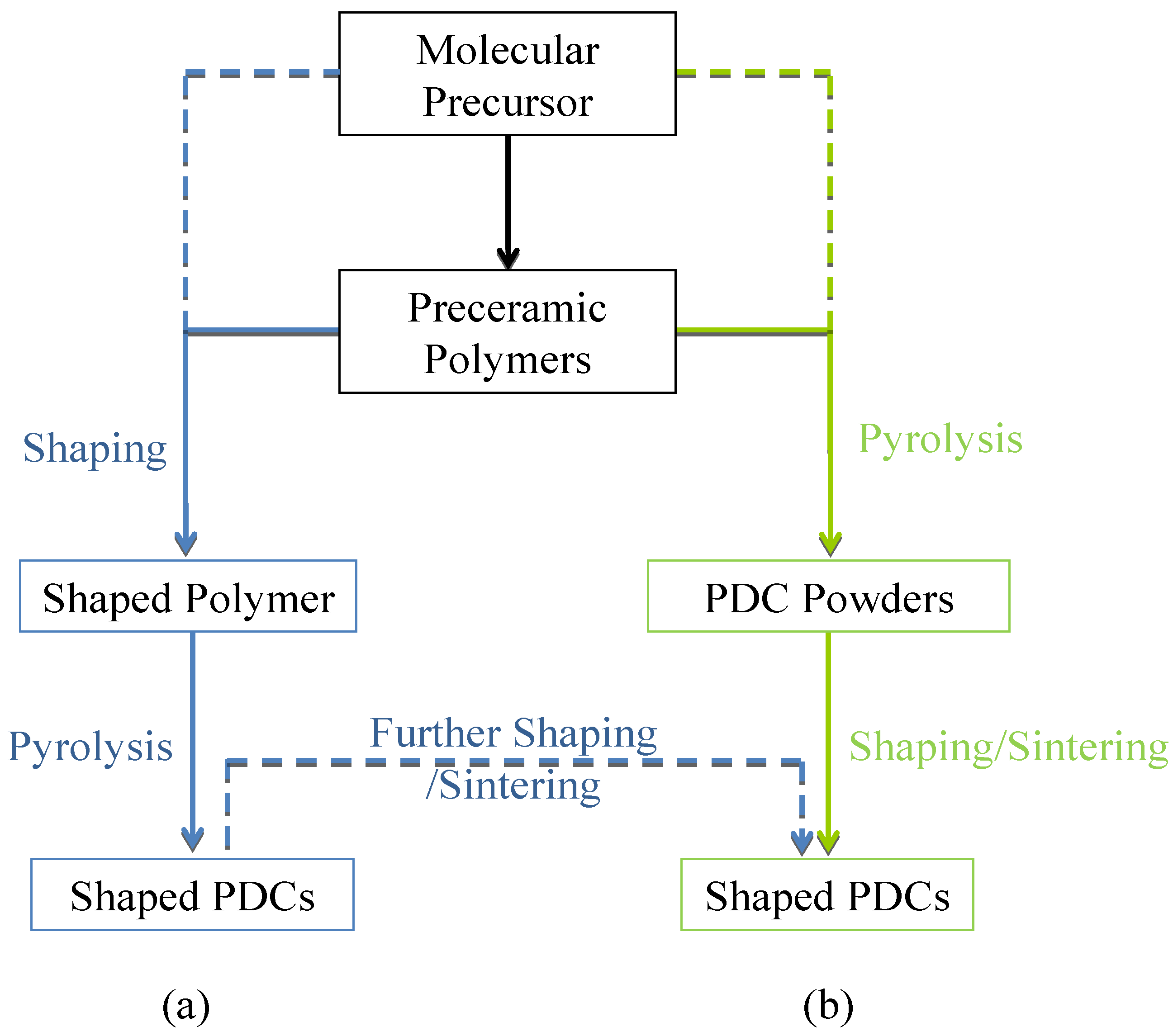
1. Introduction
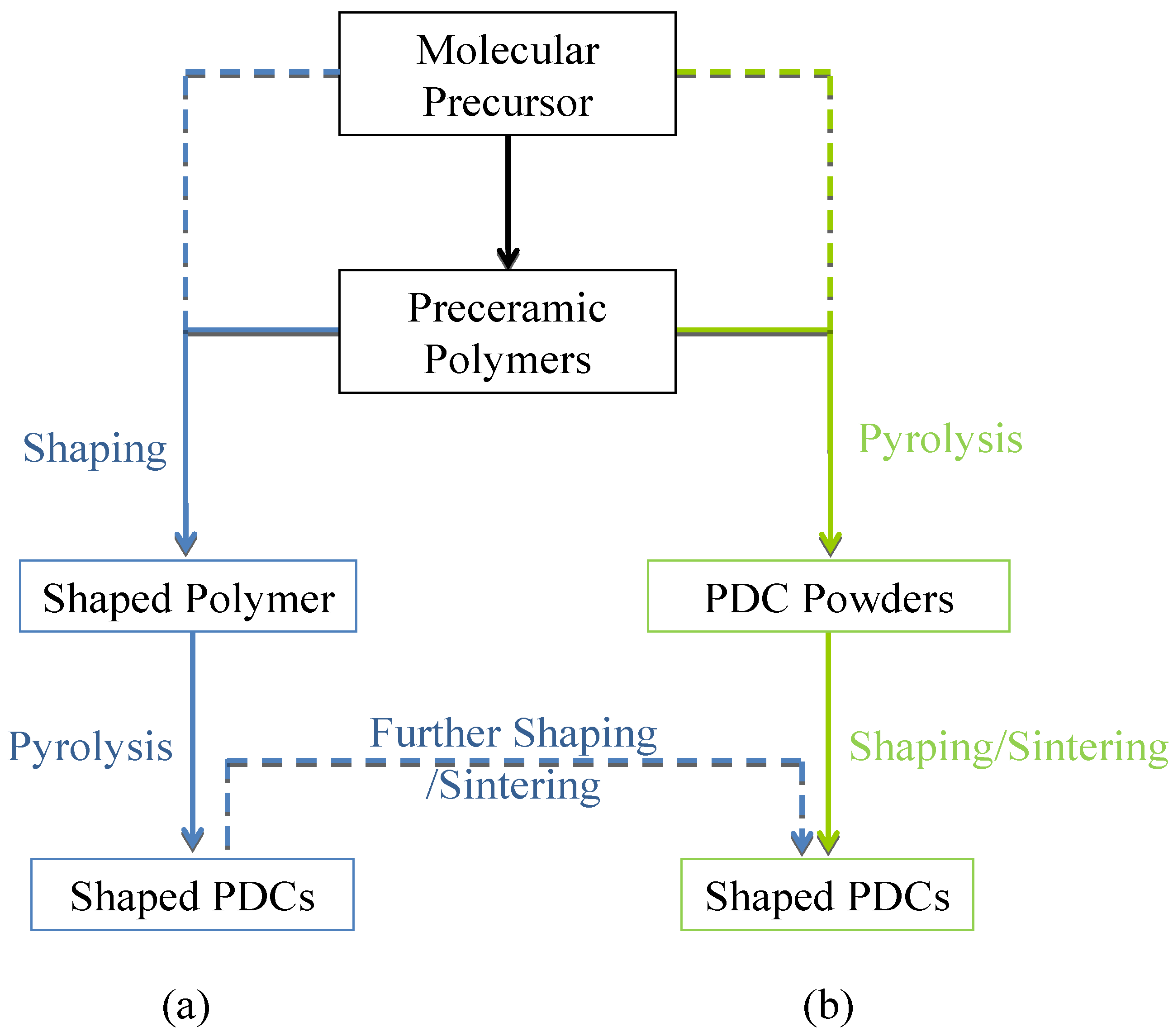
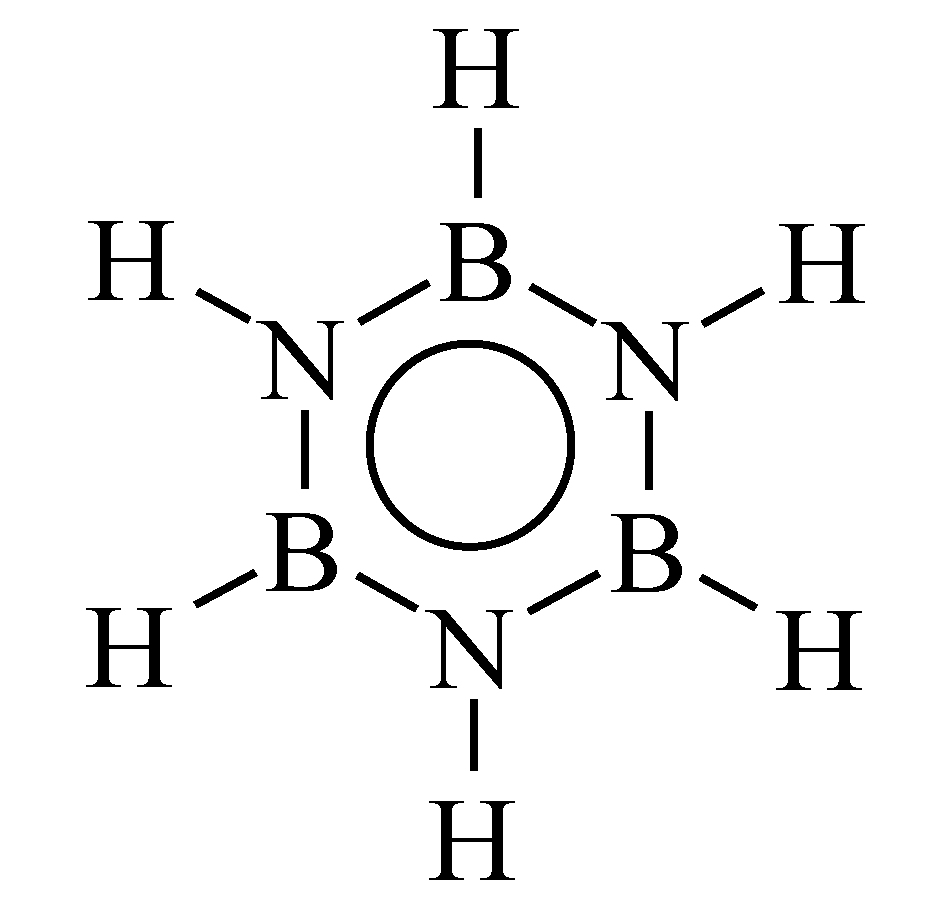
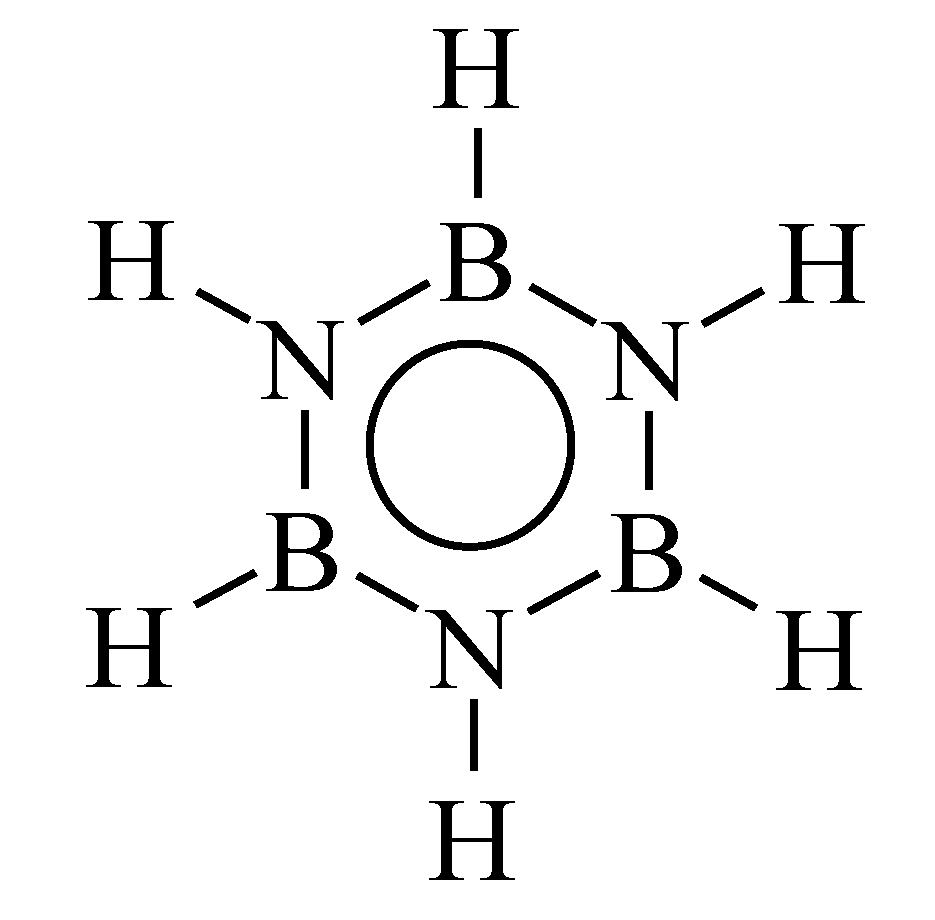
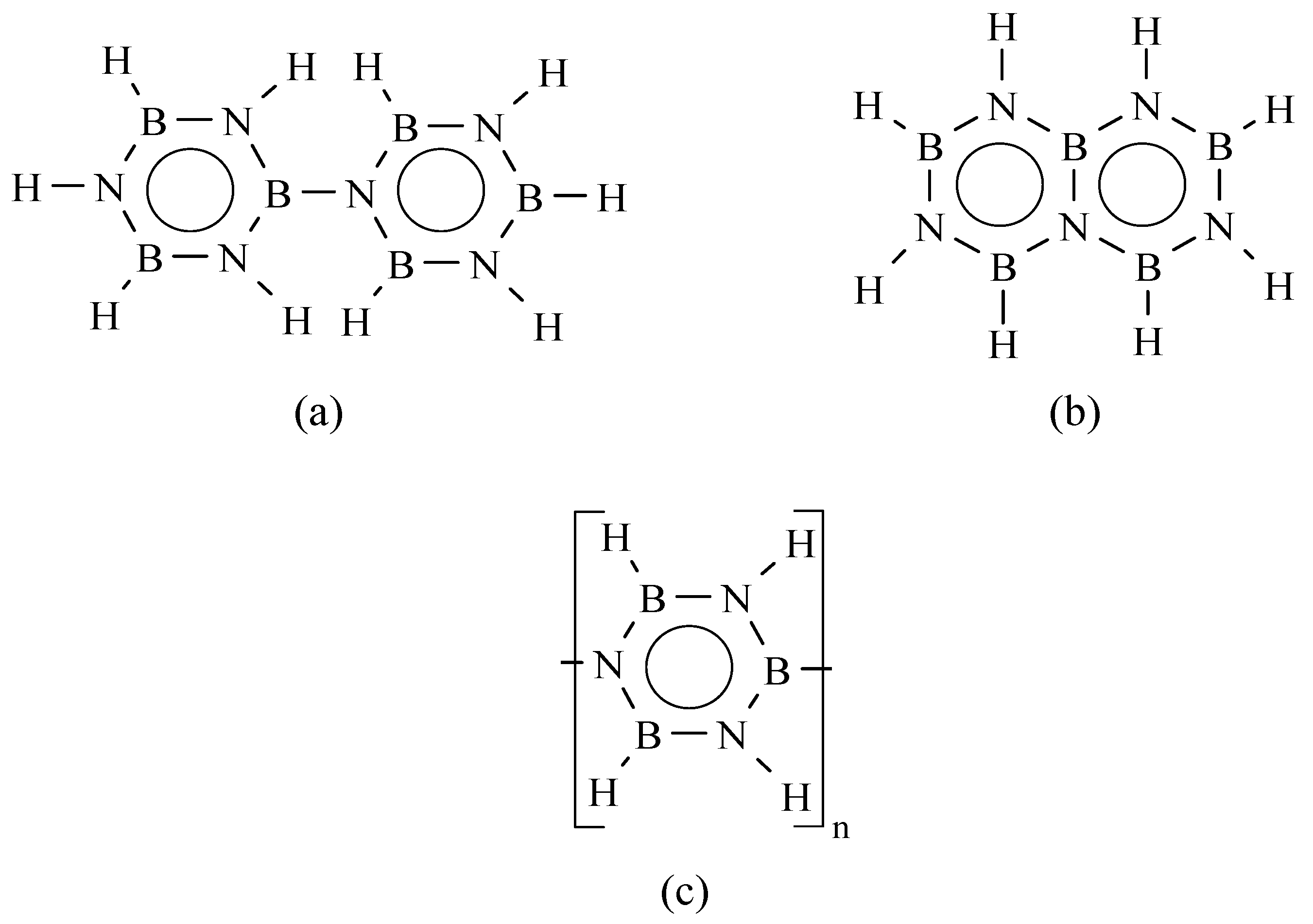
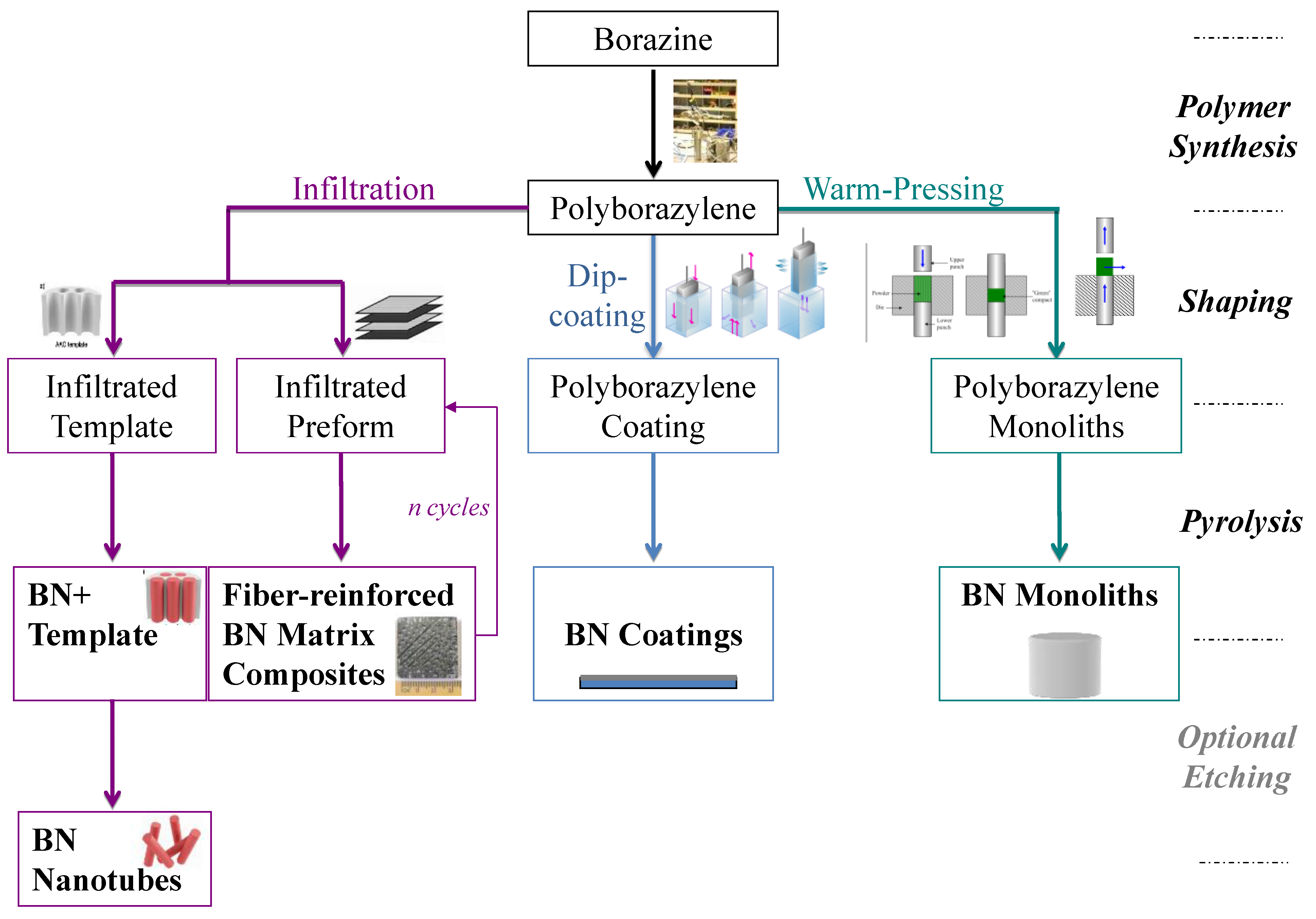
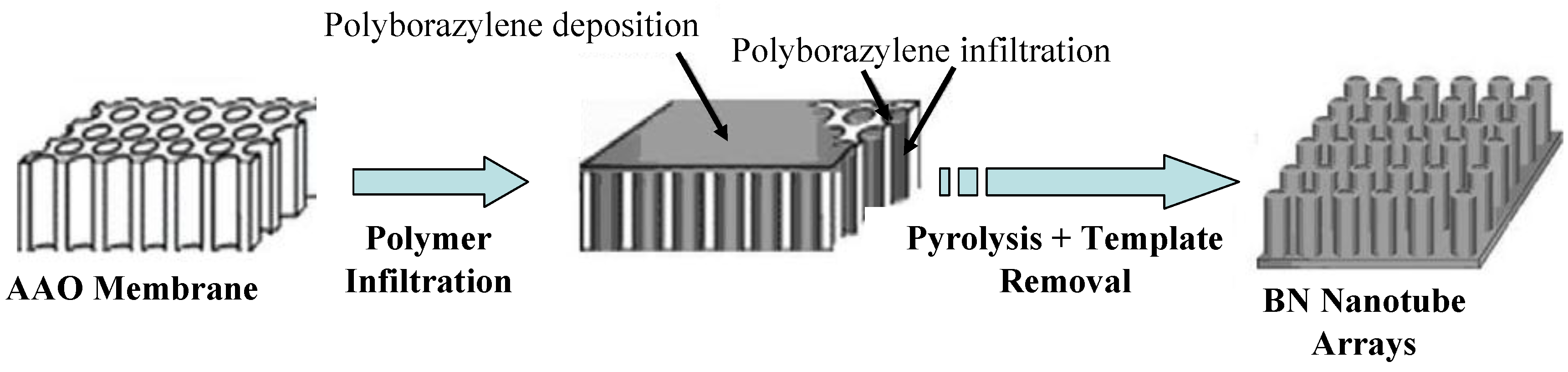
2. Borazine and Polyborazylene-Derived BN
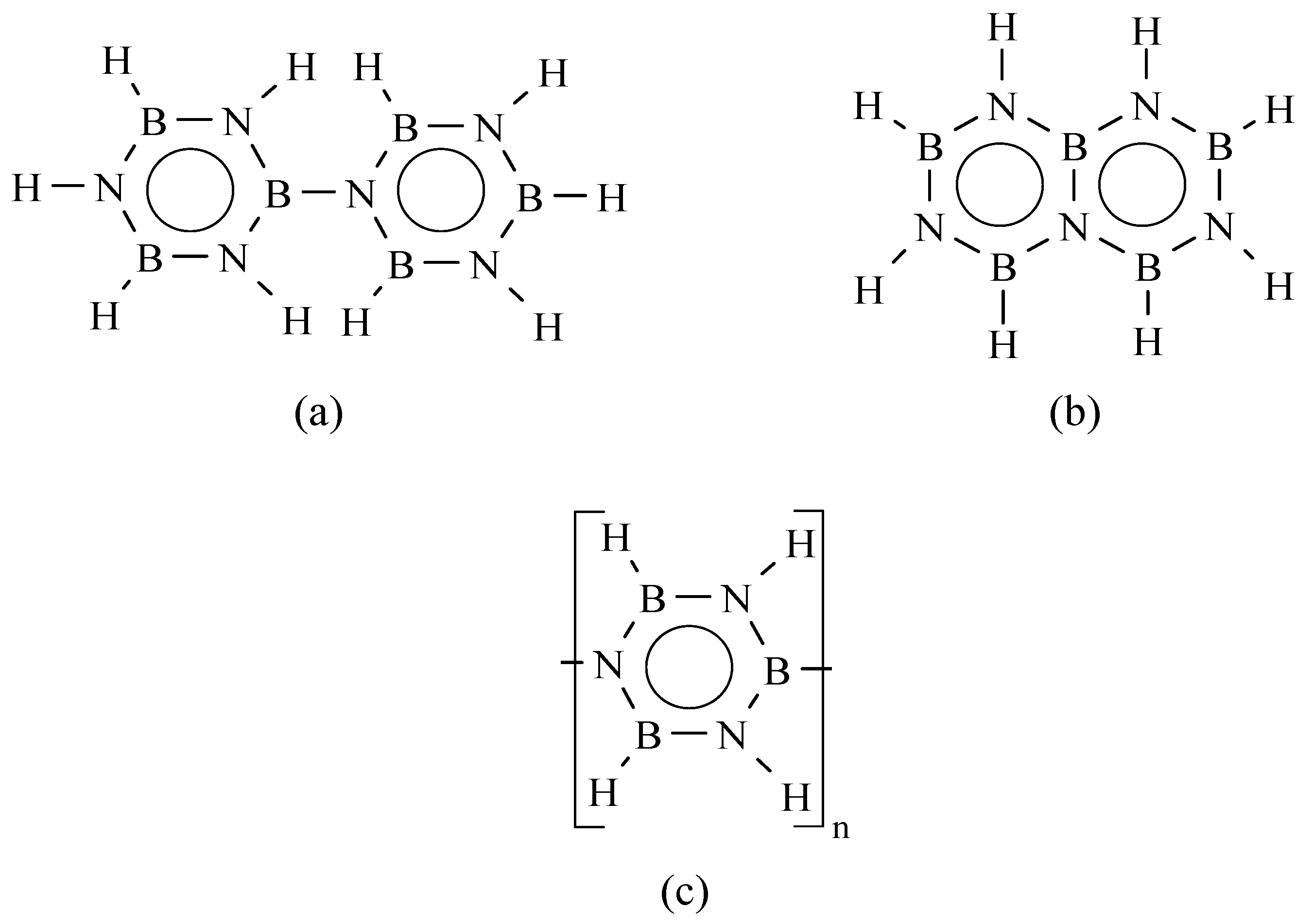
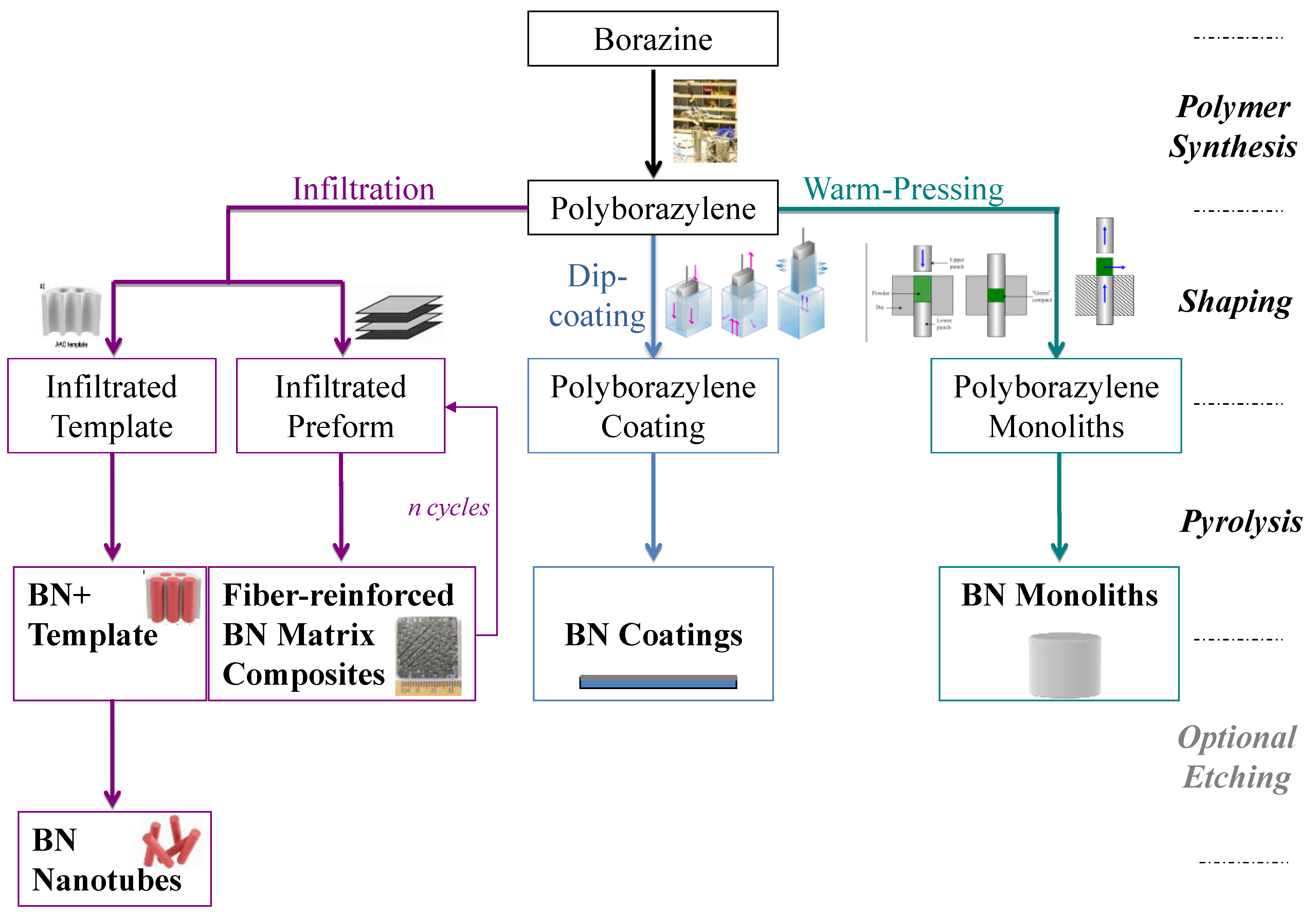

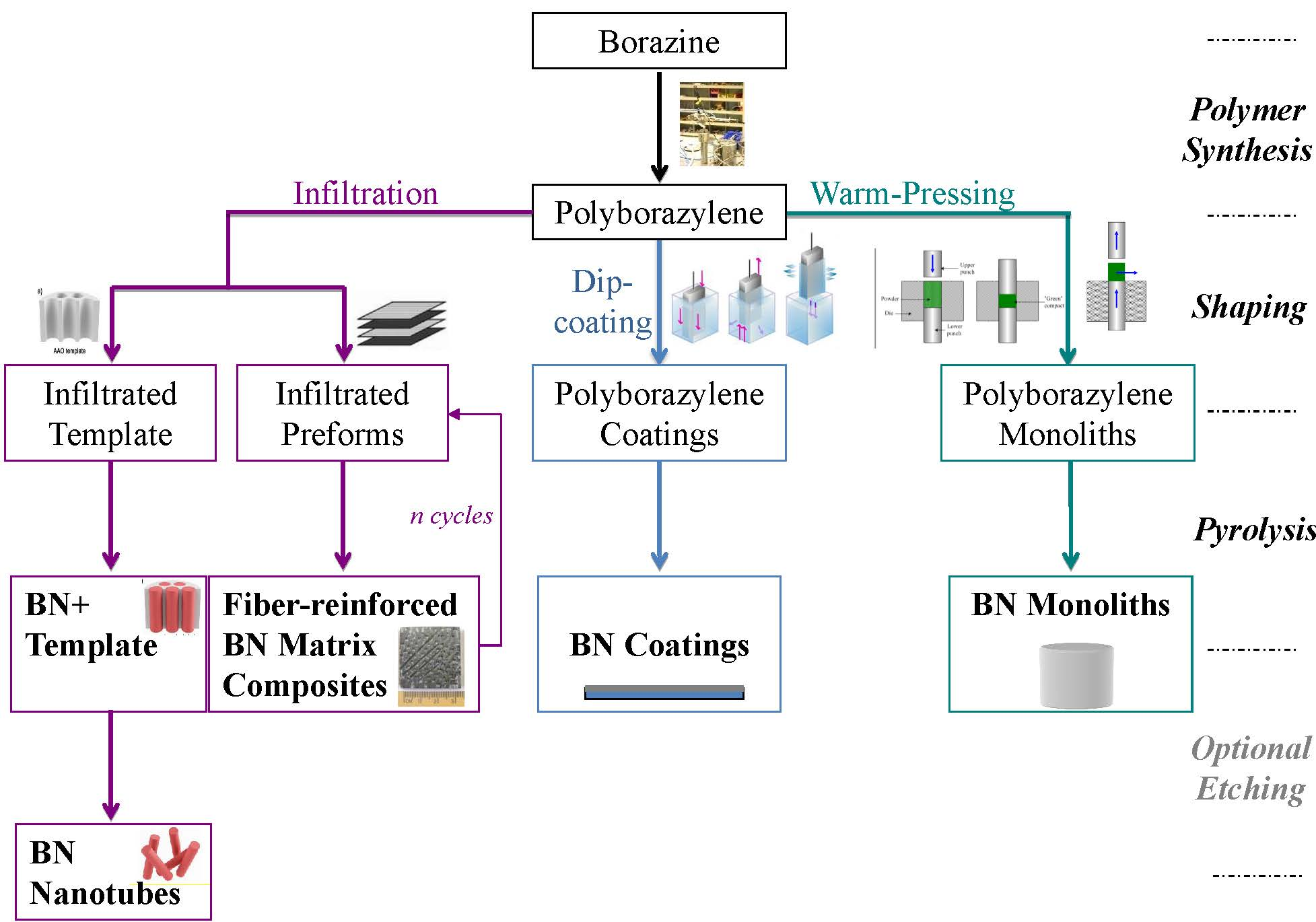{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
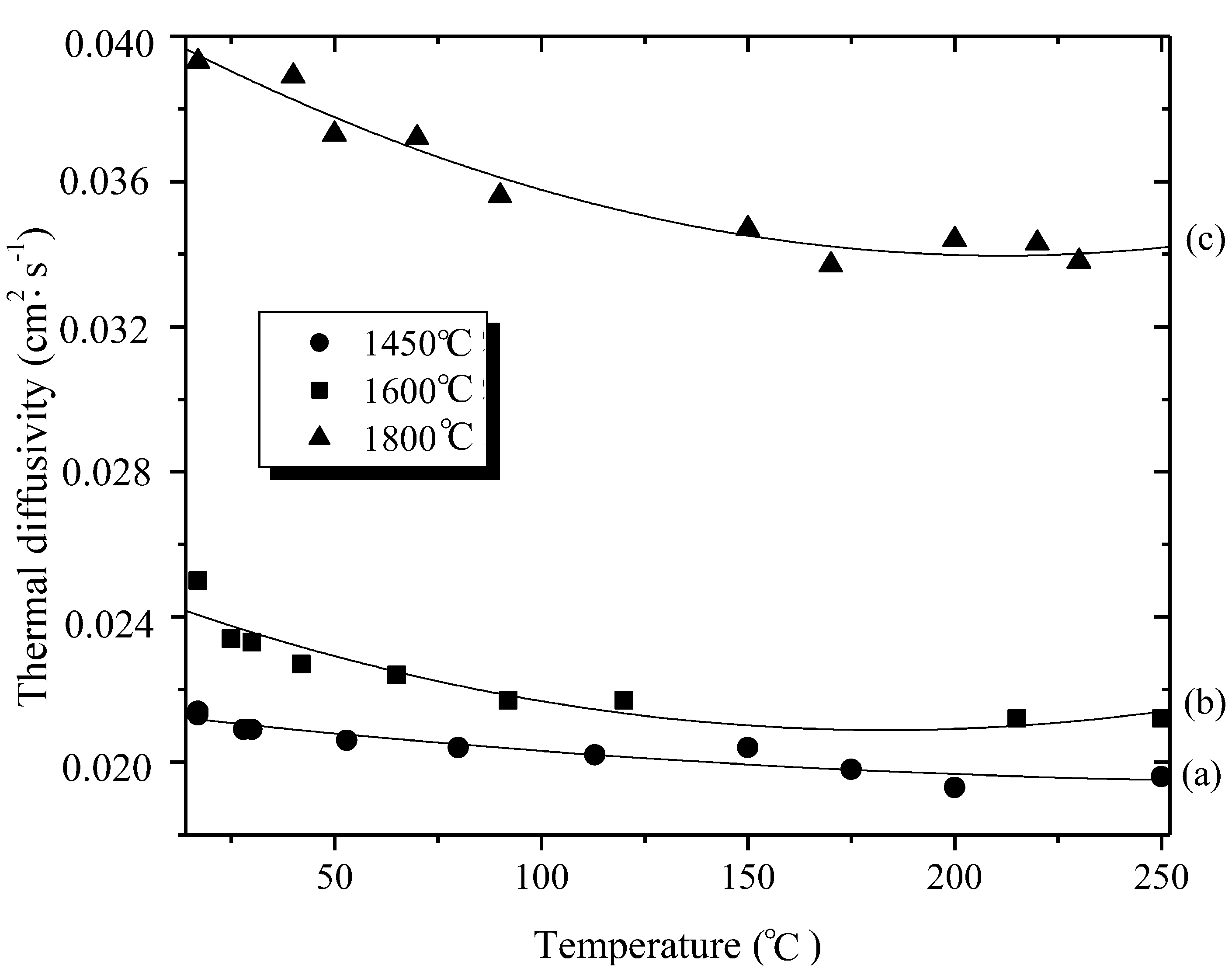{kind=link}
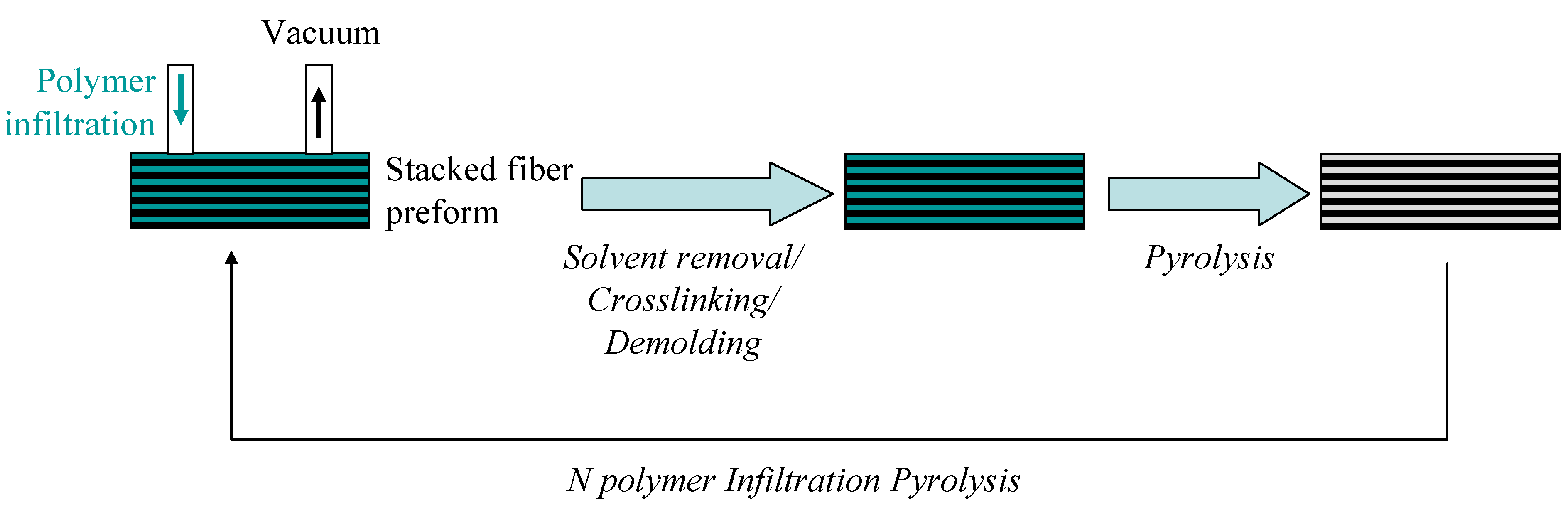{kind=link}
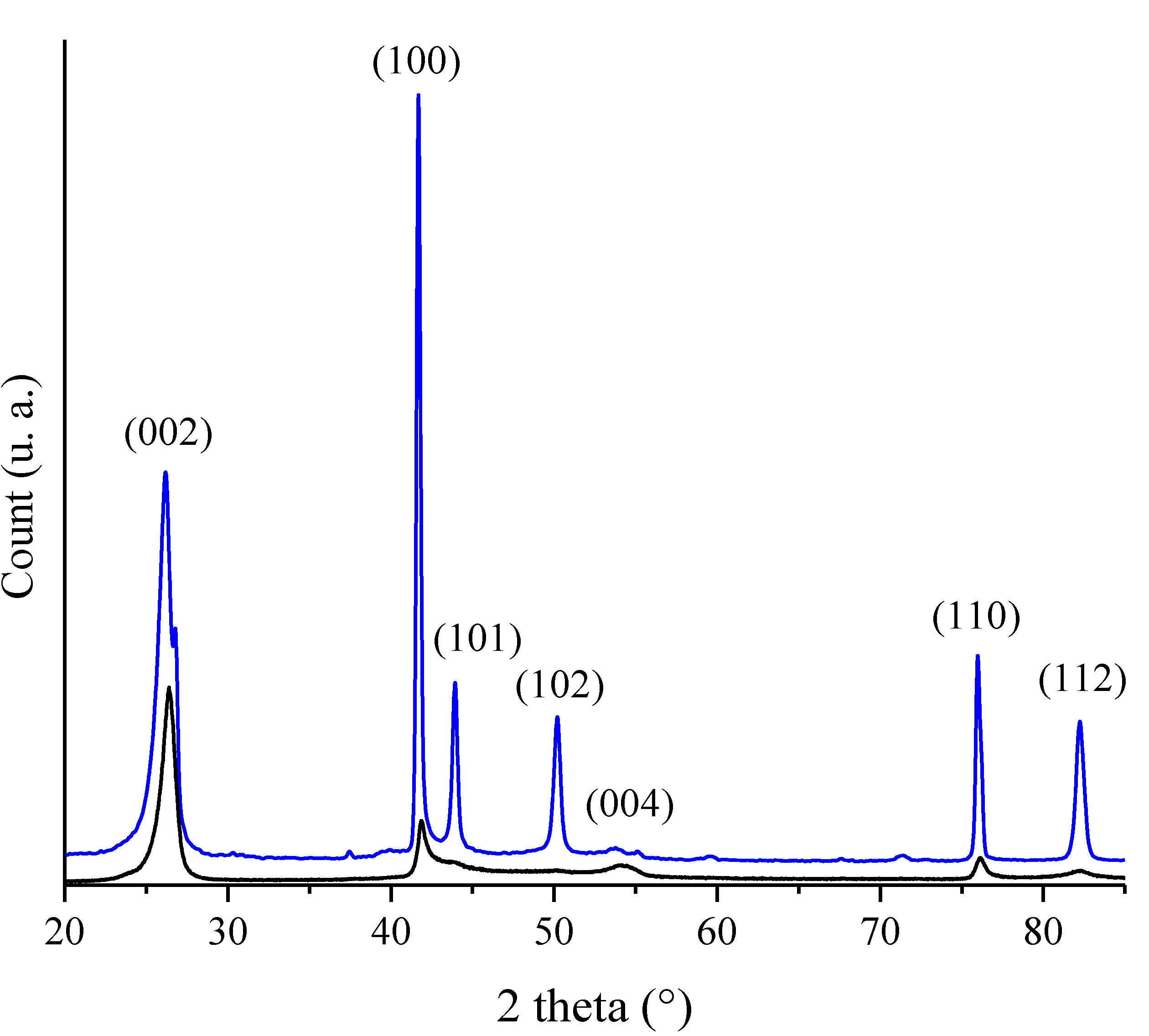{kind=link}
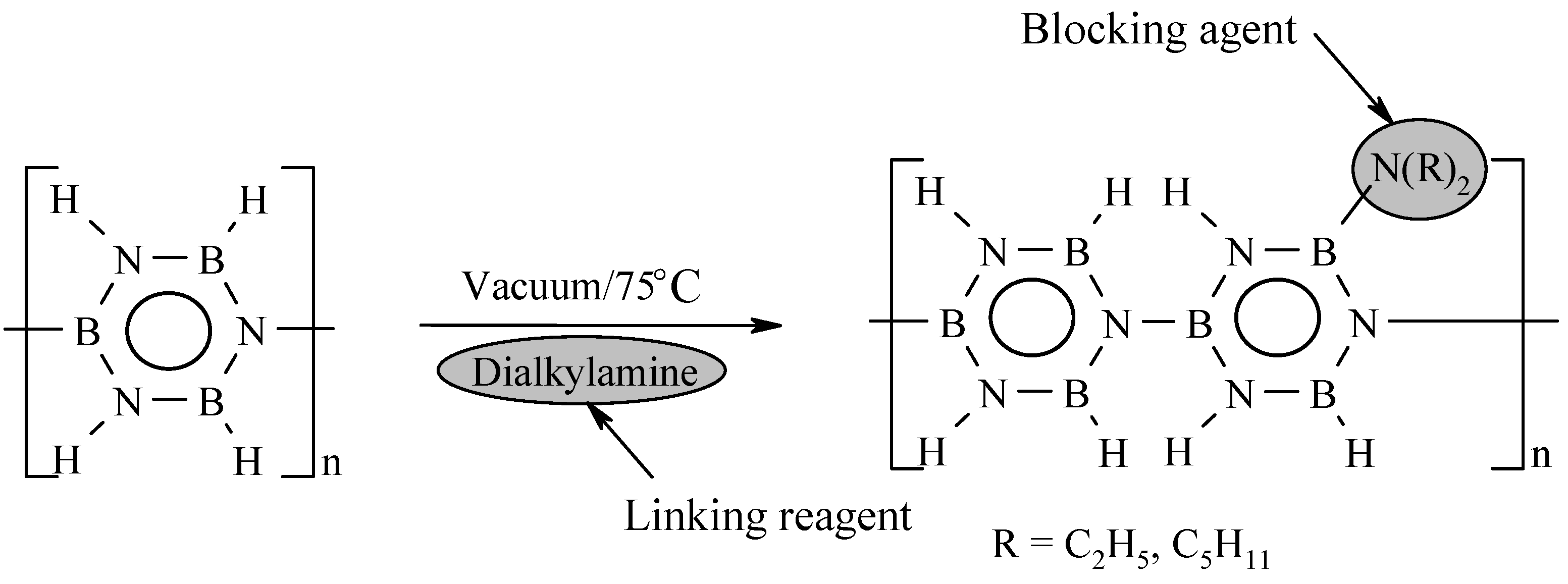{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | TThermolysis (°C) | Physical state | Shaping process type | Empirical formulae (1) | Weight loss (%) (2) |
|---|---|---|---|---|---|
| PB45 | 45 | Liquid (extremely volatile) | Solution-based shaping | [B3.0N3.0H4.8]n | 70 |
| PB50 | 50 | Liquid (volatile) | Solution-based shaping | [B3.0N3.8H4.0]n | 53.2 |
| PB60 | 60 | Solid | Plastic-forming technique | [B3.0N3.5H4.5]n | 8.8 |
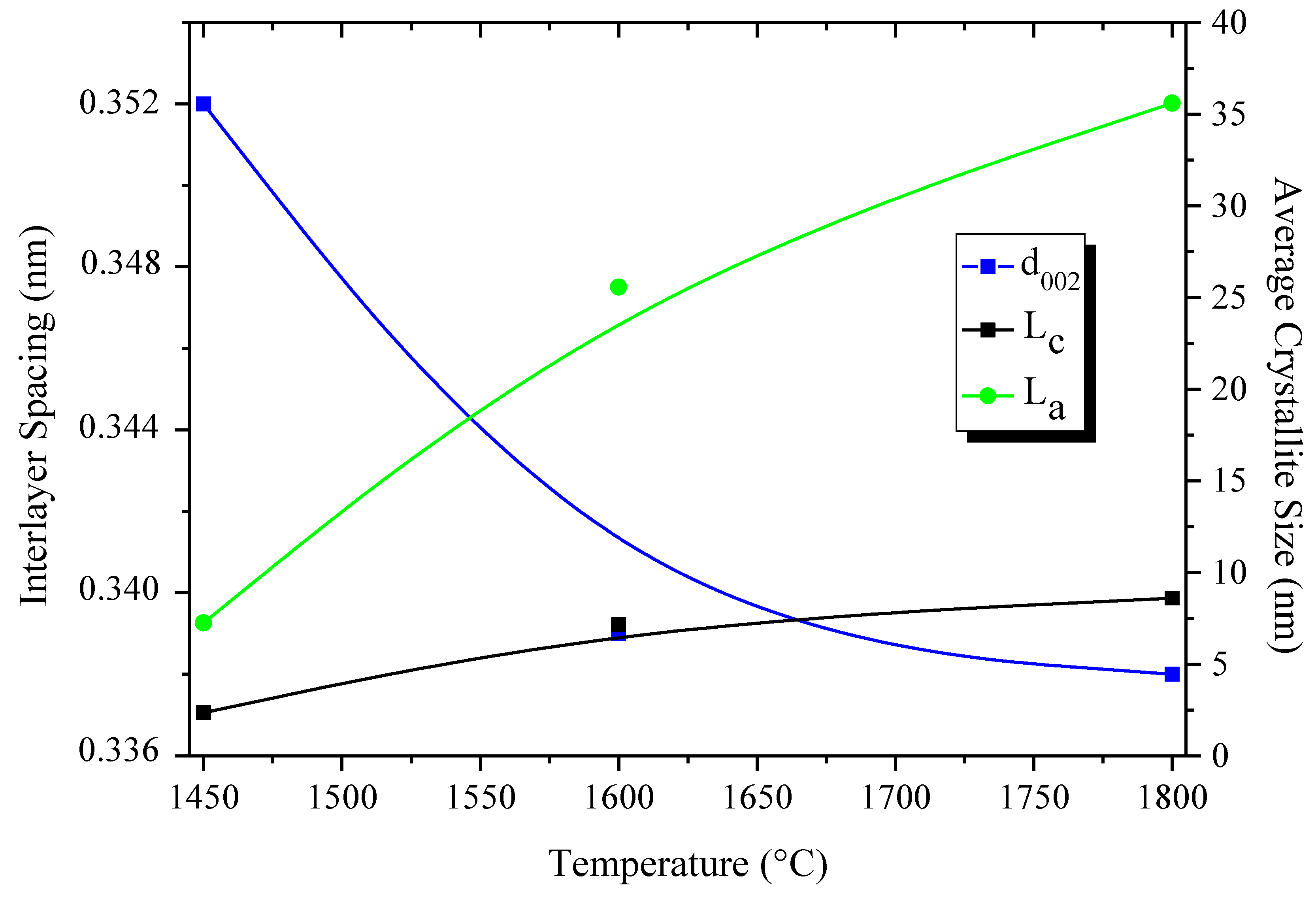
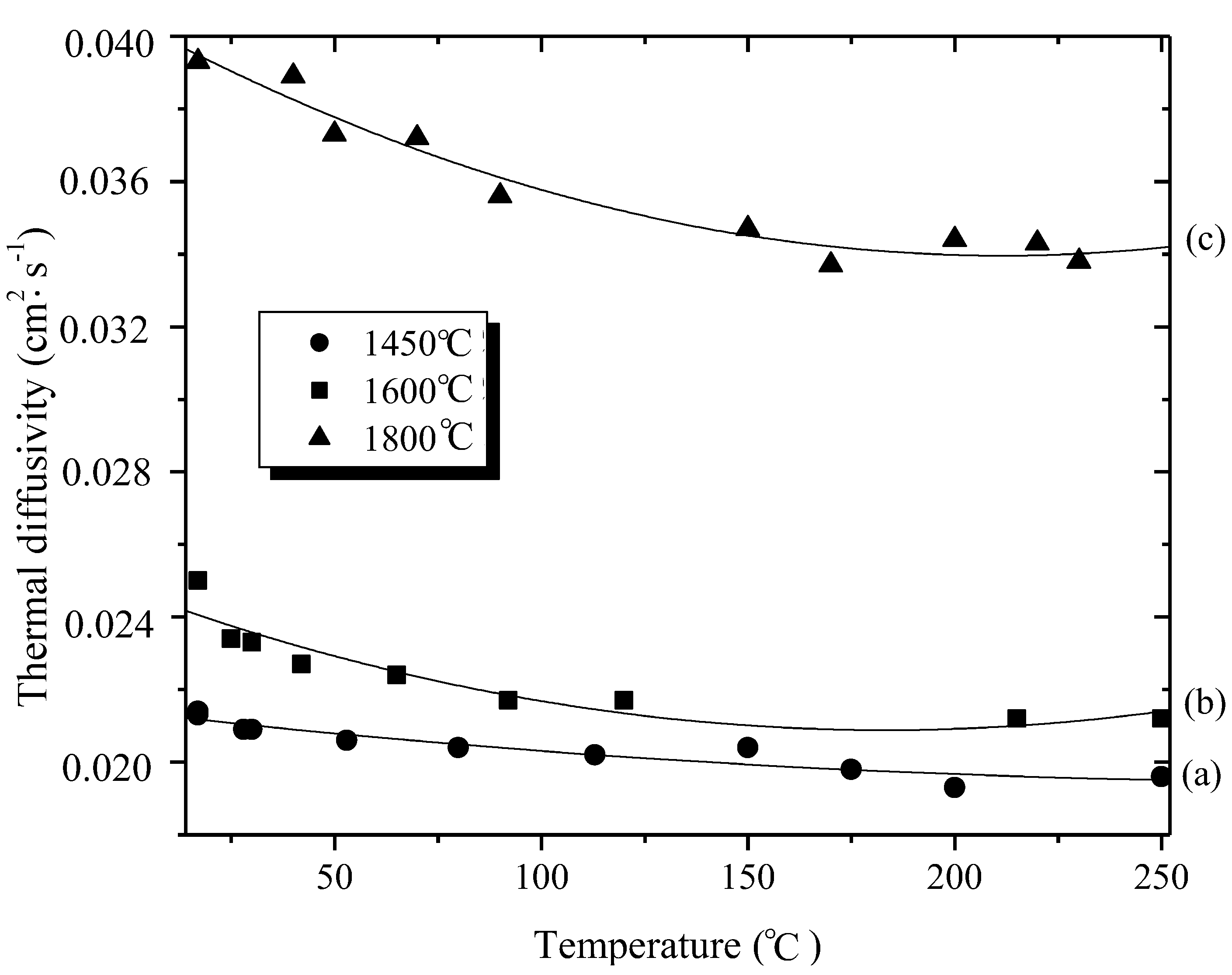
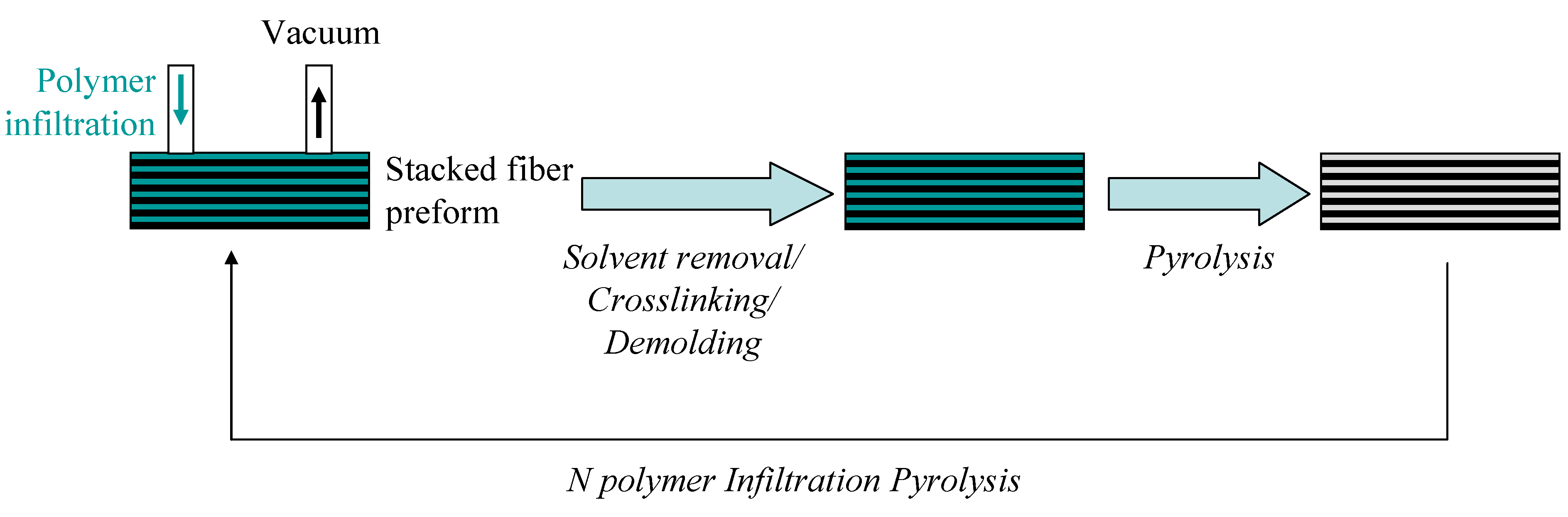

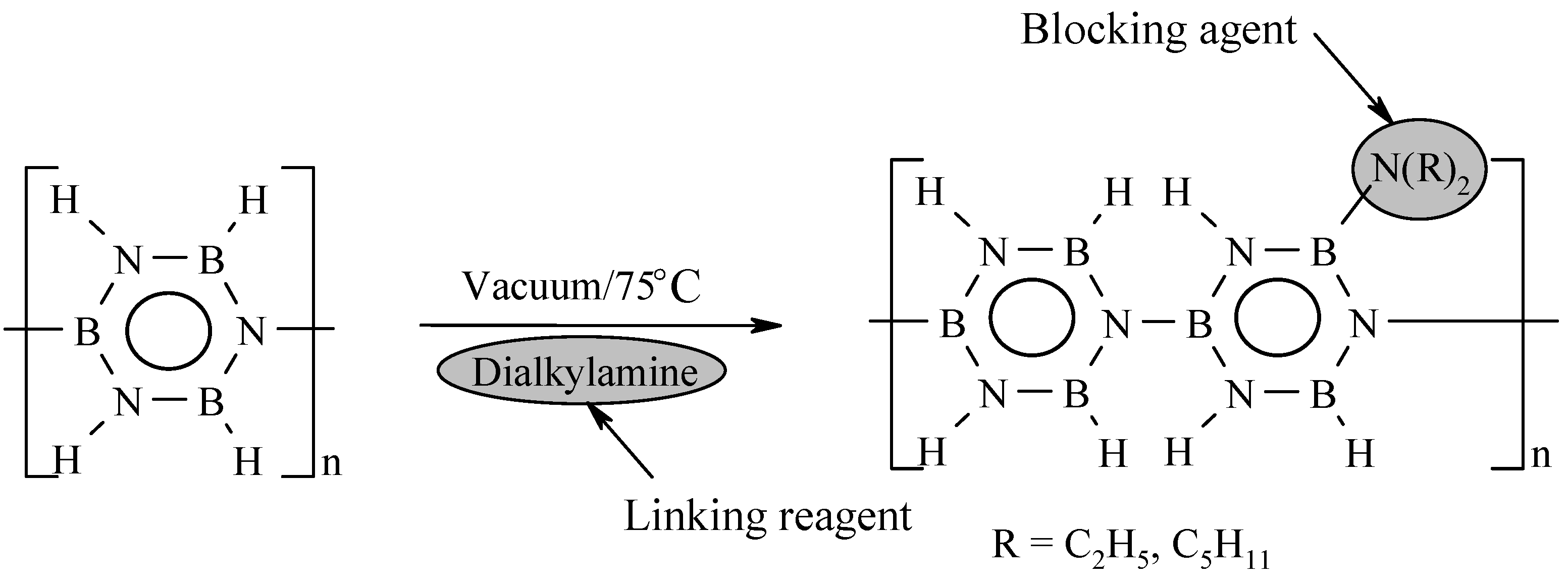
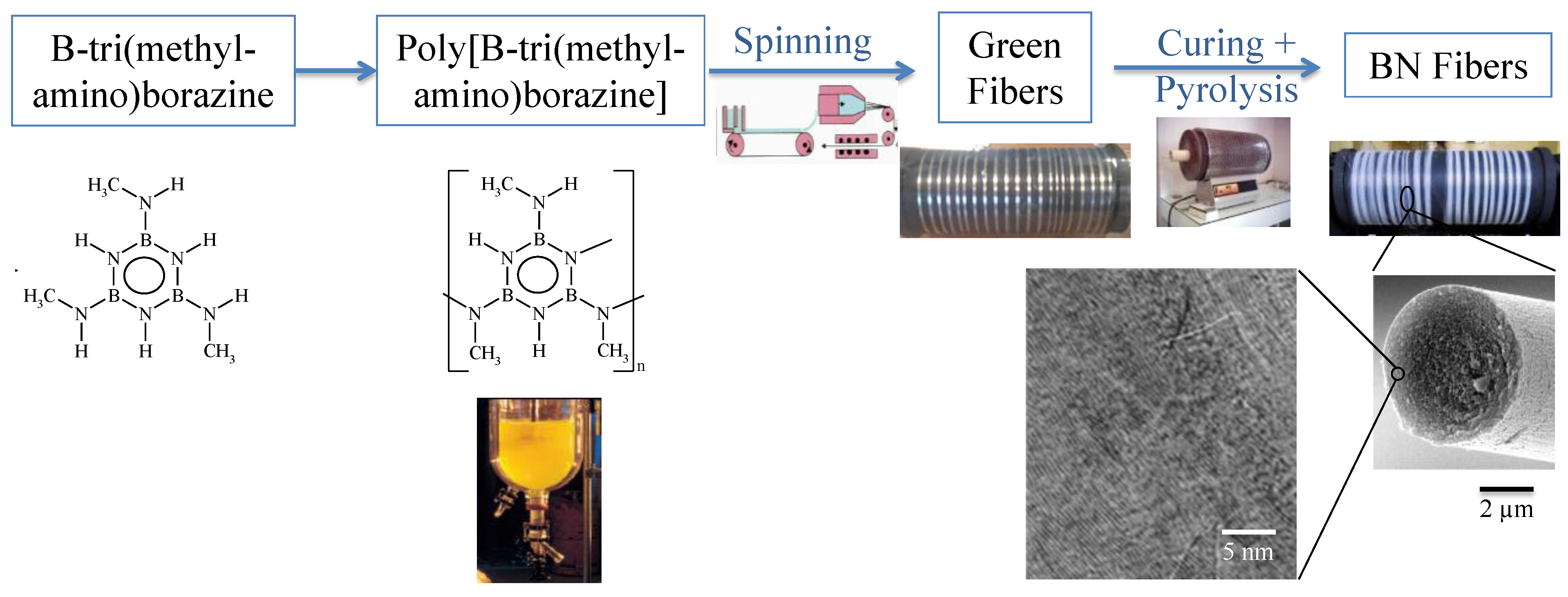
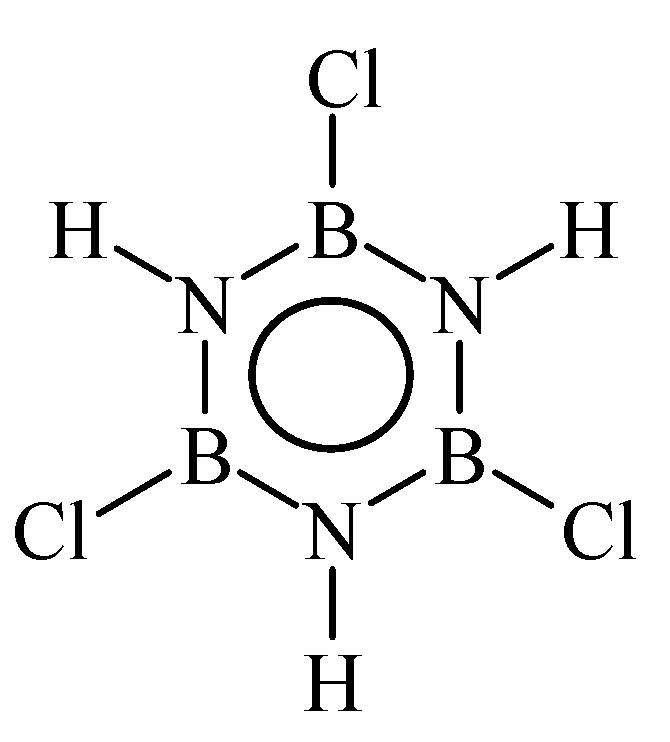
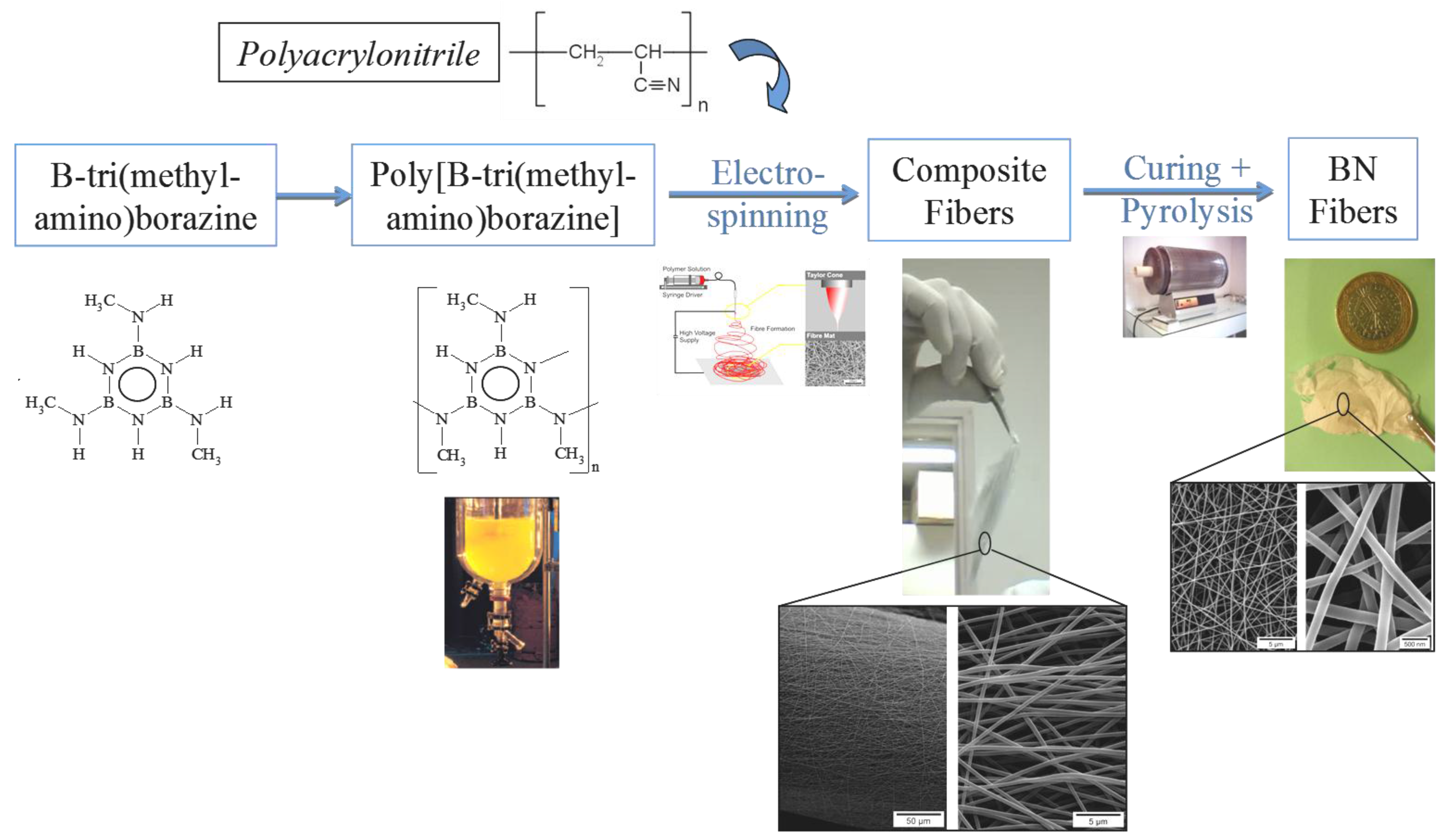
3. B-trichloro-/B-tri(methylamino)-Borazine and Poly[B-tri(methylamino)borazine-Derived BN


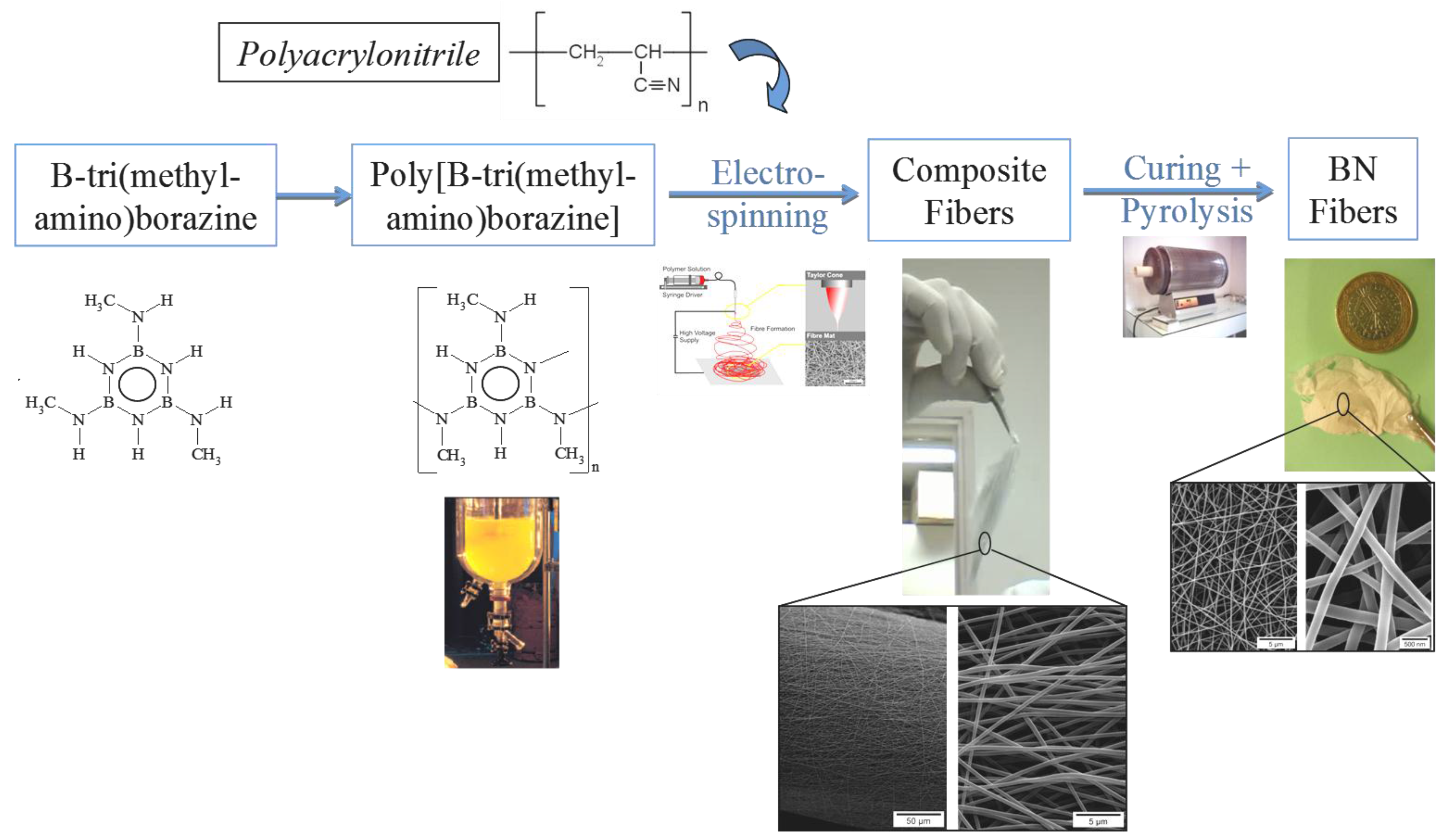
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Paine, R.T.; Narula, C.K. Synthetic route to boron nitride. Chem. Rev. 1990, 90, 73–91. [Google Scholar] [CrossRef]
- Balmain, W.H. Bemerkungen über die bildung von verbindungen des Bors und Siliciums mit Stickstoff und gewissen metallen. J. Prakt. Chem. 1842, 27, 422–430. (In German) [Google Scholar] [CrossRef]
- Han, W.-Q. Anisotropic Hexagonal Boron Nitride Nanomaterials: Synthesis and Applications; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2010. [Google Scholar]
- Kubota, Y.; Watanabe, K.; Tsuda, O.; Taniguchi, T. Deep ultraviolet light-emitting hexagonal boron nitride synthesized at atmospheric pressure. Science 2007, 317, 932–934. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Taniguchi, T.; Niiyama, T.; Miya, K.; Taniguchi, M. Far-ultraviolet plane-emission handheld device based on hexagonal boron nitride. Nat. Photonics 2009, 3, 591–594. [Google Scholar] [CrossRef]
- Yu, J.; Qin, L.; Hao, Y.; Kuang, S.; Bai, X.; Chong, Y.-M.; Zhang, W.; Wang, E. Vertically aligned boron nitride nanosheets: Chemical vapor synthesis, ultraviolet light emission and superhydrophobicity. ACS Nano 2010, 4, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Lian, G.; Si, H.; Wang, J.; Zhang, X.; Wang, Q.; Cui, D. Ultrathin BN nanosheets with zigzag edge: One-step chemical synthesis, applications in wastewater treatment and preparation of highly thermal-conductive BN-polymer composites. J. Mater. Chem. A 2013, 1, 5105–5112. [Google Scholar] [CrossRef]
- Salles, V.; Bernard, S.; Chiriac, R.; Miele, P. Structural and thermal properties of boron nitride nanoparticles. J. Eur. Ceram. Soc. 2012, 32, 1867–1871. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, H.; Liu, Y.; Craig, V.; Li, L.H.; Chen, Y. Superhydrophobic and superoleophilic boron nitride naotube-coated stainless steel meshes for oil and water separation. Adv. Mater. Interfaces 2014, 1, 1300002–1300006. [Google Scholar]
- Lei, W.; Portehault, D.; Liu, D.; Qin, S.; Chen, Y. Porous boron nitride nanosheets for effective water cleaning. Nat. Commun. 2013, 4, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xiao, X.; Xu, X.; Lin, J.; Huang, Y.; Xue, Y.; Jin, P.; Zou, J.; Tang, C. Activated boron nitride as an effective adsorbent for metal ions and organic polluants. Nature 2013, 3, 3208–3214. [Google Scholar]
- Zhang, X.; Lian, G.; Zhang, S.; Cui, D.; Wang, Q. Boron nitride nanocarpets: Controllable synthesis and their adsorption performance to organic pollutants. CrystEngComm 2012, 14, 4670–4676. [Google Scholar] [CrossRef]
- Lian, G.; Zhang, X.; Si, H.; Wang, J.; Cui, D.; Wang, Q. Boron nitride ultrathin fibrous nanonets: One-step synthesis and applications for ultrafast adsorption for water treatment and selective filtration of nanoparticles. ACS Appl. Mater. Interfaces 2013, 5, 12773–12778. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Lei, W.; Chen, Y. Template-free synthesis of functional 3D BN architecture for removal of dyes from water. Nature 2014, 4, 4453–4457. [Google Scholar]
- Rafiee, M.A.; Narayanann, T.N.; Hashimn, D.P.; Sakhavand, N.; Shahsavari, R.; Vajtai, R.; Ajayan, P.M. Hexagonal boron nitride and graphite oxide reinforced multifunctional porous cement composites. Adv. Funct. Mater. 2013, 23, 5624–5630. [Google Scholar] [CrossRef]
- Lian, G.; Zhang, X.; Zhang, S.; Liu, D.; Cui, D.; Wang, Q. Controlled fabrication of ultrathin-shell hollow spheres with excellent performance in hydrogen storage and wastewater treatment. Energy Environ. Sci. 2012, 5, 7072–7080. [Google Scholar] [CrossRef]
- Weng, Q.; Wang, X.; Zhi, C.; Bando, Y.; Golberg, D. Boron nitride porous microbelts for hydrogen storage. ACS Nano 2013, 7, 1558–1565. [Google Scholar] [CrossRef] [PubMed]
- Moussa, G.; Demirci, U.B.; Malo, S.; Bernard, S.; Miele, P. Boron nitride nanopolyhedrons with hollow core@mesoporous shell structure: From design to solid-state hydrogen storage application. J. Mater. Chem. A 2014, 2, 7717–7722. [Google Scholar]
- Haubner, R.; Wilhelm, M.; Weissenbacher, R.; Lux, B. Boron nitrides—Properties, synthesis and applications. Struct. Bond. 2002, 102, 1–45. [Google Scholar]
- Colombo, P.; Mera, G.; Riedel, R.; Soraru, G.D. Polymer-derived ceramics: 40 years of research and innovation in advanced ceramics. J. Am. Ceram. Soc. 2010, 93, 1805–1837. [Google Scholar]
- Colombo, P.; Soraru, G.D.; Riedel, R.; Kleebe, A. Polymer Derived Ceramics: Theory and Applications; DEStech Publications, Inc.: Lancaster, PA, USA, 2009. [Google Scholar]
- Bernard, S. Design, Processing and Properties of Ceramic Materials from Preceramic Precursors; Nova Science Pub Inc.: Hauppauge, NY, USA, 2012. [Google Scholar]
- Riedel, R.; Ionescu, E. Polymer Processing of Ceramics in Ceramics and Composites Processing Methods; Bansal, N.P., Boccaccini, A.R., Eds.; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2012; pp. 235–270. [Google Scholar]
- Ionescu, E. Polymer derived ceramics. In Ceramics Science and Technology; Riedel, R., Chen, I-W., Eds.; Wiley-VCH: Weinheim, Germany, 2012; Volume 3, pp. 457–500. [Google Scholar]
- Toutois, P.; Miele, P.; Jacques, S.; Cornu, D.; Bernard, S. Structural and mechanical behavior of boron nitride fibres derived from Poly[(Methylamino)Borazine] precursors: Optimization of the curing and pyrolysis procedures. J. Am. Ceram. Soc. 2006, 89, 42–49. [Google Scholar] [CrossRef]
- Bernard, S.; Ayadi, K.; Létoffé, J.-M.; Chassagneux, F.; Berthet, M.-P.; Cornu, D.; Miele, P. Evolution of structural features and mechanical properties during the conversion of poly[(methylamino)borazine] fibres into boron nitride fibres. J. Sol. State Chem. 2004, 177, 1803–1810. [Google Scholar] [CrossRef]
- Termoss, H.; Bechelany, M.; Toury, B.; Brioude, A.; Bernard, S.; Cornu, D.; Miele, P. Shaping potentialities of aluminum nitride polymeric precursors: Preparation of thin coatings and nanostructures in liquid phase. J. Eur. Ceram. Soc. 2009, 29, 857–861. [Google Scholar] [CrossRef]
- Gottardo, L.; Bernard, S.; Gervais, C.; Inzenhofer, K.; Motz, G.; Weinmann, M.; Balan, C.; Miele, P. Chemistry, structure and processability of boron-modified polysilazanes as tailored precursors of ceramic fibers. J. Mater. Chem. 2012, 22, 7739–7750. [Google Scholar] [CrossRef]
- Xie, Z.; Cao, S.; Wang, J.; Yan, X.; Bernard, S.; Miele, P. Engineering of silicon-based ceramic fibers: Novel SiTaC(O) ceramic fibers prepared from polytantalosilane. Mater. Sci. Eng. A 2010, 527, 7086–7091. [Google Scholar] [CrossRef]
- Ionescu, E.; Kleeben, H.-J.; Riedel, R. Silicon-containing polymer-derived ceramic nanocomposites (PDC-NCs): Preparative approaches and properties. Chem. Soc. Rev. 2012, 41, 5032–5052. [Google Scholar] [CrossRef]
- Bechelany, M.-C.; Proust, V.; Gervais, C.; Ghisleni, R.; Bernard, S.; Miele, P. In-situ controlled growth of titanium nitride in amorphous silicon nitride: A general route toward bulk non-oxide nitride nanocomposites with very high hardness. Adv. Mater. 2014, 26, 6548–6553. [Google Scholar] [CrossRef] [PubMed]
- Majoulet, O.; Alauzun, J.G.; Gottardo, L.; Gervais, C.; Schuster, M.E.; Bernard, S.; Miele, P. Ordered mesoporous silicoboron carbonitride ceramics from boron-modified polysilazanes: Polymer synthesis, processing and properties. Micro. Meso. Mater. 2011, 140, 40–50. [Google Scholar] [CrossRef]
- Shi, Y.; Wan, Y.; Zhao, D. Ordered mesoporous non-oxide materials. Chem. Soc. Rev. 2011, 40, 3854–3878. [Google Scholar] [CrossRef] [PubMed]
- Borchardt, L.; Hoffman, C.; Oschatz, M.; Mammitzsch, L.; Petasch, U.; Hermann, M.; Kaskel, S. Preparation and application of cellular and nanoporous carbides. Chem. Soc. Rev. 2012, 41, 5053–5067. [Google Scholar] [CrossRef]
- Wilfert, J.; Von Hagen, R.; Fiz, R.; Jansen, M.; Mathur, S. Electrospinning of preceramic polymers for the preparation of SiBNC felts and their modification with semiconductor nanowires. J. Mater. Chem. 2012, 22, 2099–2104. [Google Scholar]
- Flores, O.; Schmalz, T.; Krenkel, W.; Heymann, L.; Motz, G. Selective cross-linking of oligosilazanes to tailored meltable polysilazanes for the processing of SiCN fibers. J. Mater. Chem. A 2013, 1, 15406–15415. [Google Scholar] [CrossRef]
- Gottardo, L.; Bernard, S.; Gervais, C.; Weinmann, M.; Miele, P. Study of the intermediate pyrolysis steps and mechanism identification of polymer-derived SiBCN ceramics. J. Mater. Chem. 2012, 22, 17923–17933. [Google Scholar] [CrossRef]
- Günthner, M.; Schütz, A.; Glatze, U.; Wan, K.; Bordia, R.K.; Greißl, O.; Krenkel, W.; Motz, G. High performance environmental barrier coatings, Part I: Passive filler loaded SiCN system for steel. J. Eur. Ceram. Soc. 2011, 31, 3003–3010. [Google Scholar] [CrossRef]
- Prasad, R.M.; Iwamoto, Y.; Riedel, R.; Gurlo, A. Multilayer amorphous-Si-B-C-N/γ-Al2O3/α-Al2O3 membranes for hydrogen purification. Adv. Engineer. Mater. 2010, 12, 522–528. [Google Scholar] [CrossRef]
- Riedel, R.; Passing, G.; Schönfelder, H.; Brook, R.J. Synthesis of dense silicon-based ceramics at low temperatures. Nature 1992, 355, 714–717. [Google Scholar] [CrossRef]
- Haug, R.; Weinmann, M.; Bill, J.; Aldinger, F. Plastic forming of preceramic polymers. J. Eur. Ceram. Soc. 1999, 19, 1–6. [Google Scholar] [CrossRef]
- Motz, G.; Schmidt, S.; Beyer, S. The PIP-process: Precursor properties and applications. In Ceramic Matrix Composites—Fibre Reinforced Ceramics and Their Applications; Wiley-VCH GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 165–186. [Google Scholar]
- Liew, L.A.; Saravanan, R.A.; Bright, V.M.; Dunn, M.L.; Daily, J.W.; Raj, R. Processing and characterization of silicon carbon-nitride ceramics: Application of electrical properties towards MEMS thermal actuators. Sens. Actuators A 2003, 103, 171–181. [Google Scholar]
- Asthana, A.; Kim, K.O.; Perumal, J.; Kim, D.-M.; Kim, D.P. Facile single step fabrication of microchannels with varying size. Lab. Chip 2009, 9, 1138–1142. [Google Scholar] [PubMed]
- Ishihara, S.; Gu, H.; Bill, J.; Aldinger, F.; Wakai, F. Densification of precursor-derived Si-C-N ceramics by high-pressure hot isostatic pressing. J. Am. Ceram. Soc. 2002, 85, 1706–1712. [Google Scholar]
- Wan, J.; Duan, R.-G.; Mukherjee, A.K. Spark plasma sintering of silicon nitride/silicon carbide nanocomposites with reduced additive amounts. Scripta Mater. 2005, 53, 663–667. [Google Scholar]
- Wilfert, J.; Meier, K.; Hahn, K.; Grin, Y.; Jansen, M. SiC/BN composites by spark plasma sintering (SPS) of precursor-derived SiBNC powders. J. Ceram. Sci. Tech. 2010, 1, 1–6. [Google Scholar]
- Majoulet, O.; Bechelany, M.C.; Sandra, F.; Bonnefont, G.; Fantozzi, G.; Joly-Pottuz, L.; Malchere, A.; Bernard, S.; Miele, P. Silicon-boron-carbon-nitrogen monoliths with high, interconnected and hierarchical porosity. J. Mater. Chem. A 2013, 1, 10991–11000. [Google Scholar] [CrossRef]
- Bernard, S.; Miele, P. Ordered mesoporous polymer-derived ceramics and their processing into hierarchically porous boron nitride and silicoboron carbonitride monoliths. New J. Chem. 2014, 38, 1923–1931. [Google Scholar] [CrossRef]
- Sneddon, L.G.; Su, K.; Fazen, P.J.; Lynch, A.T.; Remsen, E.E.; Beck, J.S. Polymeric precursors to boron nitride ceramics. In Inorganic and Organometallic Oligomers and Polymers; Springer: Heidelberg, Germany, 1991; pp. 199–208. [Google Scholar]
- Bernard, S.; Cornu, D.; Duperrier, S.; Toury, B.; Miele, P. Borazine based preceramic polymers for advanced BN materials. In Inorganic and Organometallic Macromolecules Design and Applications; Abd-El-Aziz, A.S., Carraher, C.E., Jr., Pittman, C.U., Jr., Zeldin, M., Eds.; Springer: New York, NY, USA, 2008; pp. 351–71. [Google Scholar]
- Miele, P.; Bernard, S. Boron and nitrogen containing polymers for advanced materials. In Macromolecules Containing Metal and Metal-Like Elements; Abd-El-Aziz, A.S., Carraher, C.E., Jr., Pittman, C.U., Jr., Zeldin, M., Eds.; John Wiley and Sons: New York, NY, USA, 2007. [Google Scholar]
- Miele, P.; Bechelany, M.; Bernard, S. Hierarchically nanostructured porous boron nitride. In Advanced Hierarchical Nanostructured Materials; Zhang, Q., Wei, F., Eds.; Wiley-VCH Verlag GmbH and Co. kGaA: Weinheim, Germany, 2014; pp. 267–290. [Google Scholar]
- Moussa, G.; Salameh, C.; Bruma, A.; Malo, S.; Demirci, U.B.; Bernard, S.; Miele, P. From the molecular design to the hydrogen storage application of nanostructured boron nitride. Inorganics 2014, 2, 396–409. [Google Scholar] [CrossRef]
- Stock, A.; Pohland, E. Borwasserstoffe, IX: B3N3H6. Ber. Dtsch. Chem. Ges. 1926, 59, 2215–2223. (In German) [Google Scholar] [CrossRef]
- Wideman, T.; Sneddon, L.G. Convenient procedures for the laboratory preparation of borazine. Inorg. Chem. 1995, 34, 1002–1003. [Google Scholar] [CrossRef]
- Termoss, H.; Toury, B.; Pavan, S.; Brioude, A.; Bernard, S.; Cornu, D.; Valette, S.; Benayoun, S.; Miele, P. Preparation of boron nitride-based coatings on metallic substrates via infrared irradiation of dip-coated polyborazylene. J. Mater. Chem. 2009, 19, 2671–2674. [Google Scholar] [CrossRef]
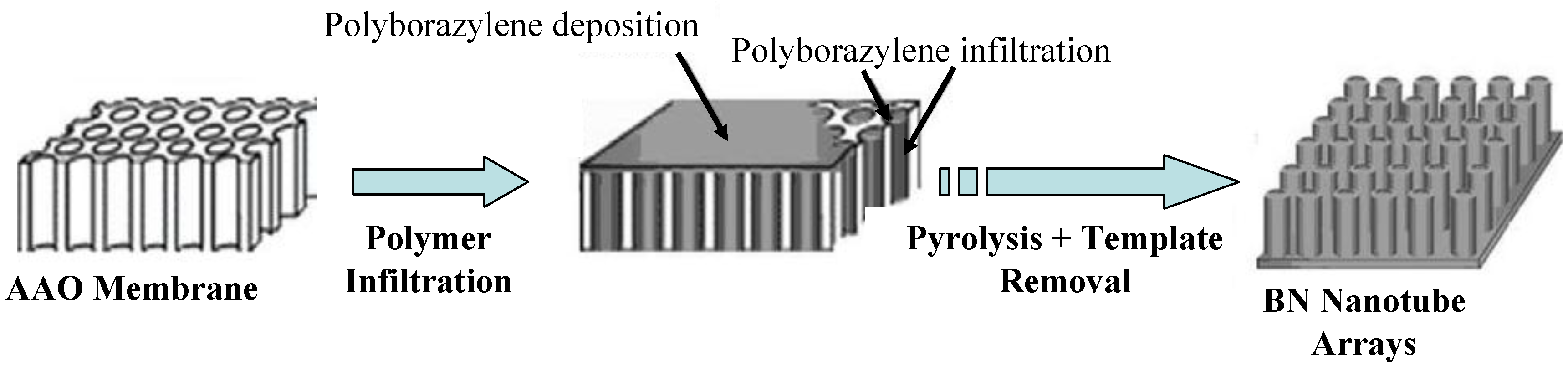
- Bechelany, M.; Bernard, S.; Brioude, A.; Stadelmann, P.; Charcosset, C.; Fiaty, K.; Cornu, D.; Miele, P. Synthesis of boron nitride nanotubes by a template-assisted polymer thermolysis process. J. Phys. Chem. C 2007, 111, 13378–13384. [Google Scholar] [CrossRef]
- Li, J.; Bernard, S.; Salles, V.; Gervais, C.; Miele, P. Preparation of polyborazylene-derived bulk boron nitride with tunable properties by warm-pressing and pressureless pyrolysis. Chem. Mater. 2010, 22, 2010–2019. [Google Scholar] [CrossRef]
- Mamantov, G.; Margrave, J.L. Decomposition of liquid borazine. J. Inorg. Nucl. Chem. 1961, 20, 348–351. [Google Scholar] [CrossRef]
- Laubengayer, A.W.; Moews, P.C.; Porter, R. The condensation of borazine to polycyclic boron-nitrogen frameworks by pyrolytic dehydrogenation. J. Am. Chem. Soc. 1961, 83, 1337–1342. [Google Scholar] [CrossRef]
- Thomas, J.; Weston, N.E.; O’Connor, T.E. Turbostratic boron nitride, thermal transformation to ordered-layer-lattice boron nitride. J. Am. Chem. Soc. 1962, 84, 4619–4622. [Google Scholar] [CrossRef]
- Schaeffer, R.; Steindler, M.; Hohnstedt, L.; Smith, H.R.; Eddy, L.B.; Schlesinger, H.I. Preparation of borazole by the reduction of trichloroborazole. J. Am. Chem. Soc. 1954, 76, 3303–3306. [Google Scholar] [CrossRef]
- Fazen, P.J.; Beck, J.S.; Lynch, A.T.; Remsen, E.E.; Sneddon, L.G. Thermally induced borazine dehydropolymerization reactions. Synthesis and ceramic conversion reactions of a new high-yield polymeric precursor to boron nitride. Chem. Mater. 1990, 2, 96–97. [Google Scholar]
- Sneddon, L.G.; Mirabelli, A.T.; Lynch, A.T.; Fazen, P.J.; Su, K.; Beck, J.S. Polymeric precursors to boron based ceramics. Pure Appl. Chem. 1991, 63, 407–410. [Google Scholar] [CrossRef]
- Fazen, P.J.; Remsen, E.E.; Caroll, P.J.; Beck, J.S.; Sneddon, L.G. Synthesis, properties, and ceramic conversion reactions of polyborazylene. A high-yield polymeric precursor to boron nitride. Chem. Mater. 1995, 7, 1942–1956. [Google Scholar]
- Kim, D.-P.; Economy, J. Fabrication of oxidation-resistant carbon fiber/boron nitride matrix composites. Chem. Mater. 1993, 5, 1216–1220. [Google Scholar] [CrossRef]
- Kim, D.-P.; Economy, J. Occurrence of liquid crystallinity in a borazine polymer. Chem. Mater. 1994, 6, 395–400. [Google Scholar] [CrossRef]
- Gervais, C.; Maquet, J.; Babonneau, F.; Duriez, C.; Framery, E.; Vaultier, M.; Florian, P.; Massiot, D. Chemically derived BN ceramics: Extensive 11B and 15N solid-state NMR study of a preceramic polyborazilene. Chem. Mater. 2001, 13, 1700–1707. [Google Scholar] [CrossRef]
- Schlienger, S.; Alauzun, J.; Michaux, F.; Vidal, L.; Parmentier, J.; Gervais, C.; Babonneau, F.; Bernard, S.; Miele, P.; Parra, J.B. Design, processing and properties of zeolite-derived boron nitride-based architectures. Chem. Mater. 2012, 24, 88–96. [Google Scholar] [CrossRef]
- Alauzun, J.G.; Ungureanu, S.; Brun, N.; Bernard, S.; Miele, P.; Backov, R.; Sanchez, C. Novel monolith-type boron nitride hierarchical foams obtained through integrative chemistry. J. Mater. Chem. 2011, 21, 14025–14030. [Google Scholar] [CrossRef]
- Kusari, U.; Bao, Z.; Cai, V.; Ahamd, G.; Sandhage, K.H.; Sneddon, L.G. Formation of nanostructured, nanocrystalline boron nitride microparticles with diatom-derived 3-D shapes. Chem. Commun. 2007, 1177–1179. [Google Scholar] [CrossRef]
- Bernard, S.; Miele, P. Nanostructured and architectured boron nitride. Mater. Today 2014, 17, 443–450. [Google Scholar] [CrossRef]
- Hubacek, M.; Ueki, M.; Sato, T. Orientation and growth of grains in copper-activated hot-pressed hexagonal boron nitride. J. Am. Ceram. Soc. 1996, 79, 283–285. [Google Scholar] [CrossRef]
- Matovic, B.; Rixecker, G.; Golczewski, J.; Aldinger, F. Thermal conductivity of pressureless sintered silicon nitride materials with LiYO2 additive. Sci. Sinter. 2004, 36, 3–9. [Google Scholar] [CrossRef]
- Kume, S.; Yamada, I.; Watari, K.; Harada, I.; Mitsuishi, K. High-thermal-conductivity AlN filler for polymer/ceramics composites. J. Am. Ceram. Soc. 2009, 92, S153–S156. [Google Scholar] [CrossRef]
- McDonald, R.A.Stull; Stull, D.R. The heat content and heat capacity of boron nitride from 298 to 1689 K. J. Phys. Chem. 1961, 65, 1918–1918. [Google Scholar]
- Cofer, C.G.; Economy, J. Oxidative and hydrolytic stability of boron nitride—A new approach to improving the oxidation resistance of carbonaceous structures. Carbon 1995, 33, 389–395. [Google Scholar] [CrossRef]
- Cofer, C.G.; Economy, J.; Xu, Y.; Zangvil, A.; Lara-Curzio, E.; Ferber, M.K.; More, K.L. Characterization of fibre/matrix interfaces in composites with boron nitride matrix. Comp. Sci. Technol. 1996, 56, 967–975. [Google Scholar] [CrossRef]
- Seghi, S.; Fabio, B.; Economy, J. Carbon/carbon-boron nitride composites with improved wear resistance compared to carbon/carbon. Carbon 2004, 42, 3043–3048. [Google Scholar] [CrossRef]
- Seghi, S.; Lee, J.; Economy, J. High density carbon fibre/boron nitride matrix composites: Fabrication of composites with exceptional wear resistance. Carbon 2005, 43, 2035–2043. [Google Scholar] [CrossRef]
- Zhong, W.; Wang, S.; Li, J.; Bechelany, M.C.; Ghisleny, R.; Rossignol, F.; Balan, C.; Chartier, T.; Bernard, S.; Miele, P. Design of carbon fibre reinforced boron nitride matrix composites by vacuum-assisted polyborazylene transfer moulding and pyrolysis. J. Eur. Ceram. Soc. 2013, 33, 2979–2992. [Google Scholar] [CrossRef]
- Salles, V.; Bernard, S.; Li, J.; Brioude, A.; Chehaidi, S.; Foucaud, S.; Miele, P. Design of highly dense boron nitride by the combination of spray-pyrolysis of borazine and additive-free sintering of derived ultrafine powders. Chem. Mater. 2009, 21, 2920–2929. [Google Scholar] [CrossRef]
- Wideman, T.; Remsen, E.E.; Cortez, E.; Chlanda, V.L.; Sneddon, L.G. Amine-modified polyborazylenes: Second-generation precursors to boron nitride. Chem. Mater. 1998, 10, 412–421. [Google Scholar] [CrossRef]
- Yajima, S.; Shishido, T.; Kayano, H. Development of high tensile strength silicon carbide fibre using an organosilicon polymer. Nature 1978, 273, 525–527. [Google Scholar] [CrossRef]
- Yajima, S.; Hayashi, J.; Omori, M.; Okamura, K. Development of a silicon carbide fiber with high tensile strength. Nature 1976, 261, 683–685. [Google Scholar] [CrossRef]
- Yajima, S.; Hasegawa, Y.; Hayashi, J.; Iimura, M. Synthesis of continuous silicon carbide fibre with high tensile strength and high Youngʼs modulus. J. Mater. Sci. 1978, 13, 2569–2576. [Google Scholar] [CrossRef]
- Yamamura, T.; Ishikawa, T.; Shibuya, M.; Hisayuki, T. Development of a new continuous Si-Ti-C-O fibre using an organometallic polymer precursor. J. Mater. Sci. 1988, 23, 2589–2594. [Google Scholar] [CrossRef]
- LeGrow, G.E.; Lim, T.F.; Lipowitz, J.; Reaoch, R.S. Ceramics from hydridopolysilazane. Am. Ceram. Soc. Bull. 1987, 66, 363–367. [Google Scholar]
- Funayama, O.; Arai, M.; Tashiro, Y.; Aoki, H.; Suzuki, T.; Tamura, K.; Kaya, H.; Nishii, H.; Isoda, T. Tensile strength of silicon nitride fibers produced from perhydrolysilazane. J. Ceram. Soc. Jpn. 1990, 98, 104–107. [Google Scholar] [CrossRef]
- Winter, G.; Verbeek, W.; Mansmann, M. Production of shaped articles of silicon carbide and silicon nitride. US Patent 3892583, 1 July 1975. [Google Scholar]
- Bunsell, A.R.; Berger, M.-H. Fine Ceramic Fibres; Marcel Dekker Inc.: New York, NY, USA, 1999. [Google Scholar]
- Okamura, K. Ceramic fibres from polyester precursors. Composites 1987, 18, 107–119. [Google Scholar] [CrossRef]
- Motz, G.; Hacker, J.; Ziegler, G.; Clauss, B.; Schawaller, D. Low-cost-ceramic SiCN fibers by an optimized polycarbosilazane and continuous processing. In Inorganic Structural Fiber Composites; Vincenzini, P., Badini, C., Eds.; Techna: Faenza, Italy, 2003; p. 47. [Google Scholar]
- Schawaller, D.; Clauss, B. Preparation of non-oxide ceramic fibers in the system Si-C-N and Si-B-C-N. In High Temperature Ceramic Matrix Composites; Krenkel, W., Naslain, R., Schneider, H., Eds.; Wiley-VCH: Weinheim, Germany, 2001; p. 56. [Google Scholar]
- Bernard, S.; Weinmann, M.; Gerstel, P.; Miele, P.; Aldinger, F. Boron-modified polysilazane as a novel single-source precursor for SiBCN ceramic fibers: Synthesis, melt-spinning, curing and ceramic conversion. J. Mater. Chem. 2005, 15, 289–299. [Google Scholar] [CrossRef]
- Bernard, S.; Weinmann, M.; Cornu, D.; Miele, P.; Aldinger, F. Preparation of high-temperature stable Si-B-C-N fibers from tailored single source polyborosilazanes. J. Eur. Ceram. Soc. 2005, 25, 251–256. [Google Scholar] [CrossRef]
- Ouyang, T.; Gottardo, L.; Bernard, S.; Balan, C.; Miele, P. Tuning of the viscoelastic properties of melt-spinnable boron- and silicon-based preceramic polymers. J. Appl. Polym. Sci. 2013, 128, 248–257. [Google Scholar] [CrossRef]
- Flores, O.; Bordia, R.K.; Nestler, D.; Krenkel, W.; Motz, G. Ceramic fibers based on SiC and SiCN systems: Current research, development, and commercial status. Adv. Engineer. Mater. 2014, 16, 621–636. [Google Scholar] [CrossRef]
- Nguyen, V.L.; Proust, V.; Quievryn, C.; Bernard, S.; Miele, P.; Soraru, G.D. Processing, mechanical characterization and alkali resistance of SiliconBoronOxycarbide (SiBOC) glass fibers. J. Am. Ceram. Soc. 2014, 97, 3143–3149. [Google Scholar] [CrossRef]
- Bernard, S.; Fiati, K.; Miele, P.; Cornu, D.; Laurent, P. Kinetic modelling of the polymer-derived ceramics (PDCs) route: Investigations of the thermal decompostion kinetics of poly[B-(methylamino)borazine] precursors into boron nitride. J. Phys. Chem. B 2006, 110, 9048–9060. [Google Scholar] [CrossRef] [PubMed]
- Bernard, S.; Duperrier, S.; Cornu, D.; Miele, P.; Weinmann, M.; Balan, C.; Aldinger, F. Chemical tailoring of Single-Source molecular and polymeric precursors for the preparation of ceramic fibers. J. Optoelectron. Adv. Mater. 2006, 8, 648–653. [Google Scholar]
- Miele, P.; Bernard, S.; Cornu, D.; Toury, B. Recent develoment in polymer-derived ceramic fibers. (PDCFs): Preparation, properties, applications—A review. Soft. Mater. 2006, 4, 249–286. [Google Scholar]
- Cornu, D.; Bernard, S.; Duperrier, S.; Toury, B.; Miele, P. Alkylaminoborazine-based precursors for the preparation of boron nitride fibers by the polymer-derived-ceramics (PDCs) route. J. Eur. Ceram. Soc. 2005, 25, 111–121. [Google Scholar] [CrossRef]
- Miele, P.; Toury, B.; Cornu, D.; Bernard, S. Borylborazines as new precursors of boron nitride fibres. J. Organomet. Chem. 2005, 690, 2809–2814. [Google Scholar] [CrossRef]
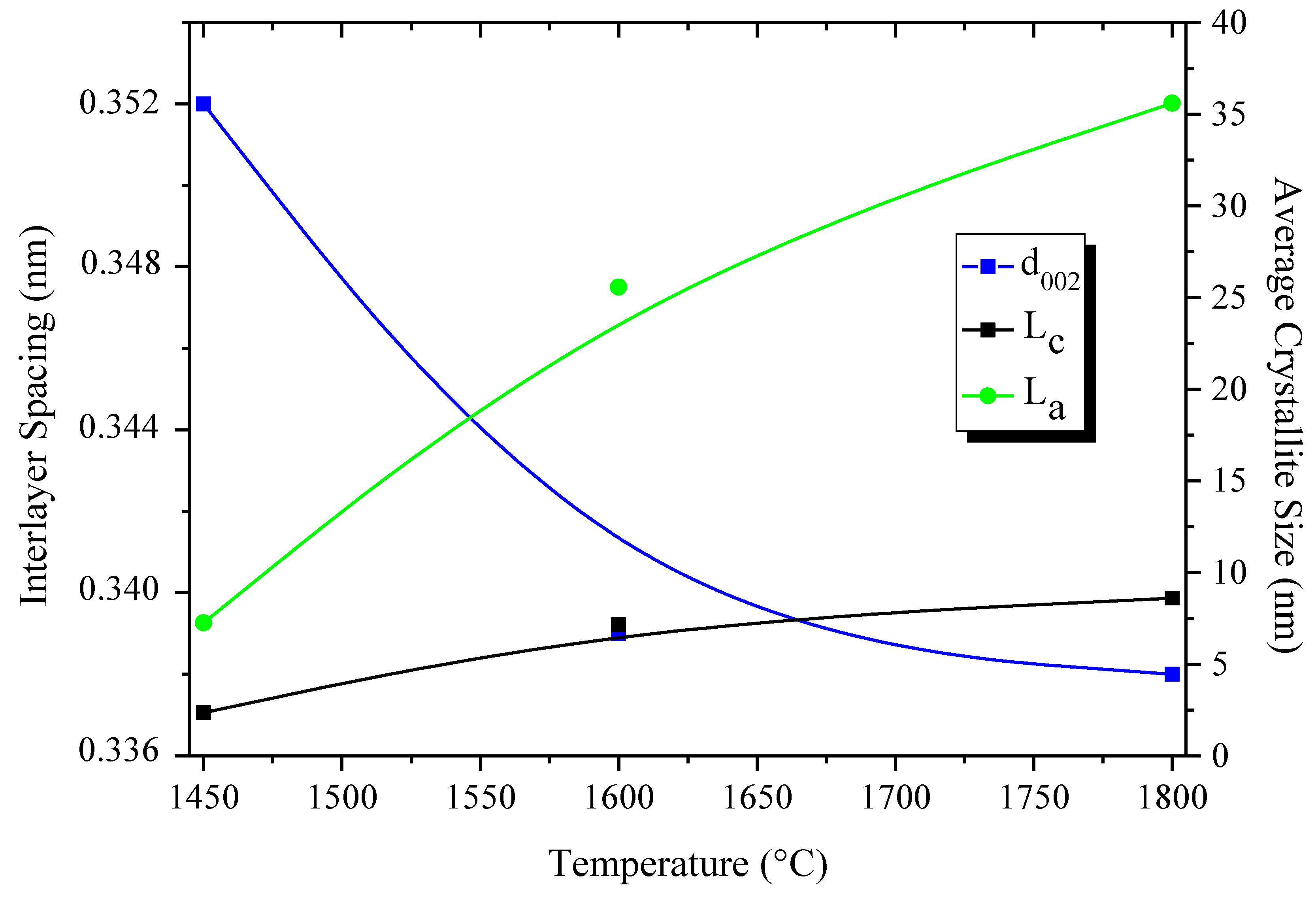
- Bernard, S.; Chassagneux, F.; Berthet, M.-P.; Cornu, D.; Miele, P. Crystallinity, crystalline quality and microstructural ordering in boron nitride fibers. J. Am. Ceram. Soc. 2005, 88, 1607–1614. [Google Scholar] [CrossRef]
- Bernard, S.; Cornu, D.; Miele, P.; Vincent, H.; Bouix, J. Pyrolysis of poly[2,4,6-tri(methylamino)borazine] and its conversion into BN fibres. J. Organomet. Chem. 2002, 657, 91–97. [Google Scholar] [CrossRef]
- Bernard, S.; Chassagneux, F.; Berthet, M.-P.; Vincent, H.; Bouix, J. Structural and mechanical properties of a high-performance BN fibre. J. Eur. Ceram. Soc. 2002, 22, 2047–2059. [Google Scholar] [CrossRef]
- Duperrier, S.; Chiriac, R.; Sigala, C.; Gervais, C.; Bernard, S.; Cornu, D.; Miele, P. Thermal and thermo-mechanical behaviours of a series of B-(methylamino)borazine-based polymers for fiber preparation. Application to boron nitride fibers. J. Eur. Ceram. Soc. 2009, 29, 851–855. [Google Scholar]
- Duperrier, S.; Gervais, C.; Bernard, S.; Cornu, D.; Babonneau, F.; Balan, C.; Miele, P. Design of a series of preceramic B-tri(methylamino)borazine-based polymers as fiber precursors: Architecture, thermal behavior, and melt-spinnability. Macromolecules 2007, 40, 1018–1027. [Google Scholar] [CrossRef]
- Duperrier, S.; Bernard, S.; Calin, A.; Sigala, C.; Chiriac, R.; Miele, P.; Balan, C. Design of a series of preceramic B-tri(methylamino)borazine-based polymers as fiber precursors: Shear rheology investigations. Macromolecules 2007, 40, 1028–1034. [Google Scholar] [CrossRef]
- Duperrier, S.; Calin, A.; Bernard, S.; Balan, C.; Miele, P. Rheological behaviour of poly[(B-alkylamino)borazine] in a fiber spinning process. Soft. Mater. 2006, 4, 123–142. [Google Scholar] [CrossRef]
- Duperrier, S.; Gervais, C.; Bernard, S.; Cornu, D.; Babonneau, F.; Miele, P. Controlling the chemistry, morphology and structure of boron nitride-based ceramic fibers through a comprehensive mechanistic study of the reactivity of spinnable polymers with ammonia. J. Mater. Chem. 2006, 16, 3126–3138. [Google Scholar] [CrossRef]
- Lindquist, D.A.; Janik, J.F.; Datye, A.K.; Paine, R.T. Boron nitride fibers processed from poly(borazinylamine) solutions. Chem. Mater. 1992, 4, 17–19. [Google Scholar] [CrossRef]
- Paciorek, K.J.L.; Kratzer, R.H.; Harris, D.H.; Krone-Schmidt, W. Boron Nitride Polymeric Precursors. US Patent 4707556, 17 November 1987. [Google Scholar]
- Tanigushi, L.; Harada, K.; Maeda, T. Boron Nitride in Filament, Film, or Other Forms. Japan Kokai 76 53,000, 11 May 1976. [Google Scholar]
- Kimura, Y.; Kubo, Y.; Hayashi, N. High-performance boron-nitride fibers from poly(borazine) preceramics. Compos. Sci. Technol. 1994, 51, 173–179. [Google Scholar] [CrossRef]
- Rothgery, E.F.; Hohnstedt, L.F. Convenient preparation of B-trichloroborazine. Inorg. Chem. 1967, 6, 1065–1066. [Google Scholar] [CrossRef]
- Salles, V.; Bernard, S.; Brioude, A.; Cornu, D.; Miele, P. A new class of boron nitride fibers with tunable properties by combining an electrospinning process and the Polymer-Derived Ceramics route. Nanoscale 2010, 2, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Greil, P. Polymer derived engineering ceramics. Adv. Eng. Mater. 2000, 2, 339–348. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernard, S.; Miele, P. Polymer-Derived Boron Nitride: A Review on the Chemistry, Shaping and Ceramic Conversion of Borazine Derivatives. Materials 2014, 7, 7436-7459. https://doi.org/10.3390/ma7117436
Bernard S, Miele P. Polymer-Derived Boron Nitride: A Review on the Chemistry, Shaping and Ceramic Conversion of Borazine Derivatives. Materials. 2014; 7(11):7436-7459. https://doi.org/10.3390/ma7117436
Chicago/Turabian StyleBernard, Samuel, and Philippe Miele. 2014. "Polymer-Derived Boron Nitride: A Review on the Chemistry, Shaping and Ceramic Conversion of Borazine Derivatives" Materials 7, no. 11: 7436-7459. https://doi.org/10.3390/ma7117436
APA StyleBernard, S., & Miele, P. (2014). Polymer-Derived Boron Nitride: A Review on the Chemistry, Shaping and Ceramic Conversion of Borazine Derivatives. Materials, 7(11), 7436-7459. https://doi.org/10.3390/ma7117436