New Approaches to the Computer Simulation of Amorphous Alloys: A Review

Abstract
:1. Preamble: Atomic Topology versus Properties
- ➢
- Small nanoscopic defects
- ➢
- Short and middle range ordering
2. Antecedents [1]
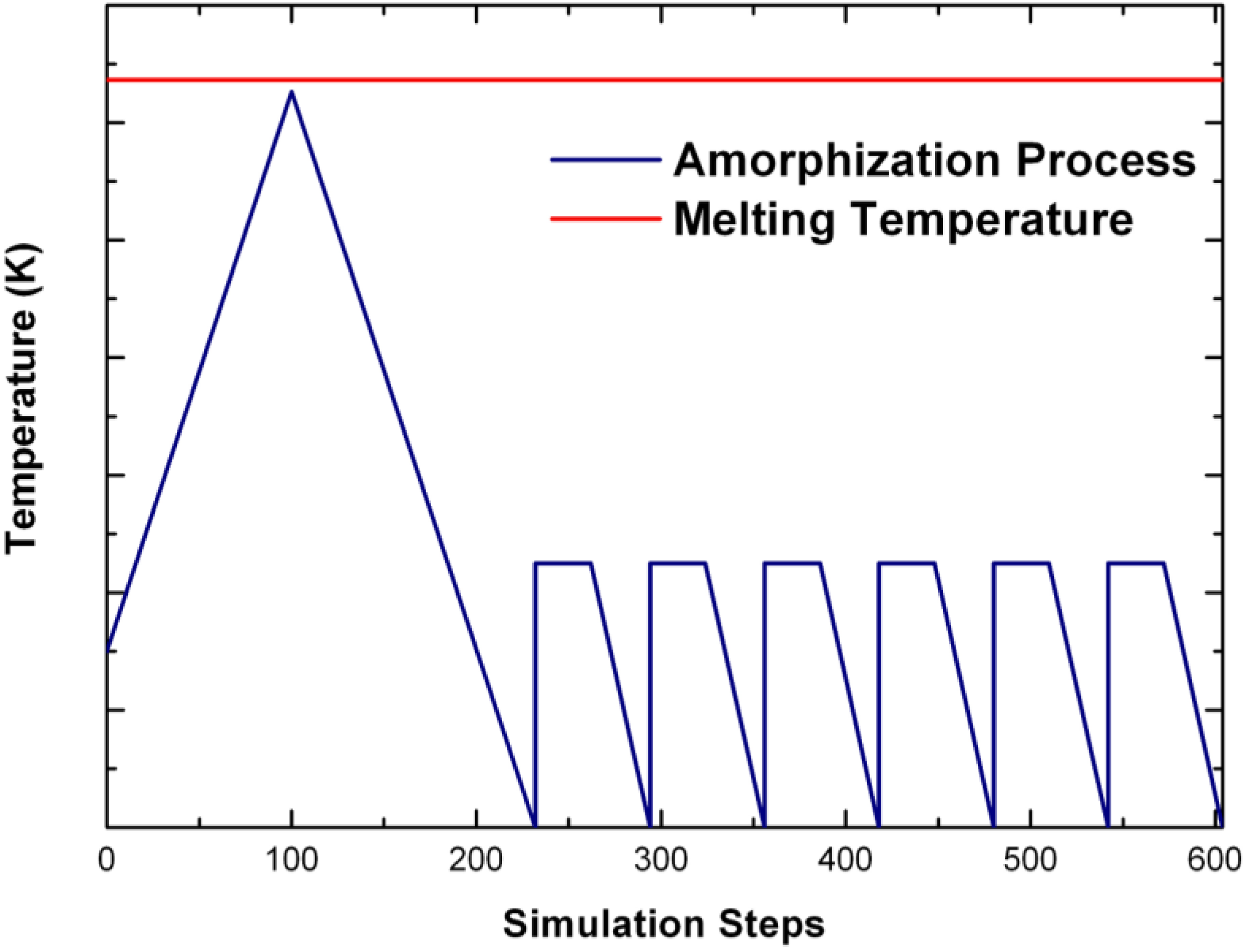
3. The Undermelt-Quench Approach [1], Its Variants, the Method and the Bonding Criterion
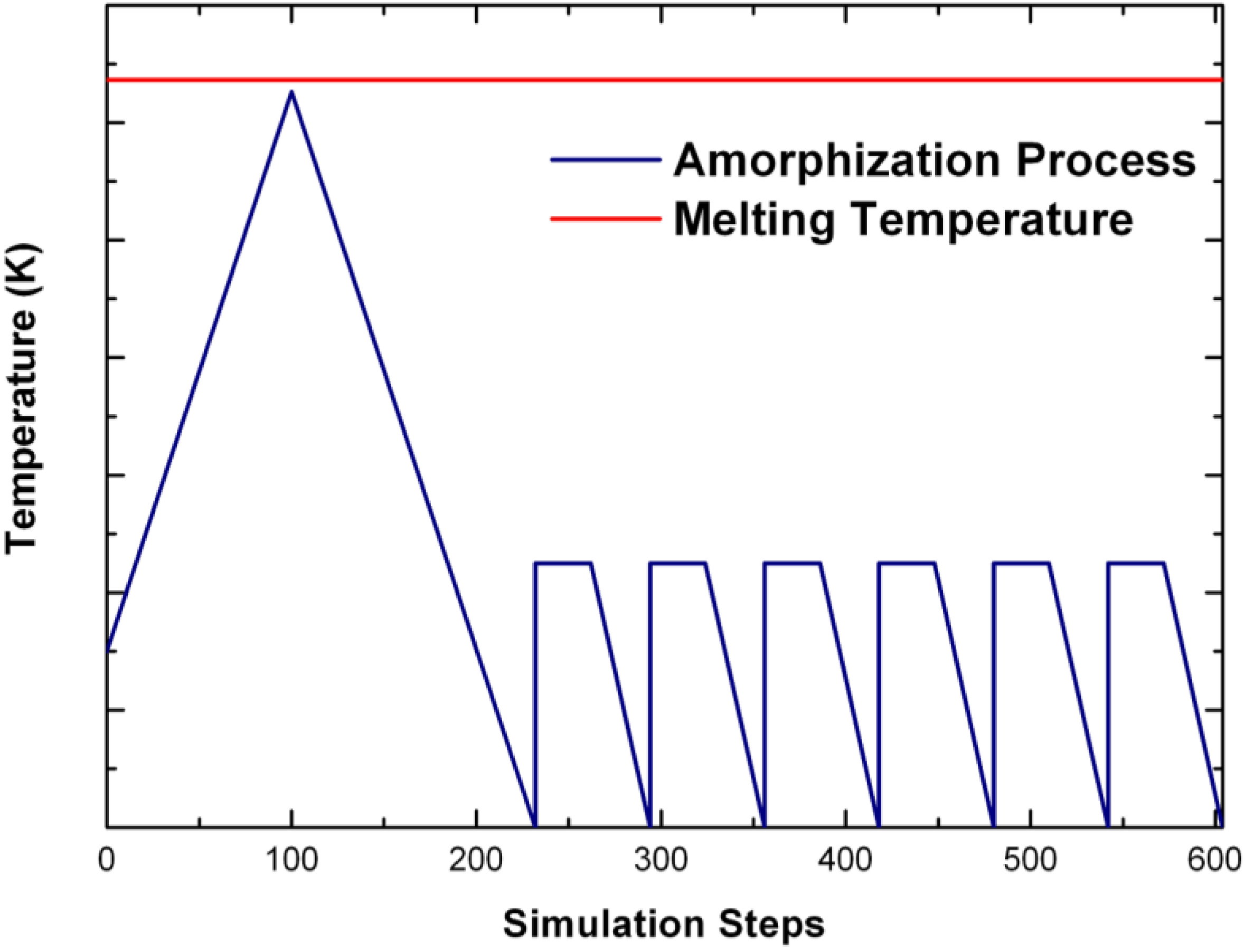
4. Amorphous Alloys: Their Atomic Topologies
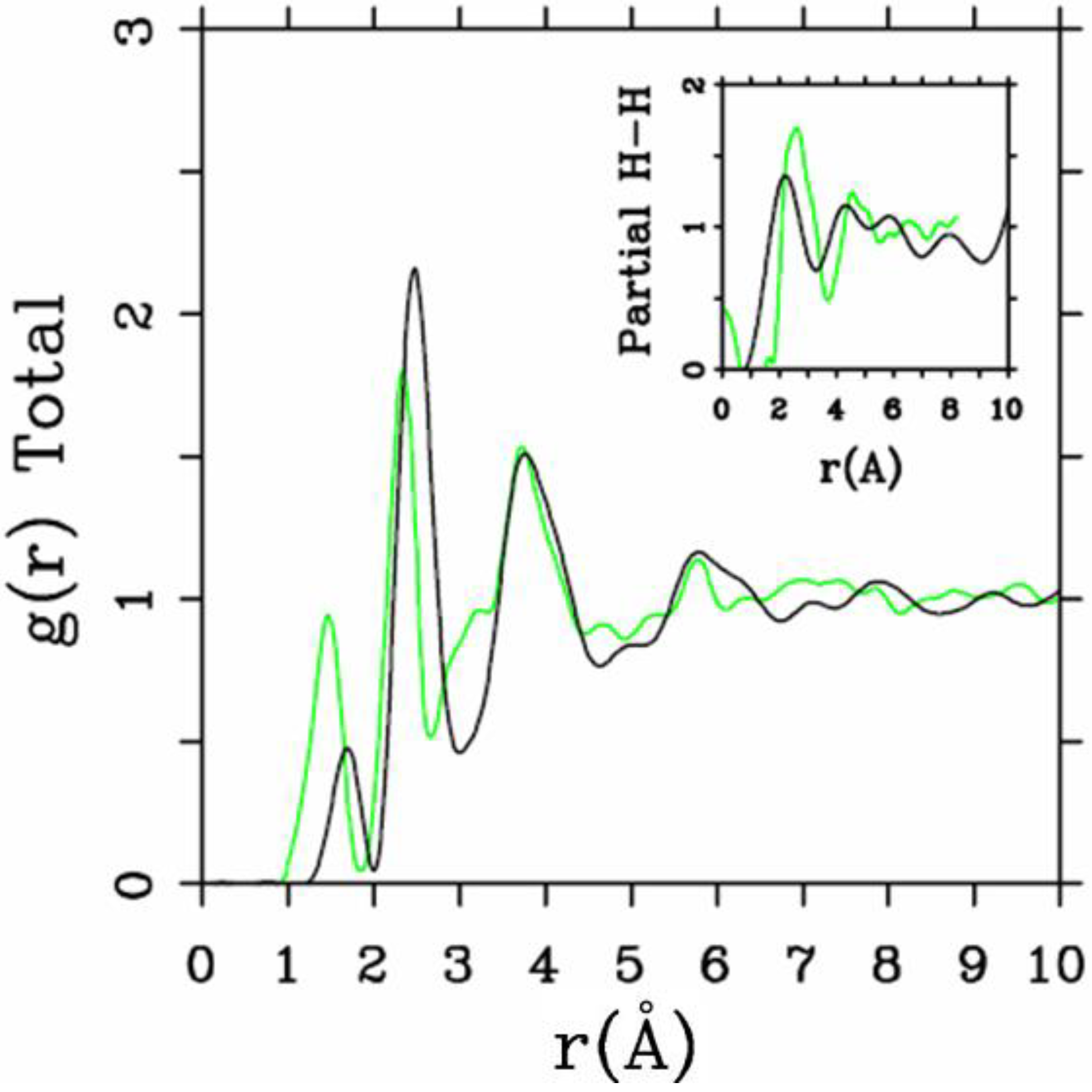
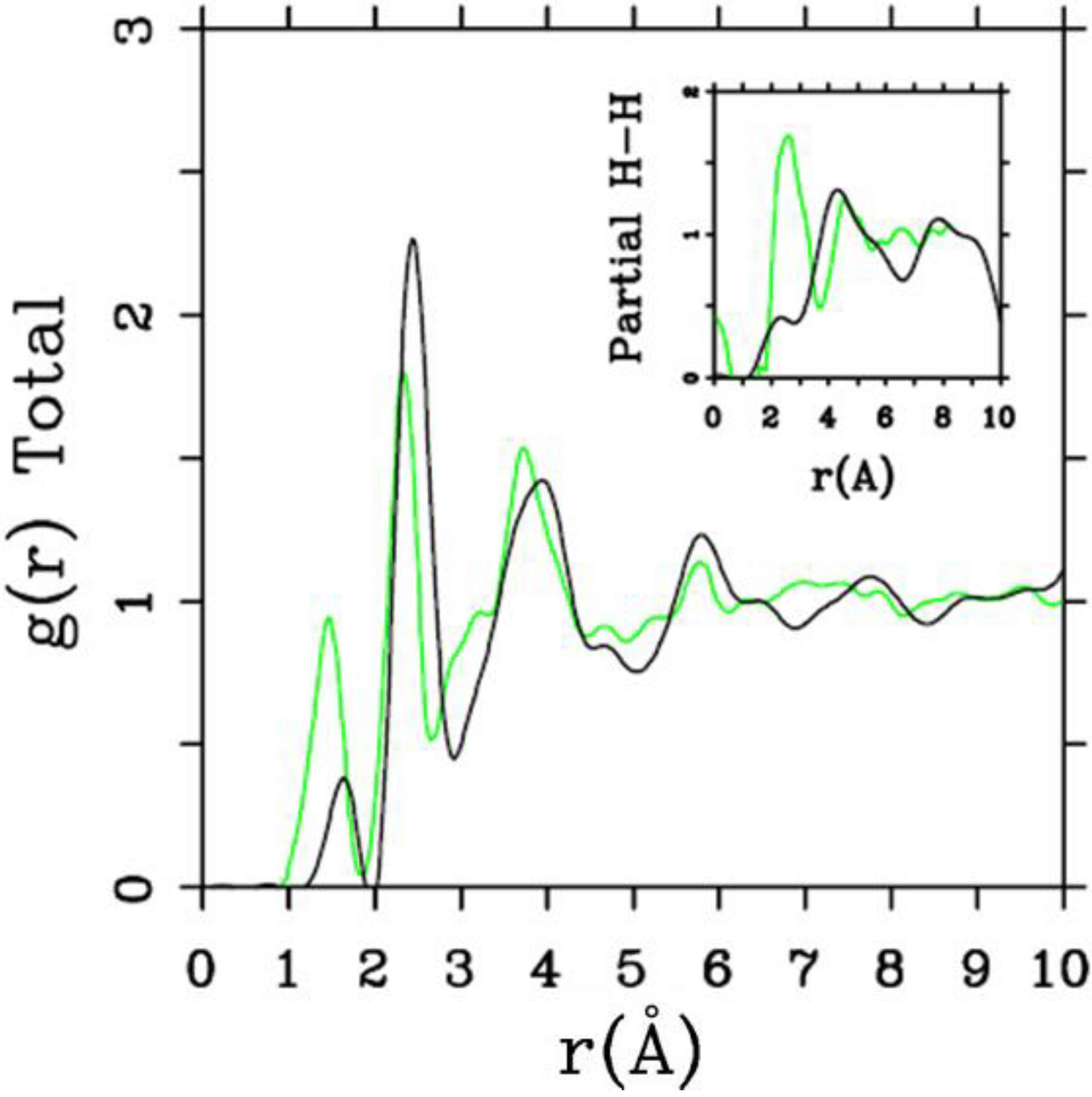
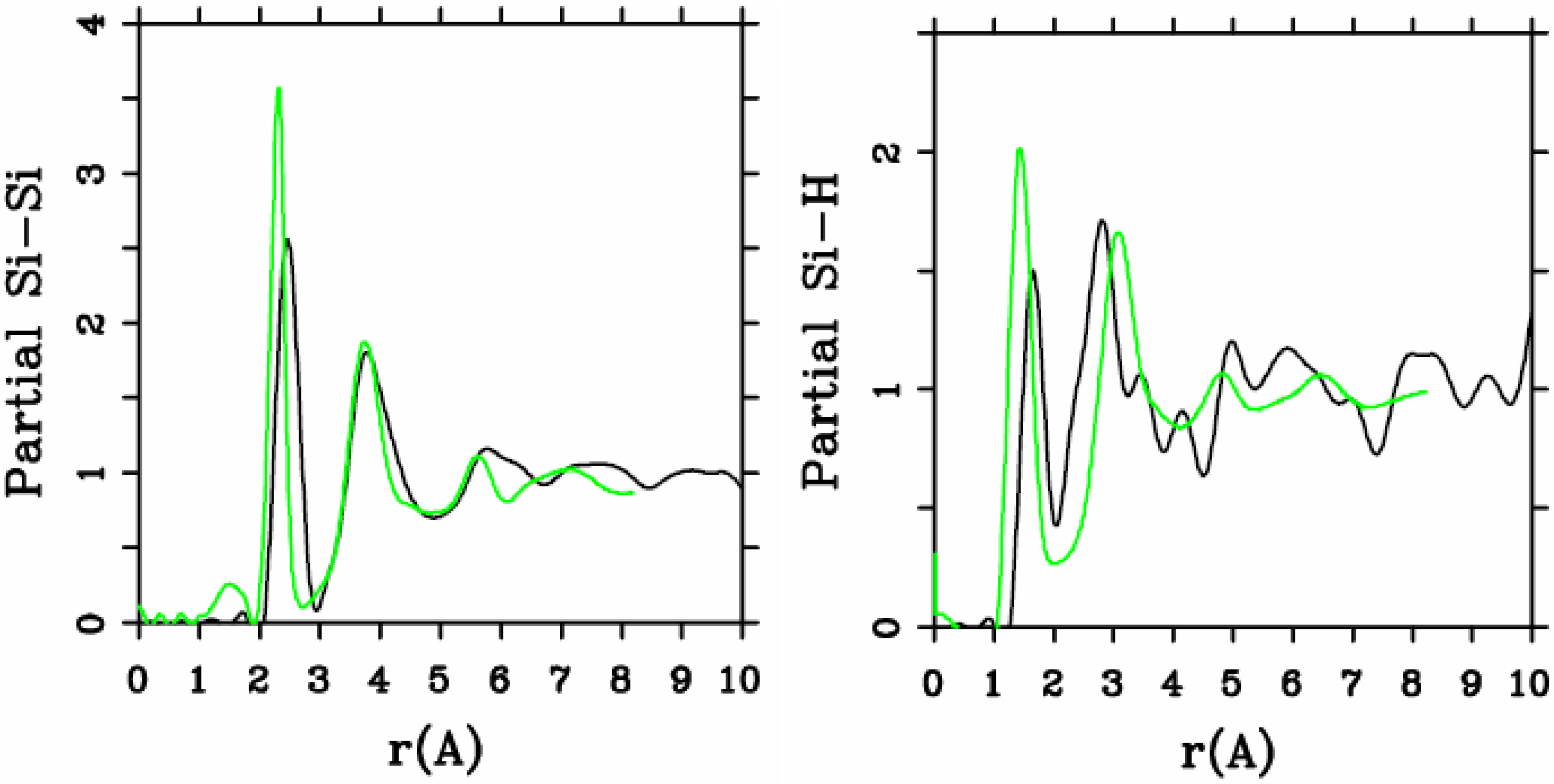
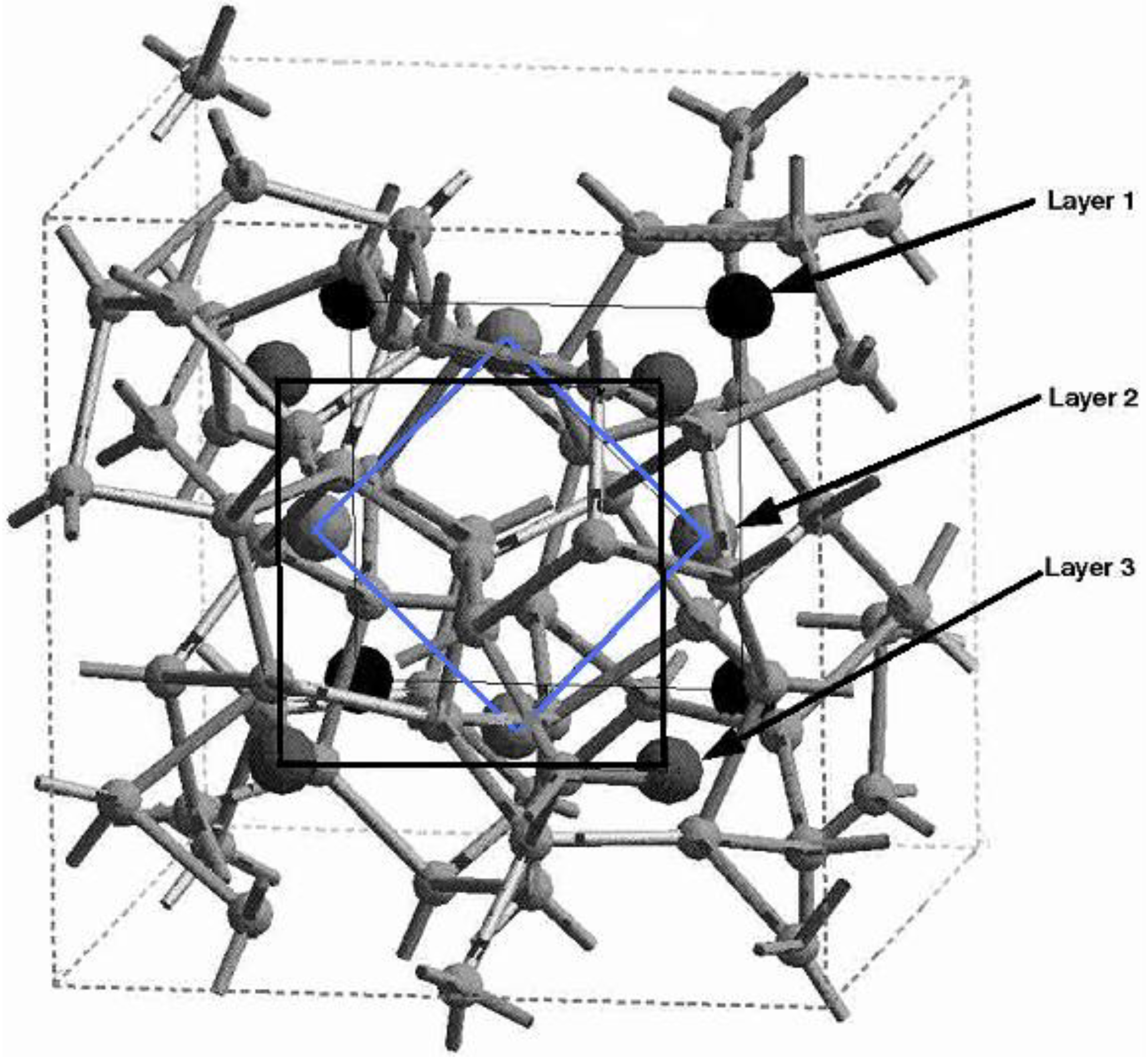
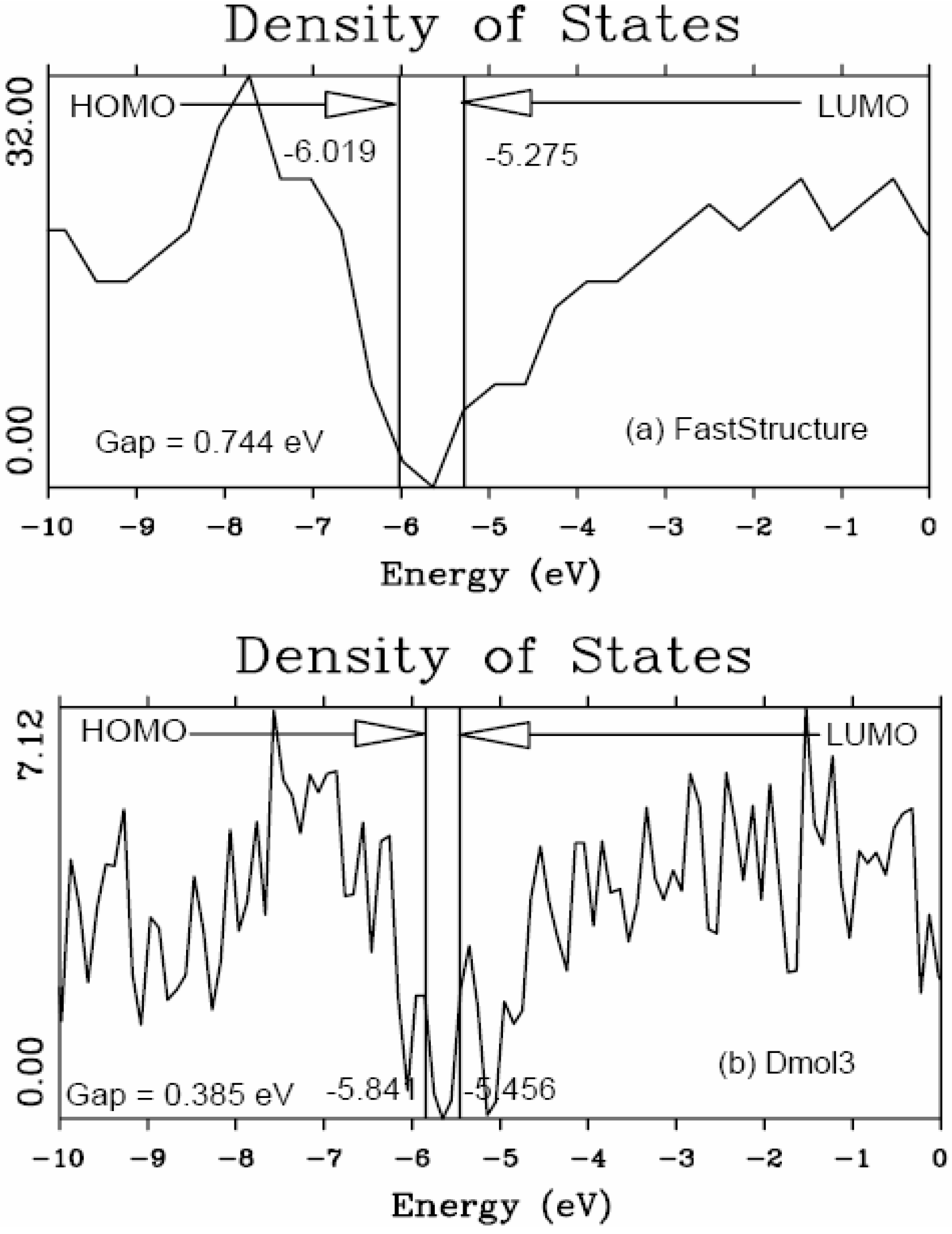
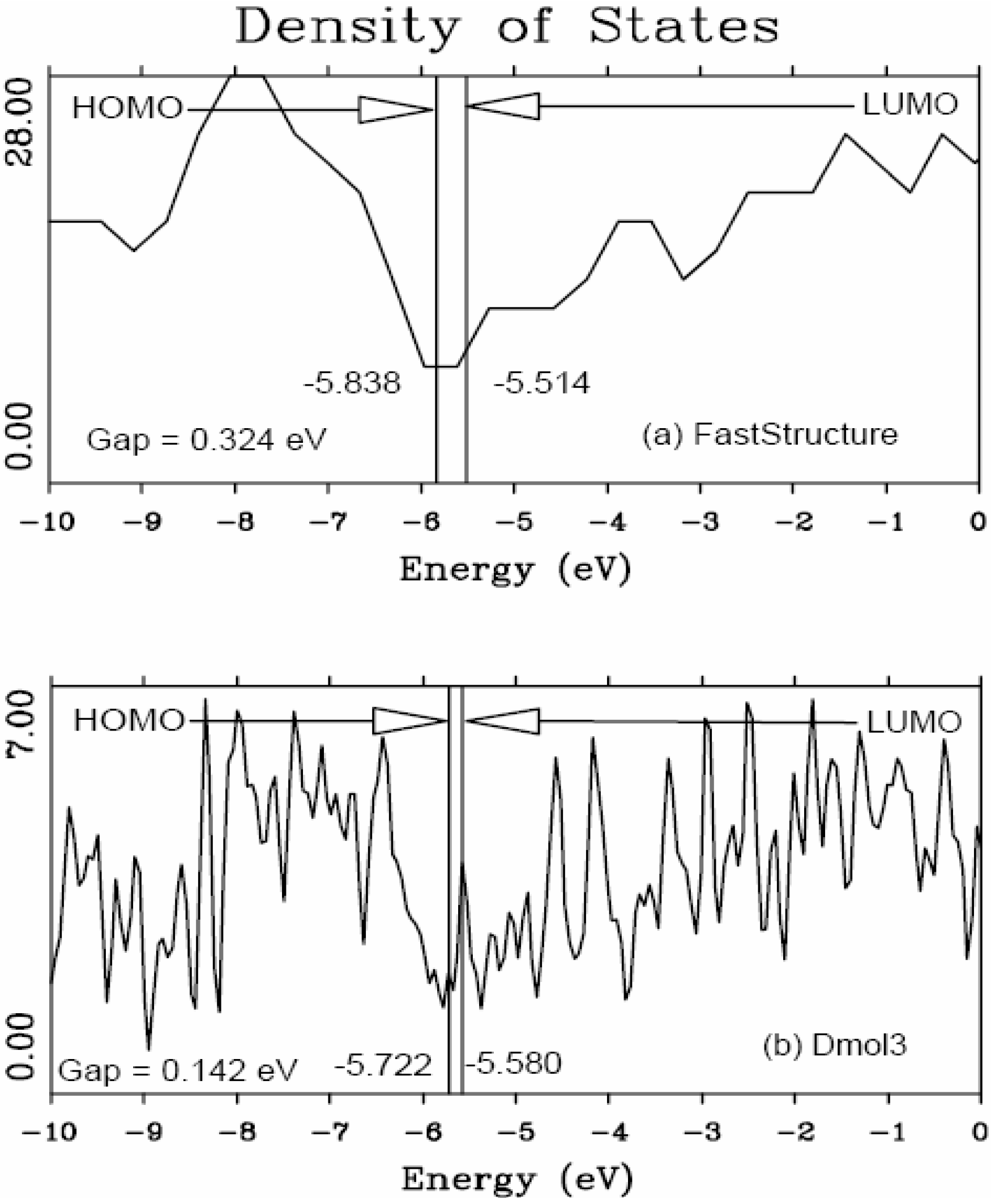
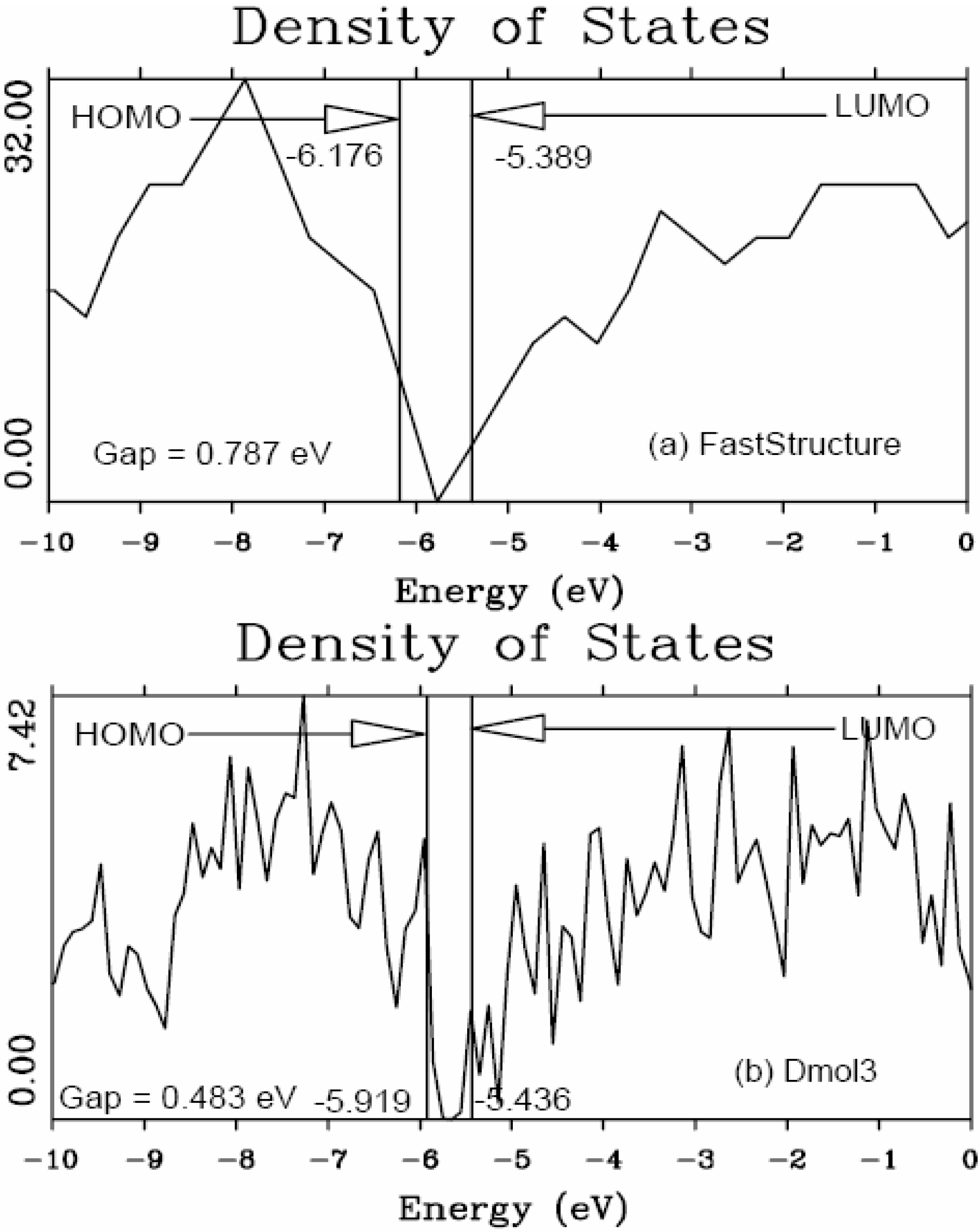
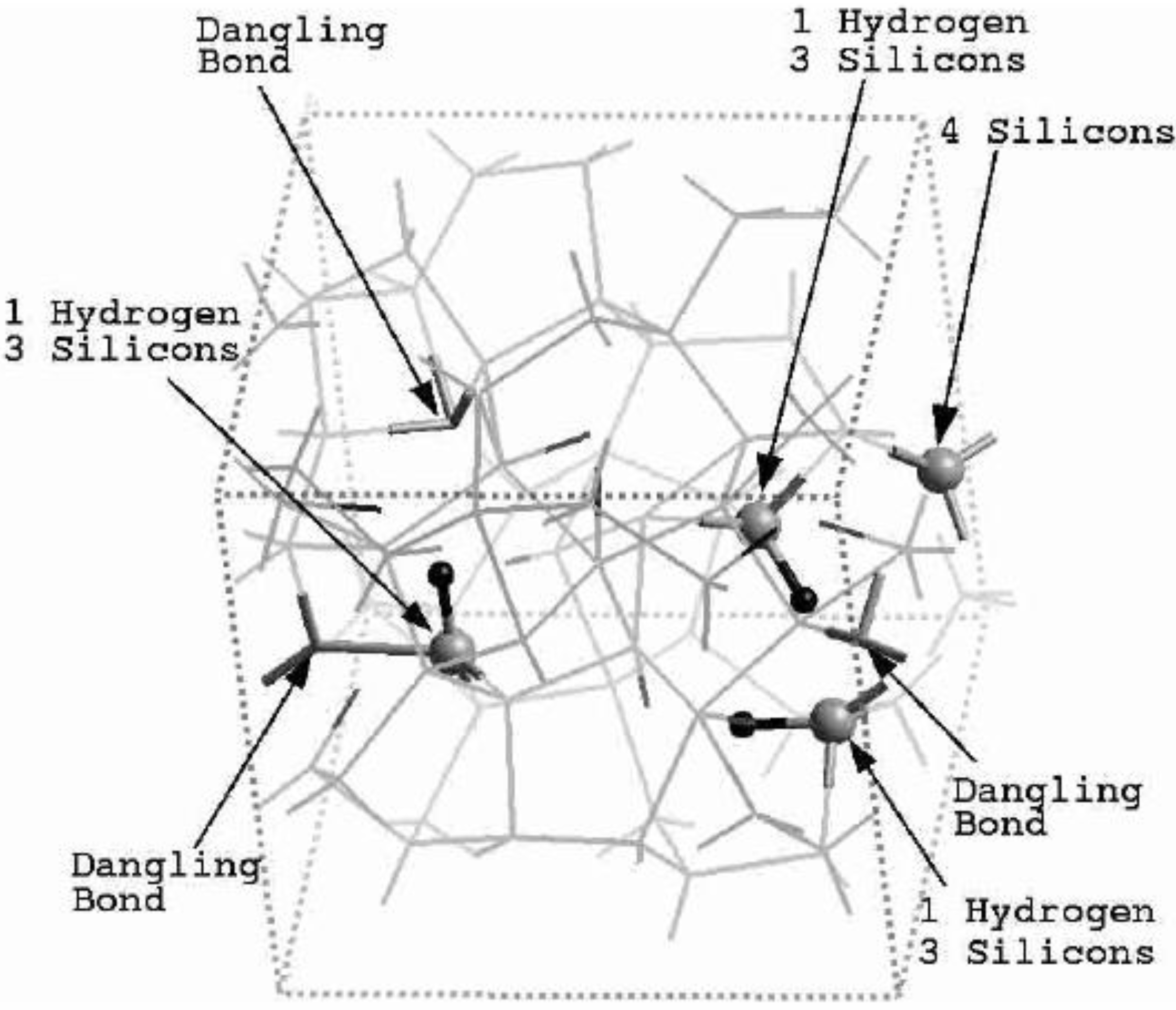
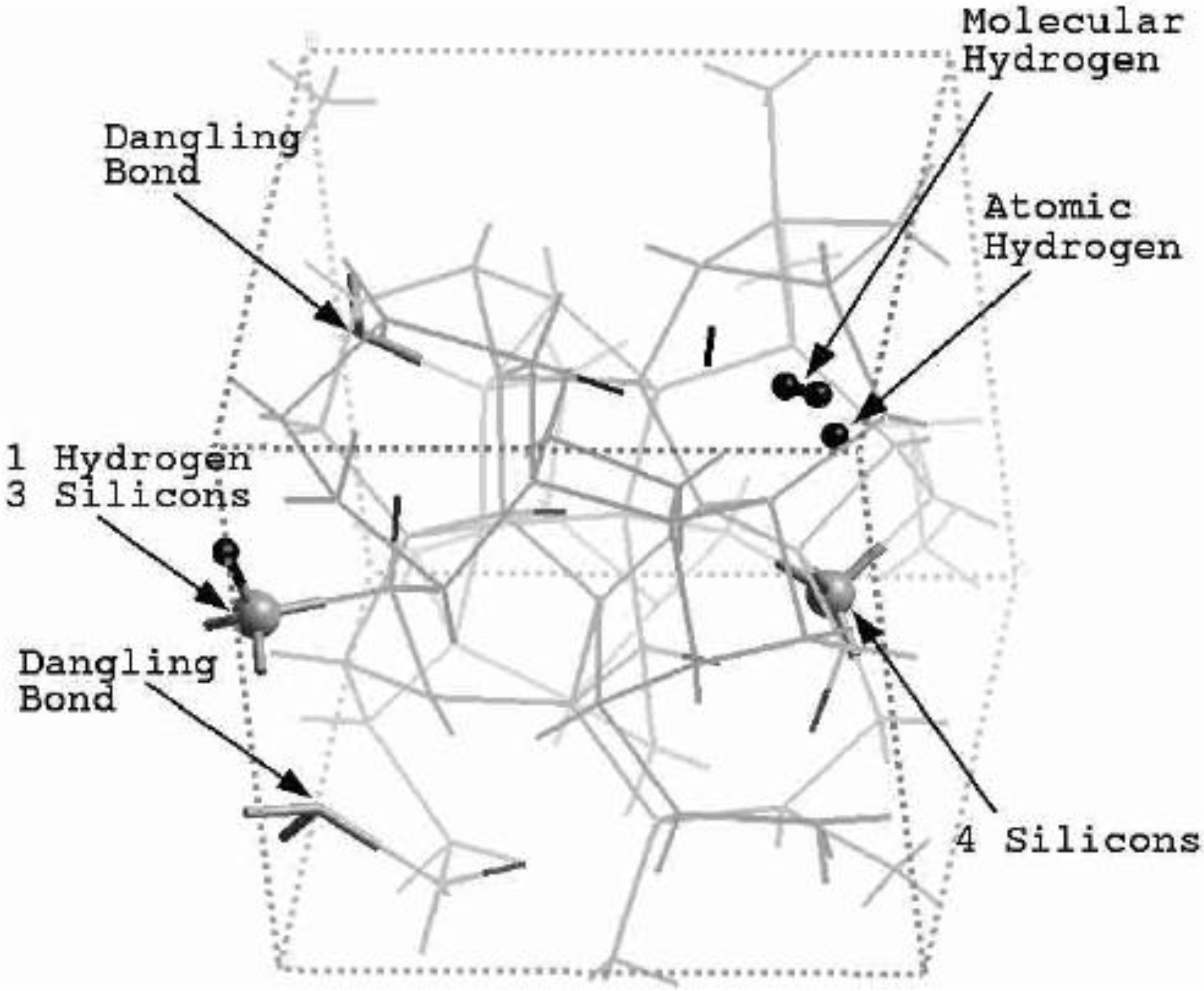
4.1. Amorphous Hydrogenated Silicon [1,48]
4.1.1. Preamble

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Relative starting positions of the hydrogen atoms in the amorphous silicon cells | |
|---|---|
| H1 | (1/4, 1/4, 1/4) |
| H2 | (3/4, 1/4, 1/4) |
| H3 | (3/4, 3/4, 1/4) |
| H4 | (1/4, 3/4, 1/4) |
| H5 | (1/2, 1/4, 1/2) |
| H6 | (3/4, 1/2, 1/2) |
| H7 | (1/2, 3/4, 1/2) |
| H8 | (1/4, 1/2, 1/2) |
| H9 | (1/4, 1/4, 3/4) |
| H10 | (3/4, 1/4, 3/4) |
| H11 | (3/4, 3/4, 3/4) |
| H12 | (1/4, 3/4, 3/4) |
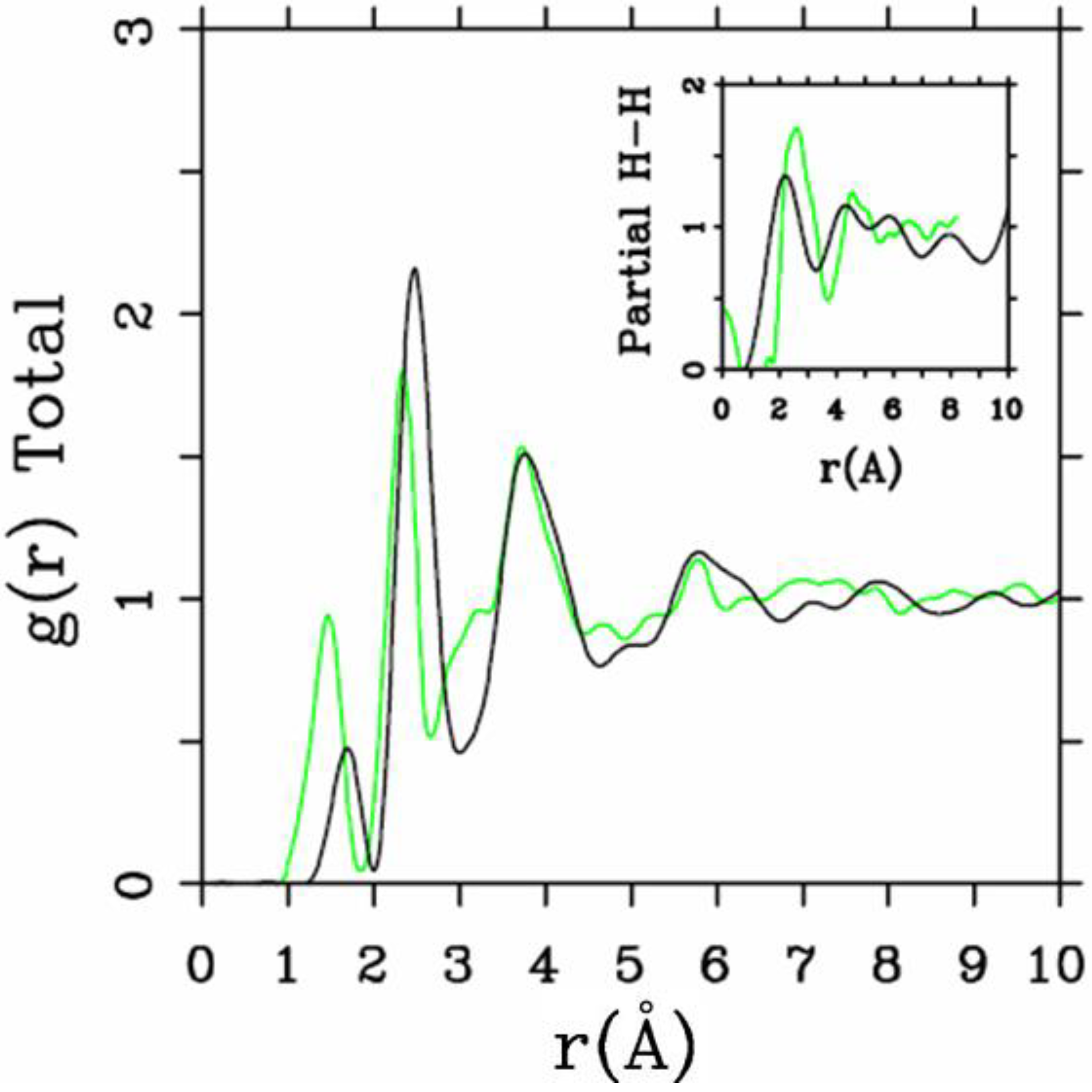
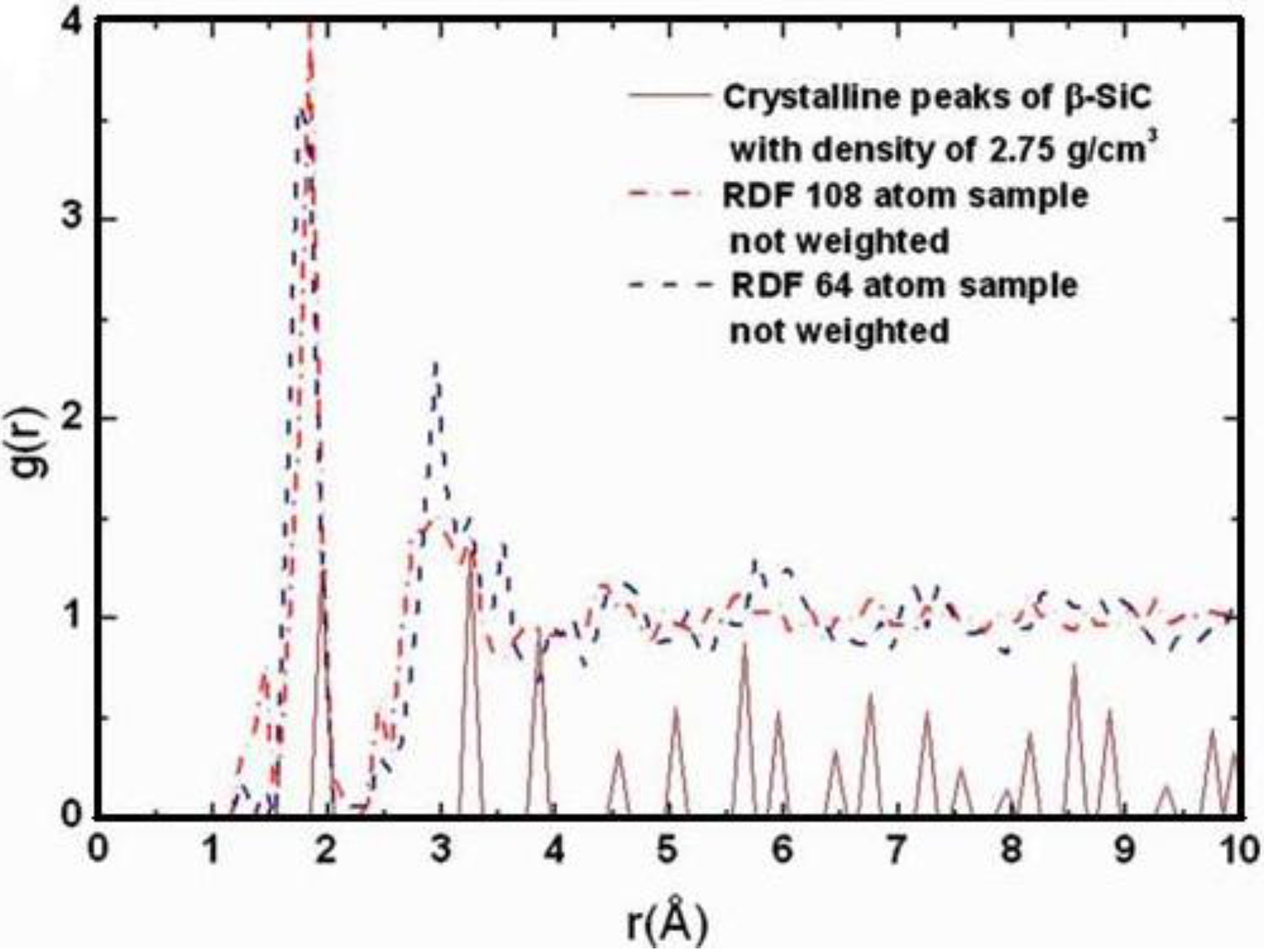
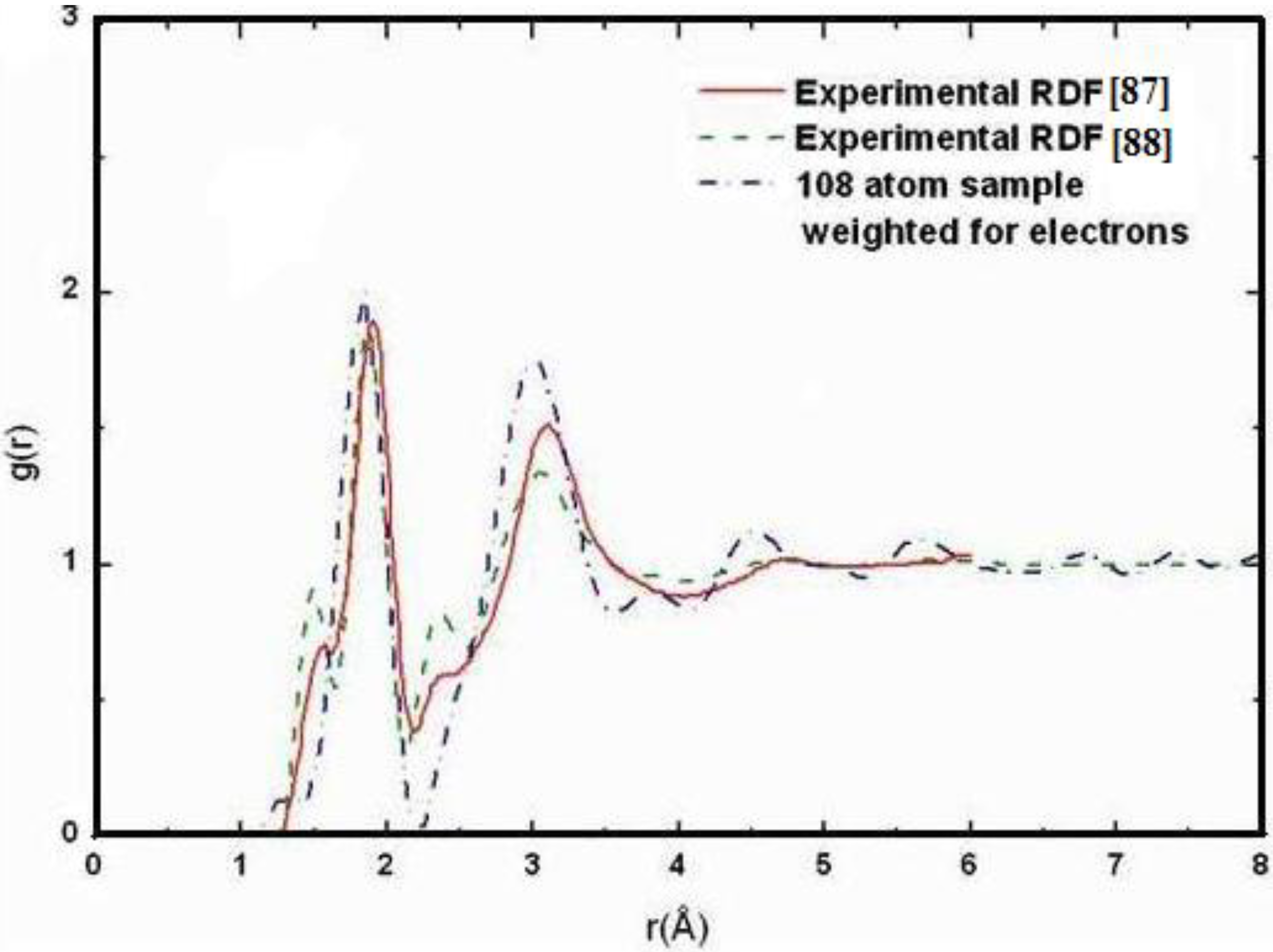
4.1.2. Results and Analysis
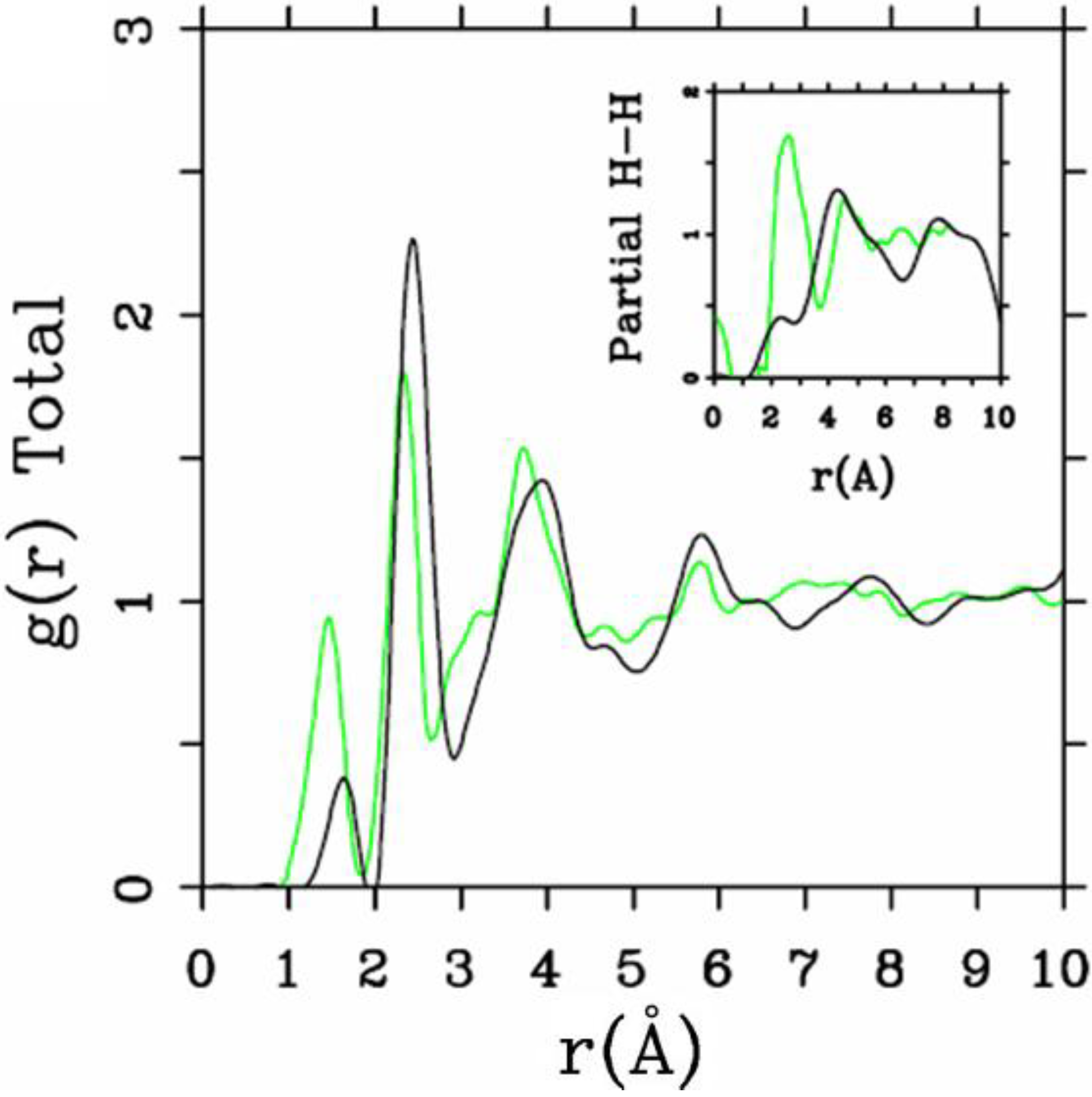
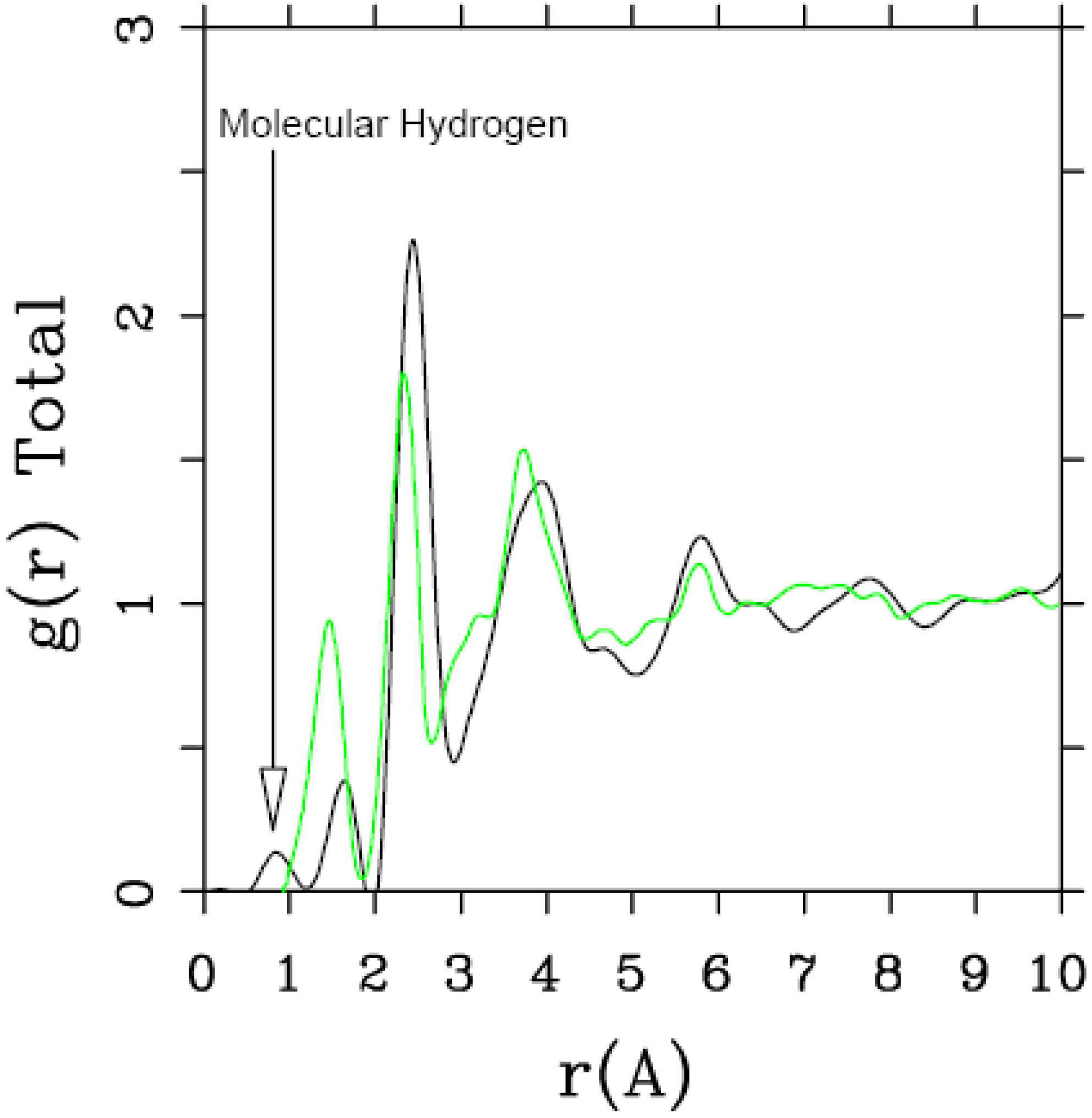
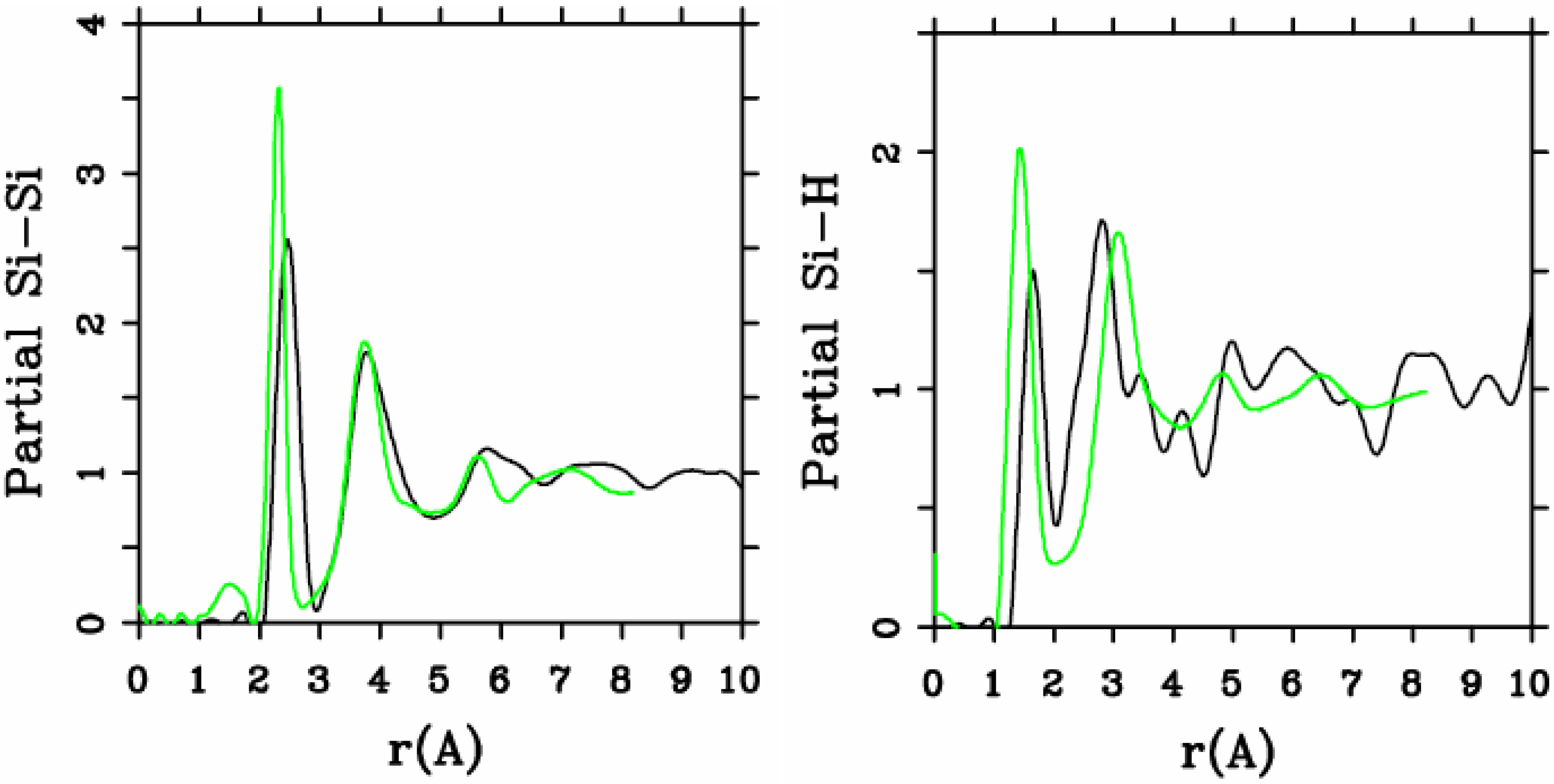
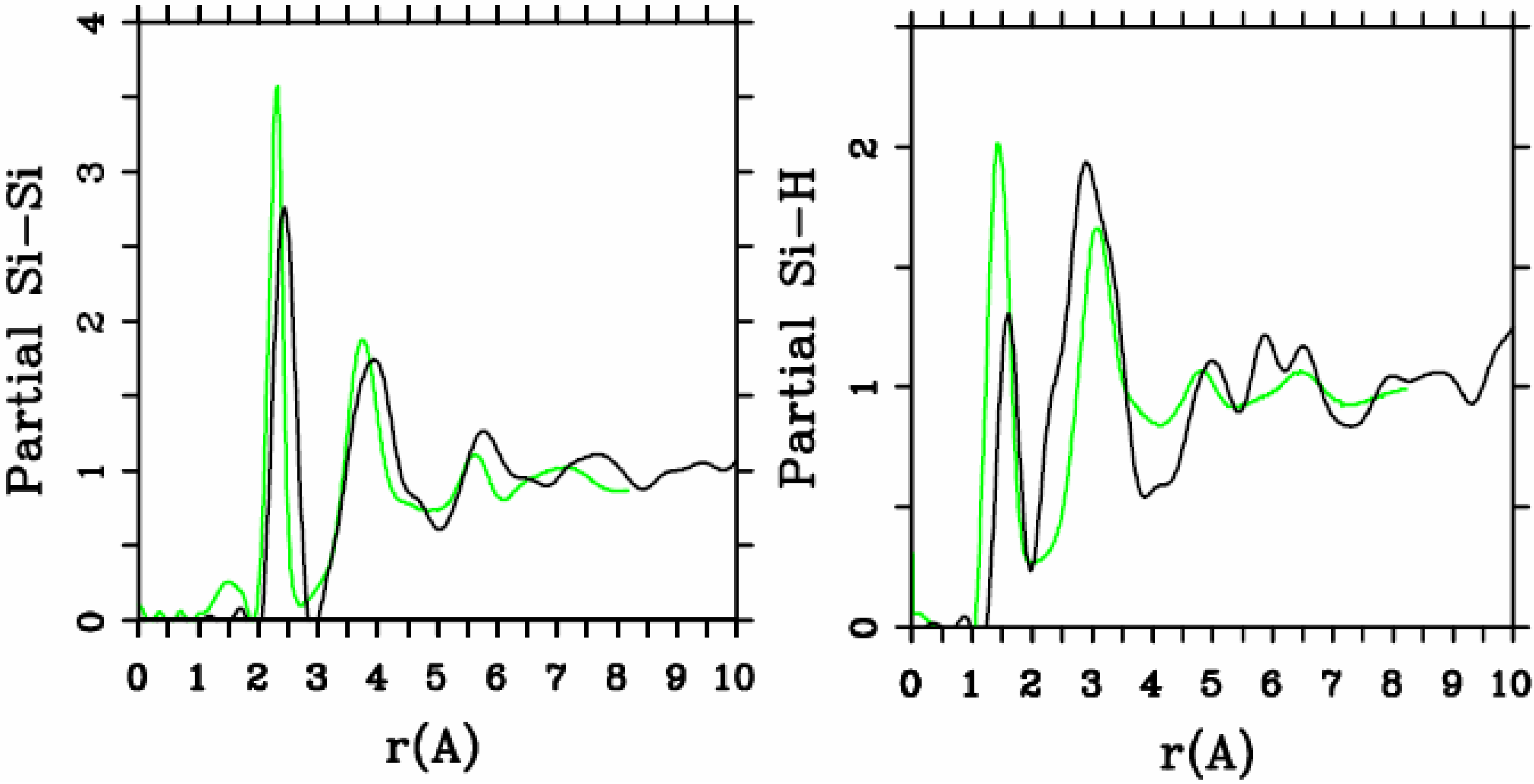
4.1.3. Summary
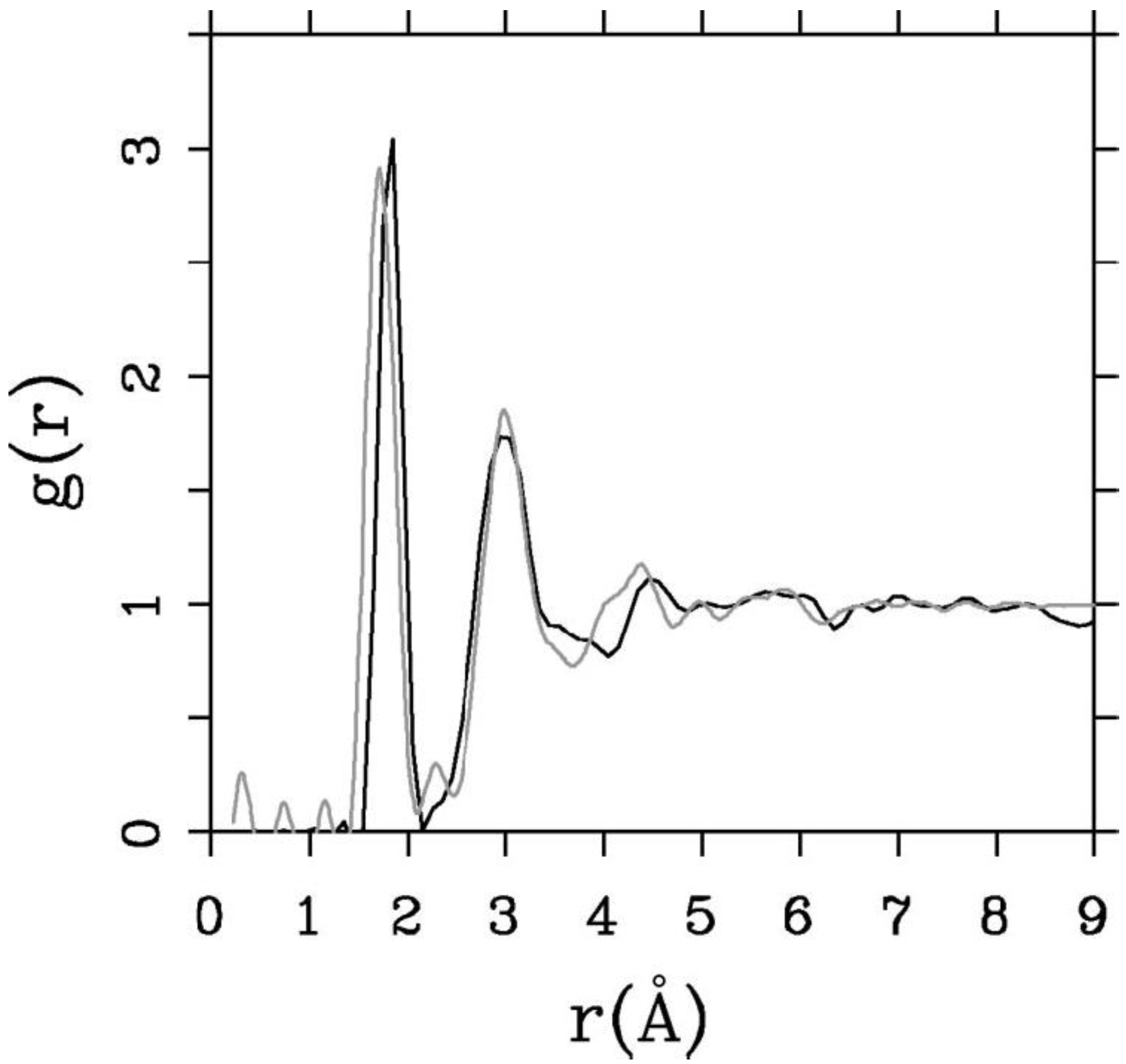
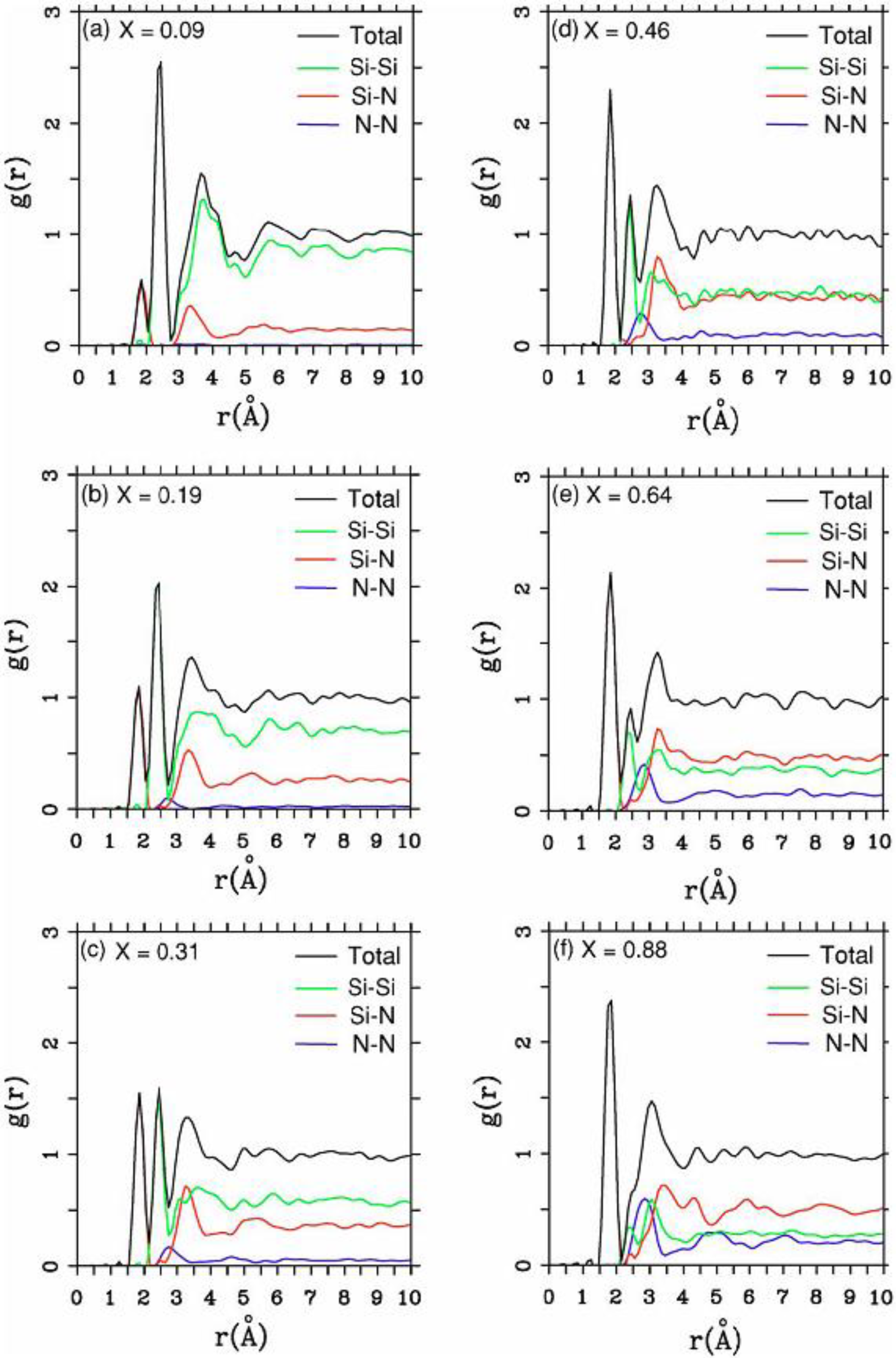
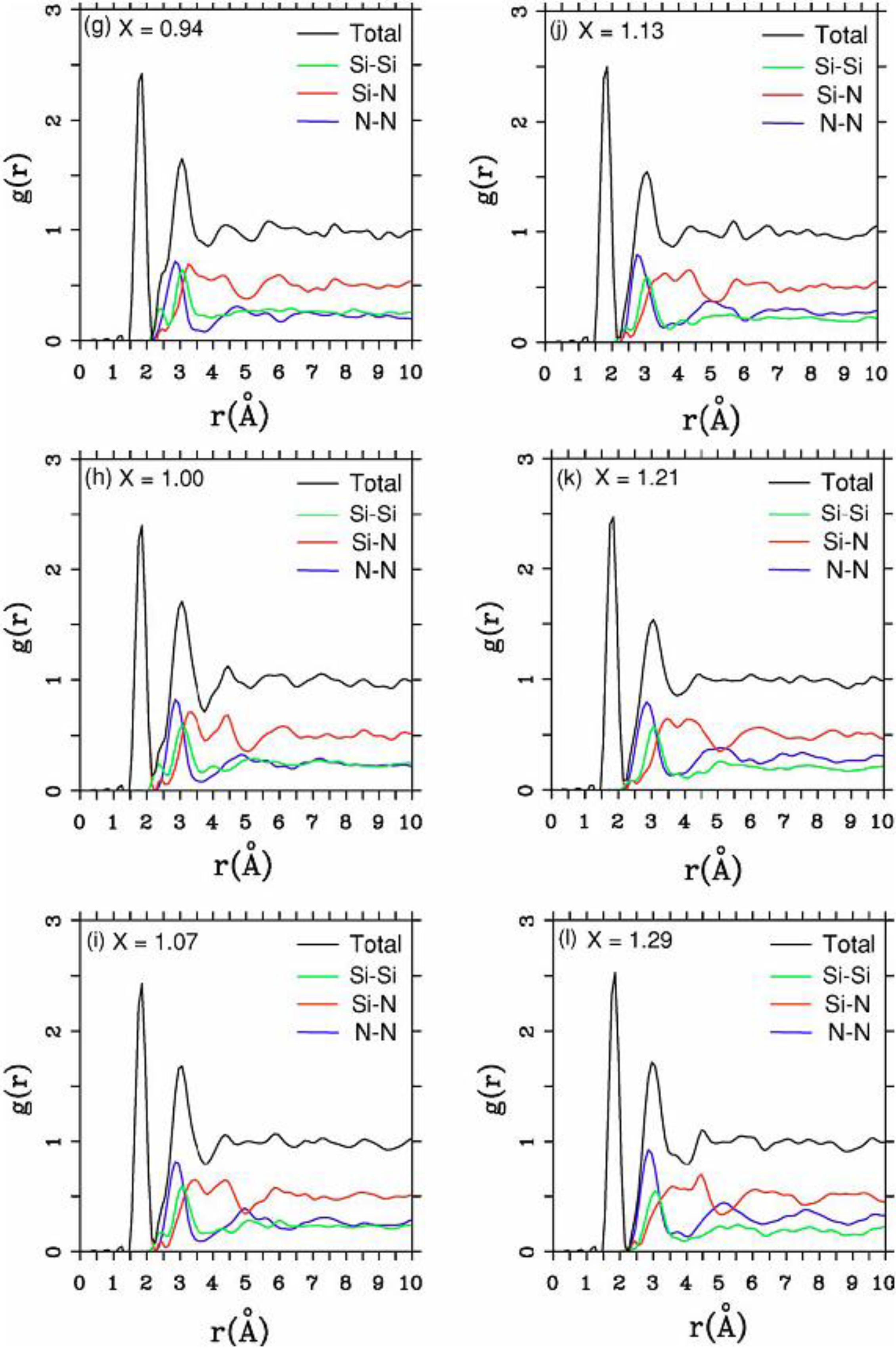
4.2. Amorphous Silicon-Nitrogen Alloys [1,51,52,53,54,55]
4.2.1. Preamble
| Contents, amorphization temperatures, densities and gaps for a-SiNx | ||||
|---|---|---|---|---|
| Sample | x | Amorphous temp. (K) | Density (g/cm3) | Average Gap (eV) |
| Si64N0 | 0.000 | 1680 | 2.329 | 1.09 |
| Si59N5 | 0.085 | 1747 | 2.435 | 1.11 |
| Si54N10 | 0.185 | 1814 | 2.512 | 1.05 |
| Si49N15 | 0.306 | 1881 | 2.600 | 1.19 |
| Si44N20 | 0.455 | 1948 | 2.694 | 1.69 |
| Si39N25 | 0.641 | 2015 | 2.803 | 2.49 |
| Si34N30 | 0.882 | 2082 | 2.931 | 3.41 |
| Si33N31 | 0.939 | 2095 | 2.957 | 3.73 |
| Si32N32 | 1.000 | 2108 | 2.988 | 4.10 |
| Si31N33 | 1.065 | 2122 | 3.017 | 4.31 |
| Si30N34 | 1.133 | 2136 | 3.048 | 4.28 |
| Si29N35 | 1.207 | 2149 | 3.081 | 4.69 |
| Si28N36 | 1.286 | 2162 | 3.115 | 4.95 |
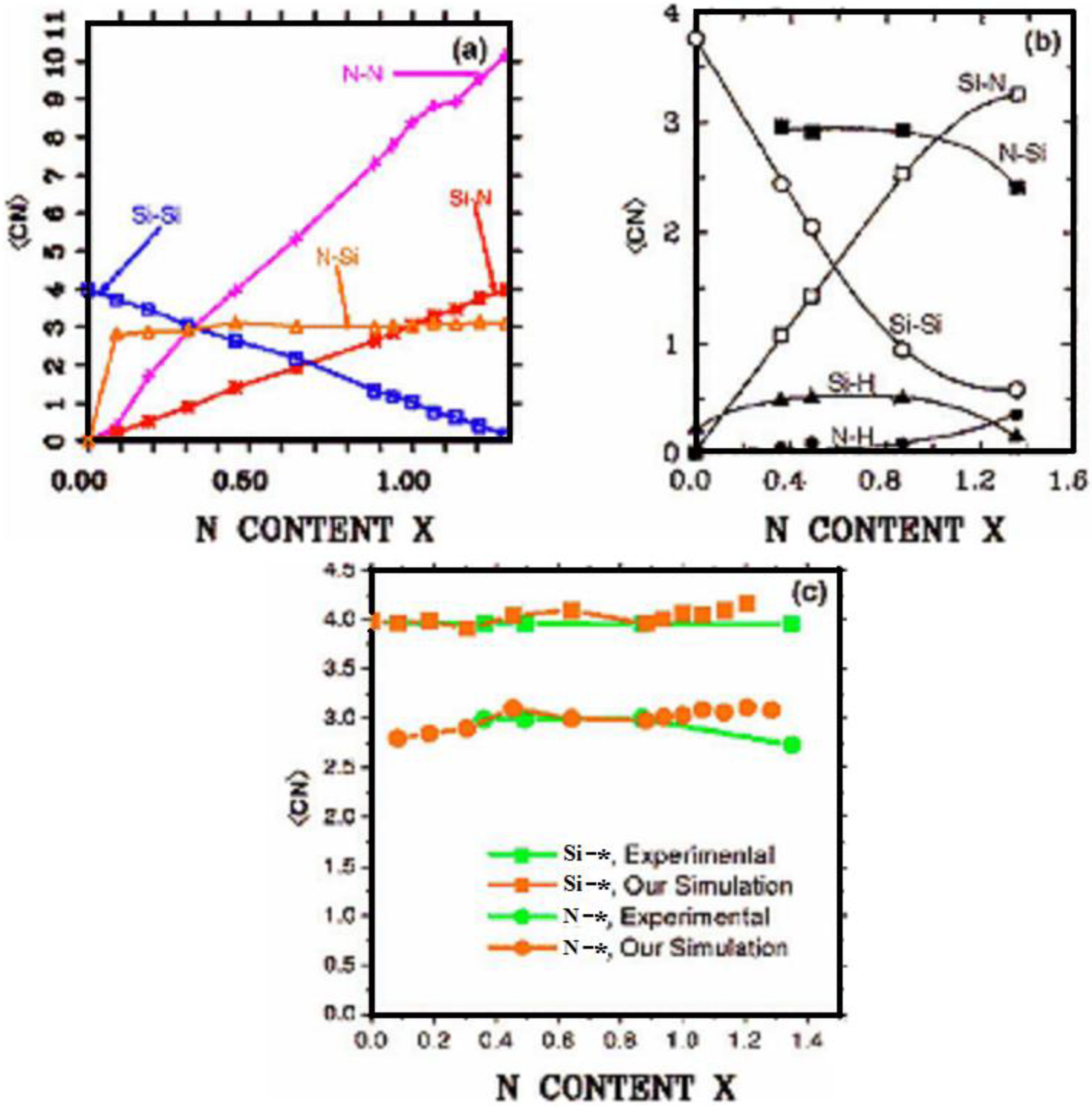
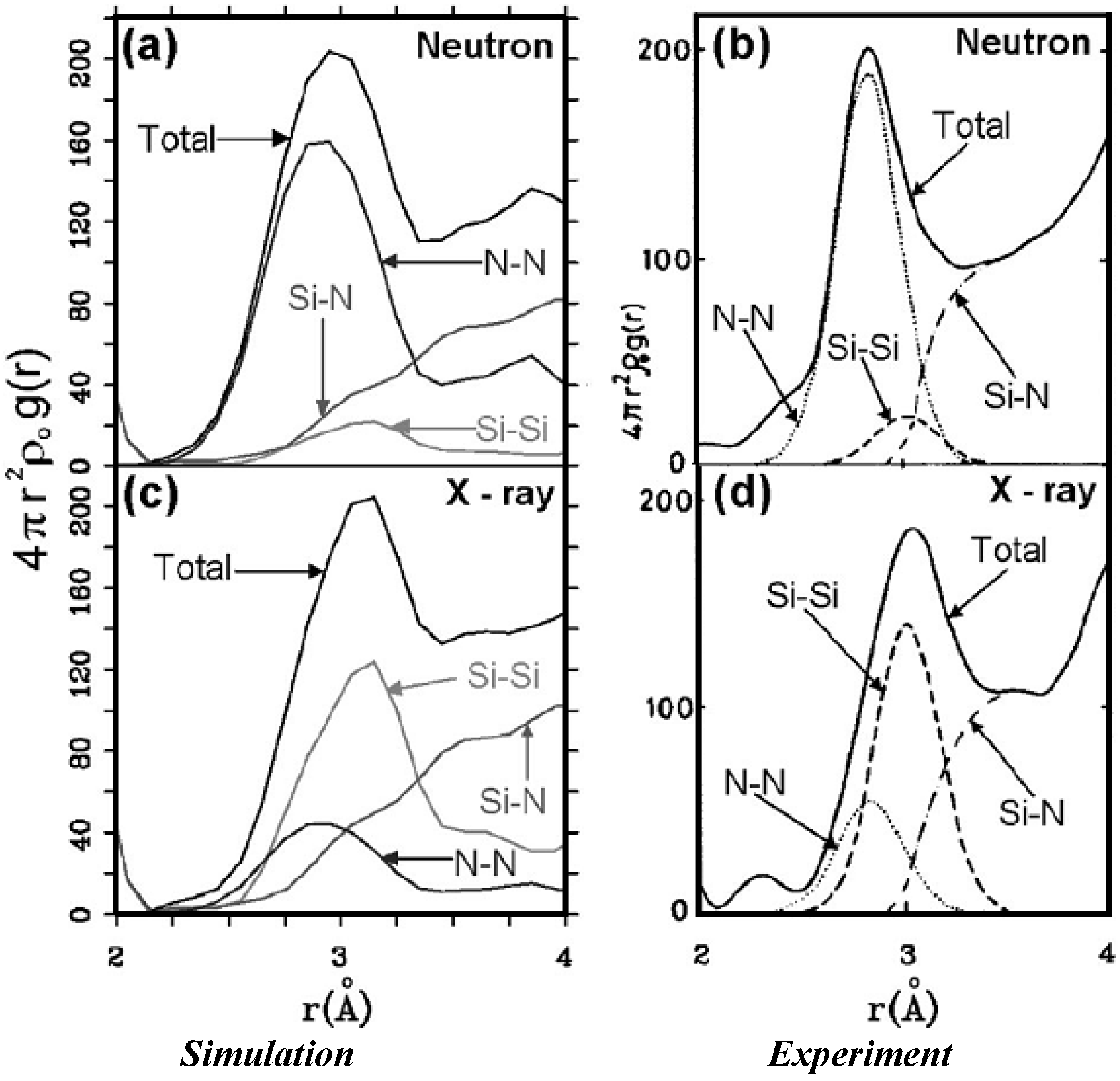
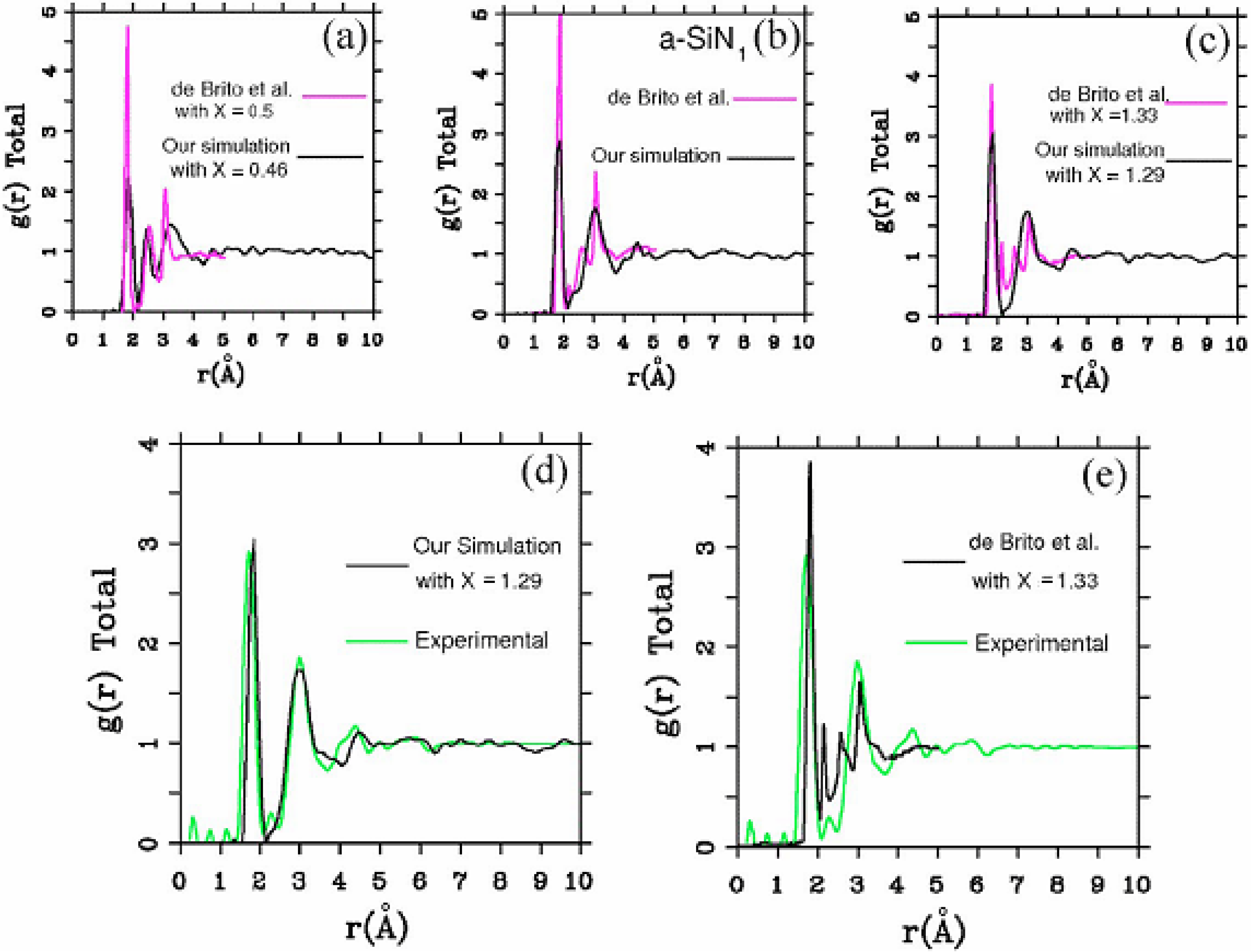
4.2.2. Results and Analysis

| Height and position of the maxima of radial peaks and nearest neighbors <nn> for a-silicon nitride (x = 1.29) | |||||
|---|---|---|---|---|---|
| Material | FIRST PEAK | SECOND PEAK | <nn> | ||
| Position | Height | Position | Height | ||
| SiN1.29 | 1.85 Å | 3.04 | 2.95 Å | 1.73 | 3.47 |
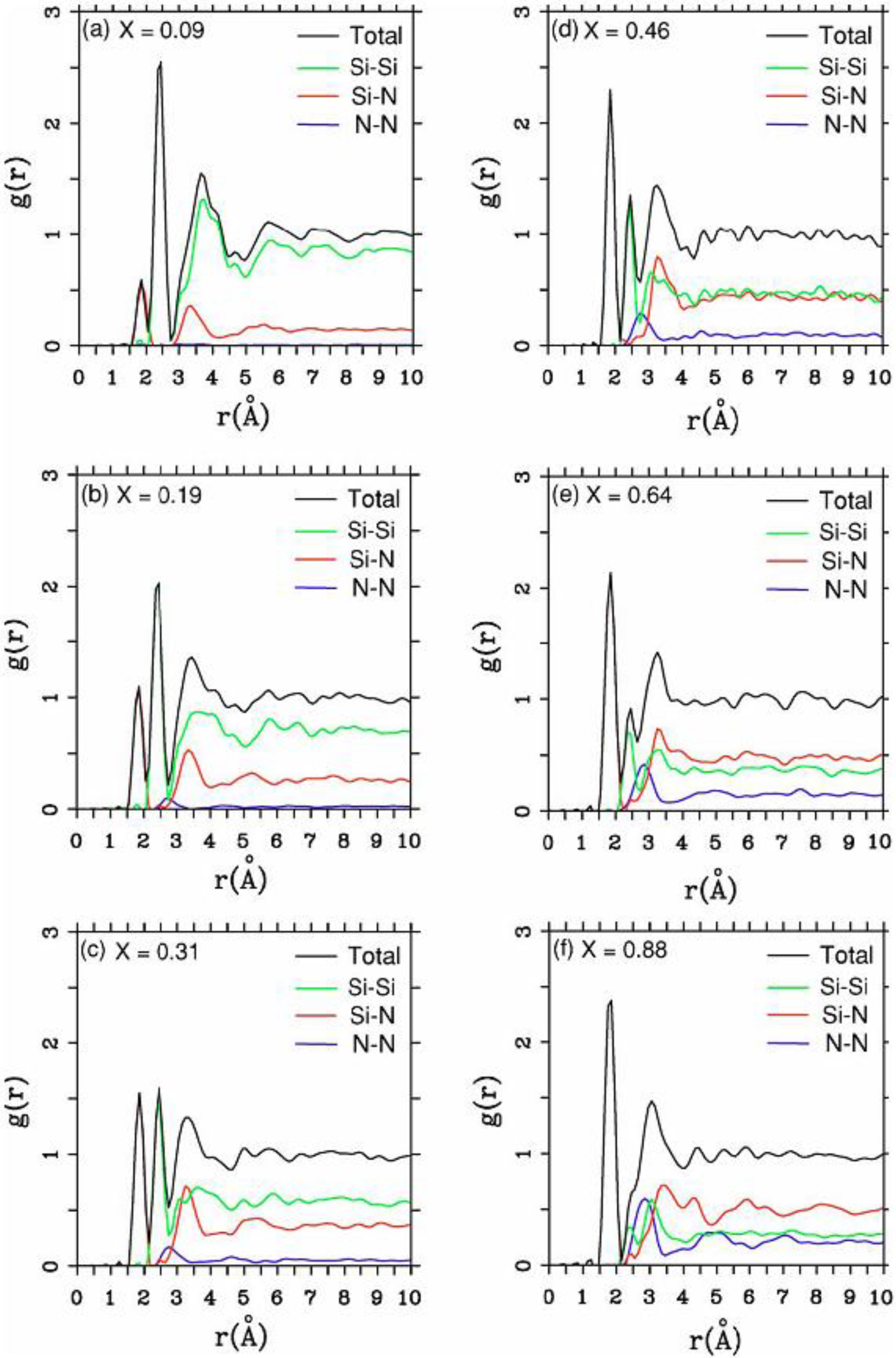
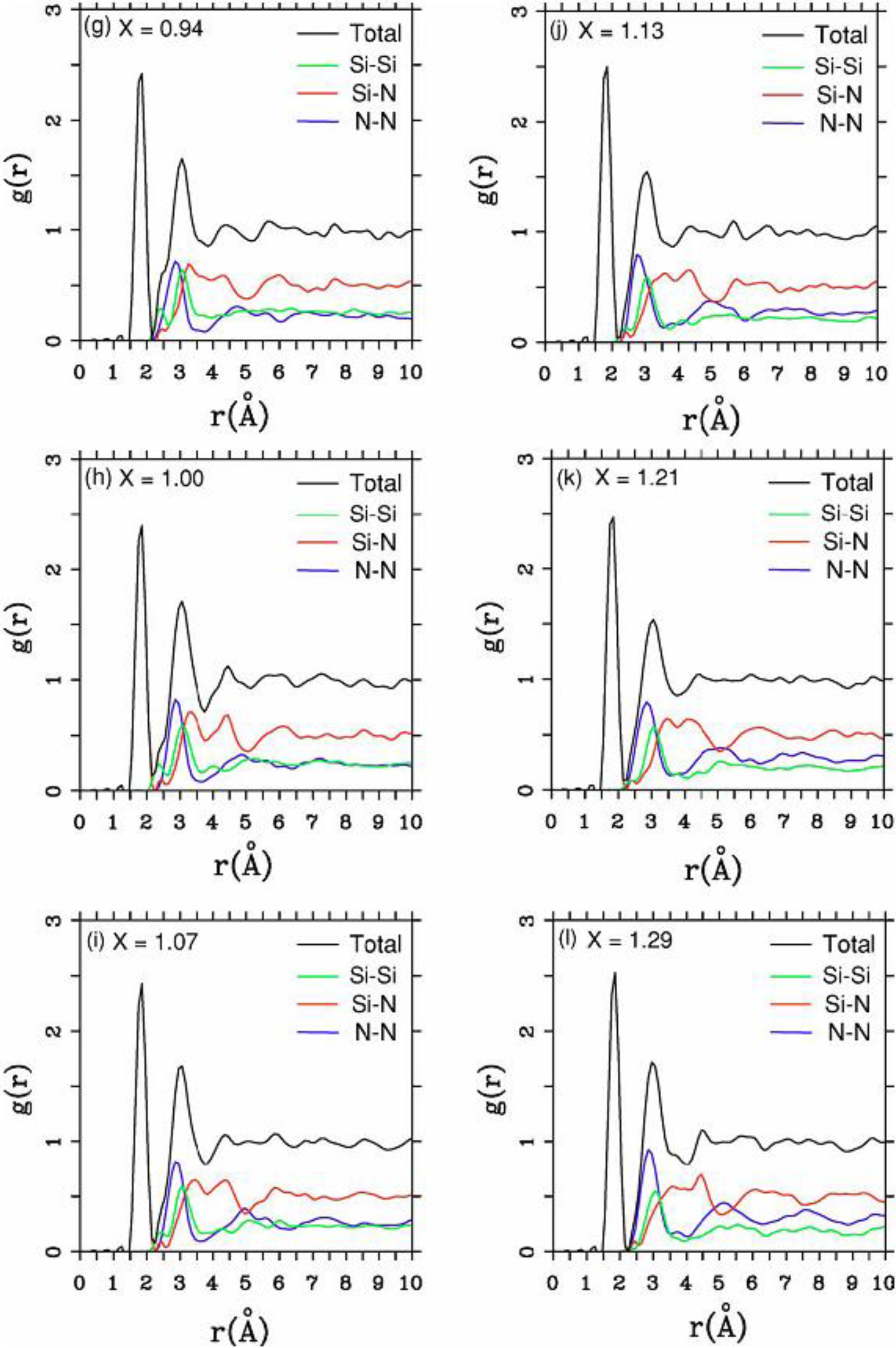
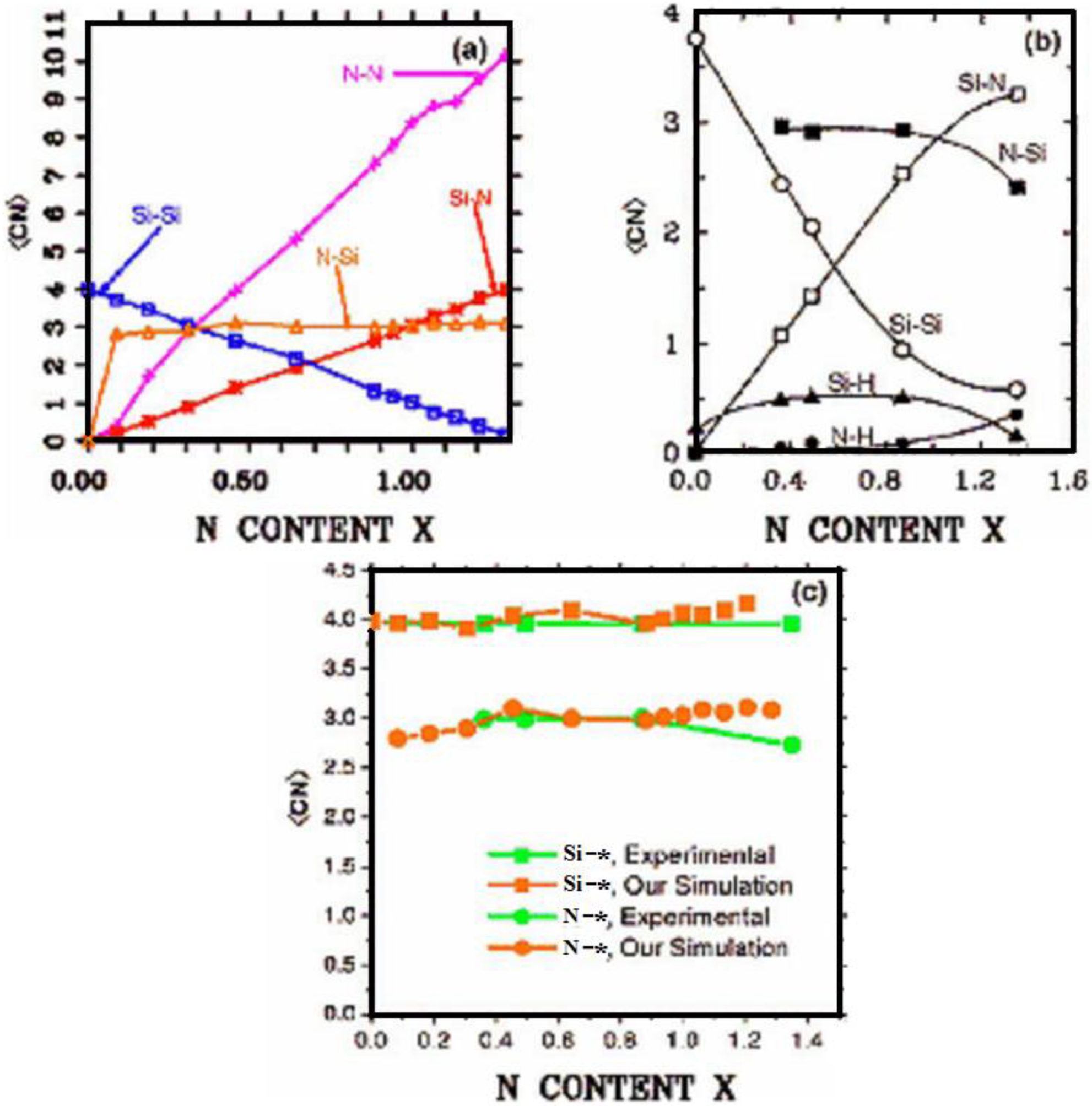
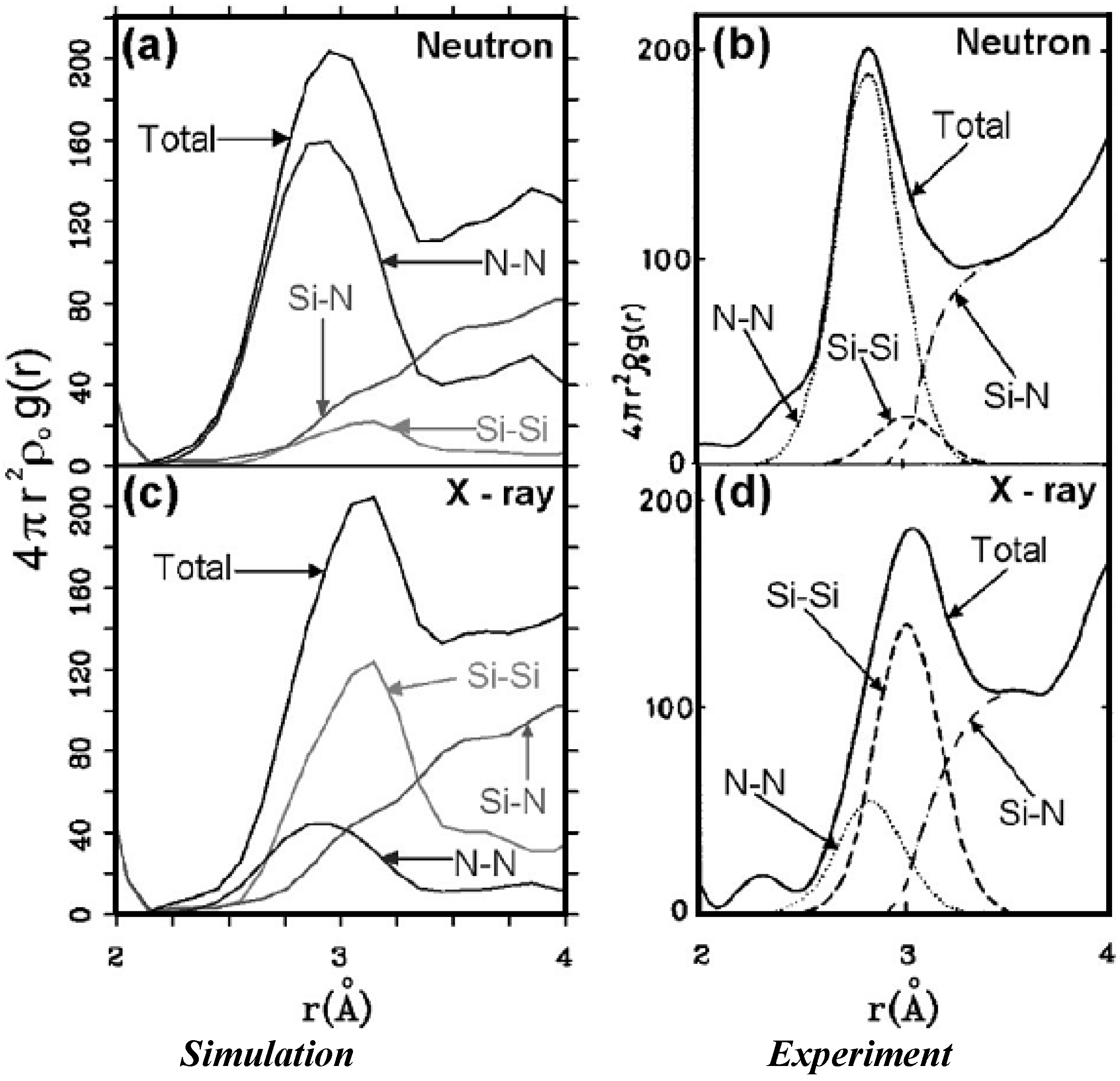
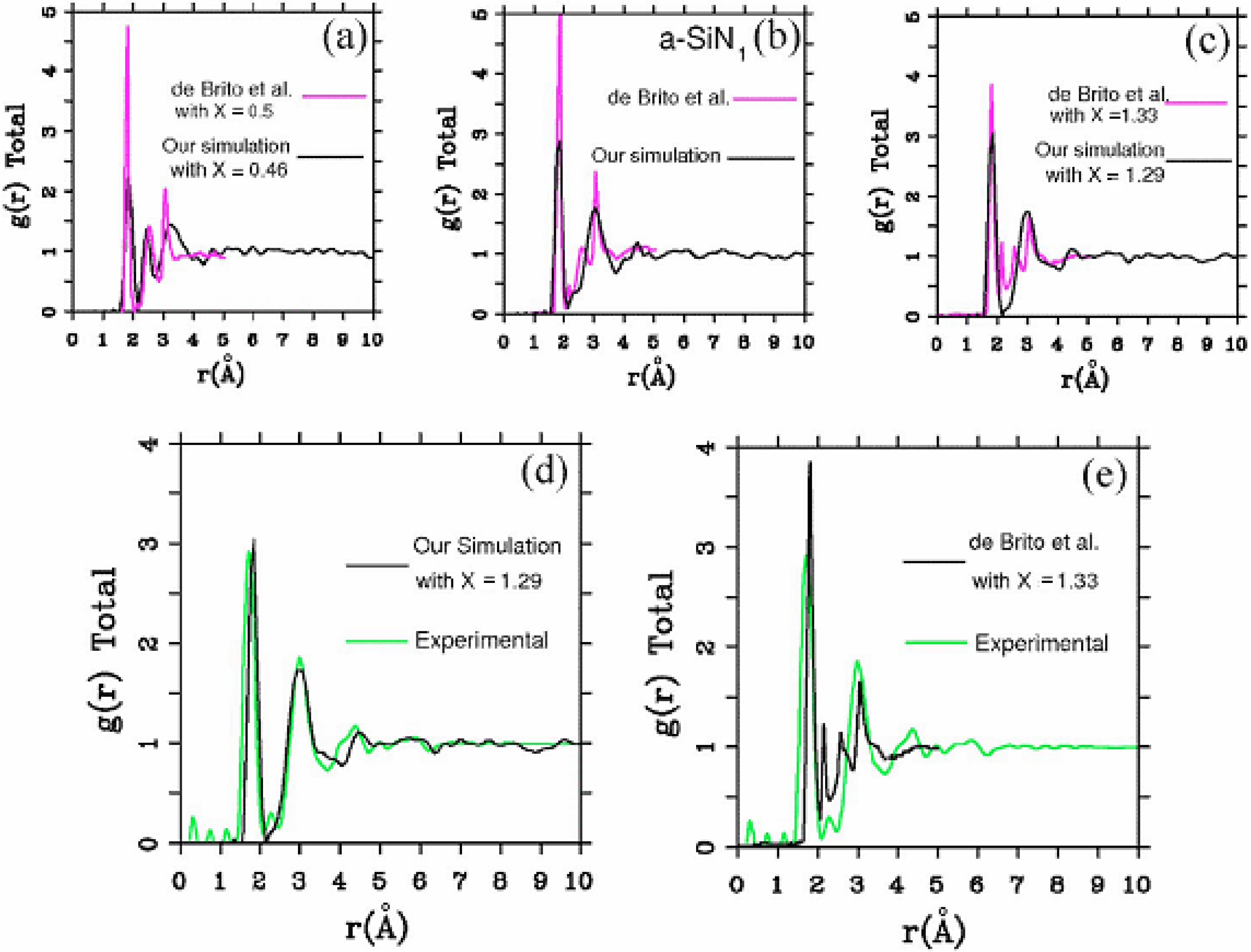
4.2.3. Summary
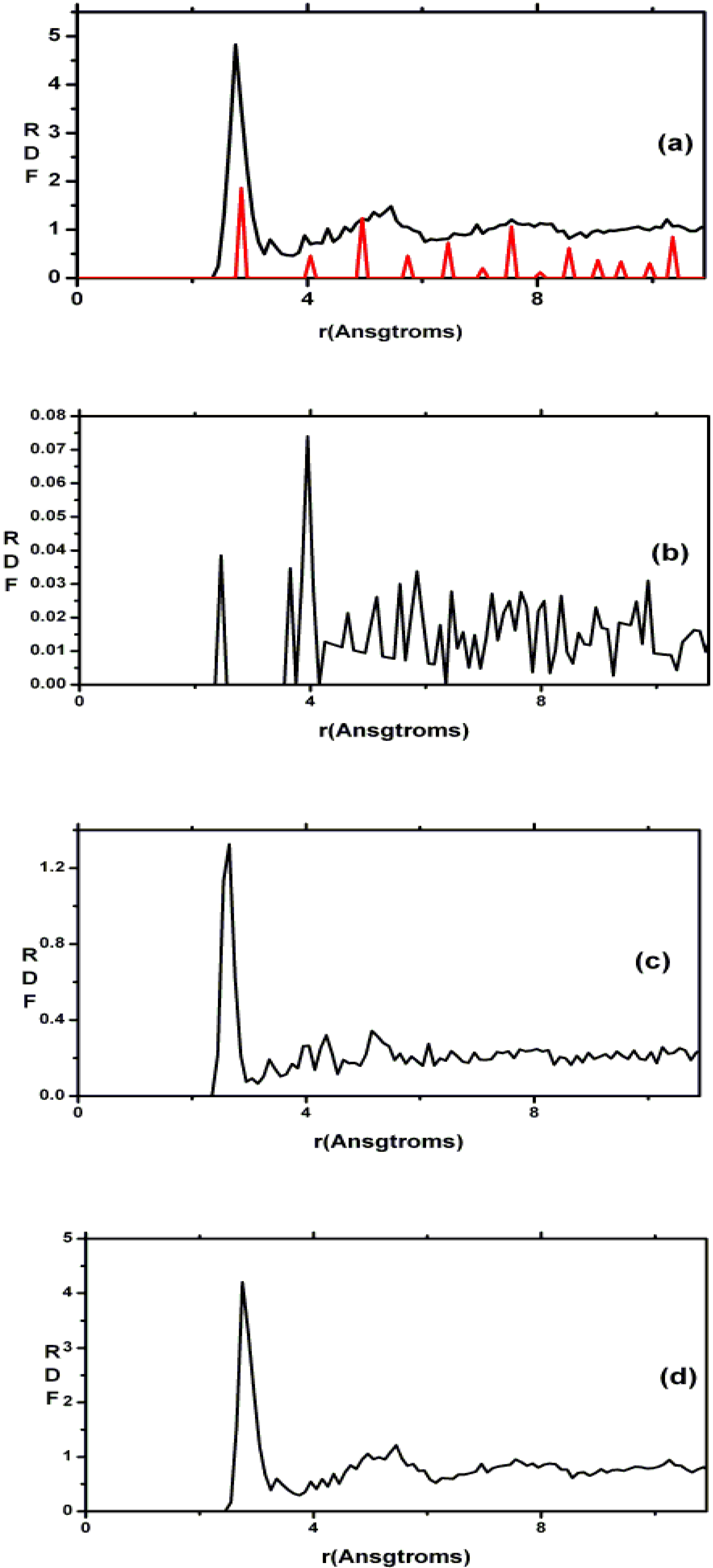
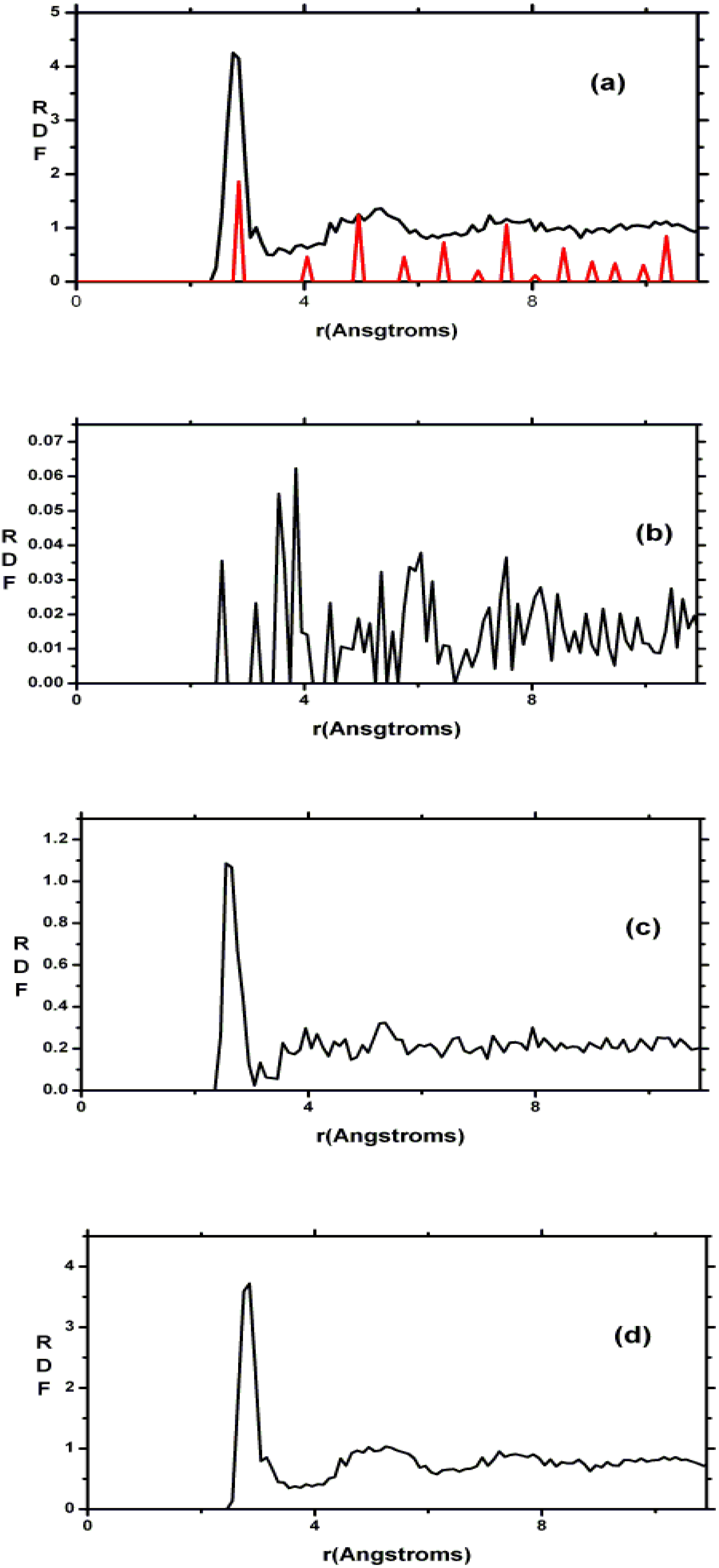
4.3. Amorphous Carbon-Nitrogen Alloys [1,68]
4.3.1. Preamble
| Concentrations and densities for a-CN | ||
|---|---|---|
| Sample | N concentration c (%) | Density ρ = [2.897 − 1.784c] (g/cm3) |
| C216N00 | 0.0 | 2.90 |
| C205N11 | 5.1 | 2.81 |
| C194N22 | 10.2 | 2.72 |
| C184N32 | 14.8 | 2.63 |
| C173N43 | 19.9 | 2.54 |
| C162N54 | 25.0 | 2.45 |
| C151N65 | 30.1 | 2.36 |
| C140N76 | 35.2 | 2.27 |
| C130N86 | 39.8 | 2.19 |
| C119N97 | 44.9 | 2.10 |
4.3.2. Results and Analysis
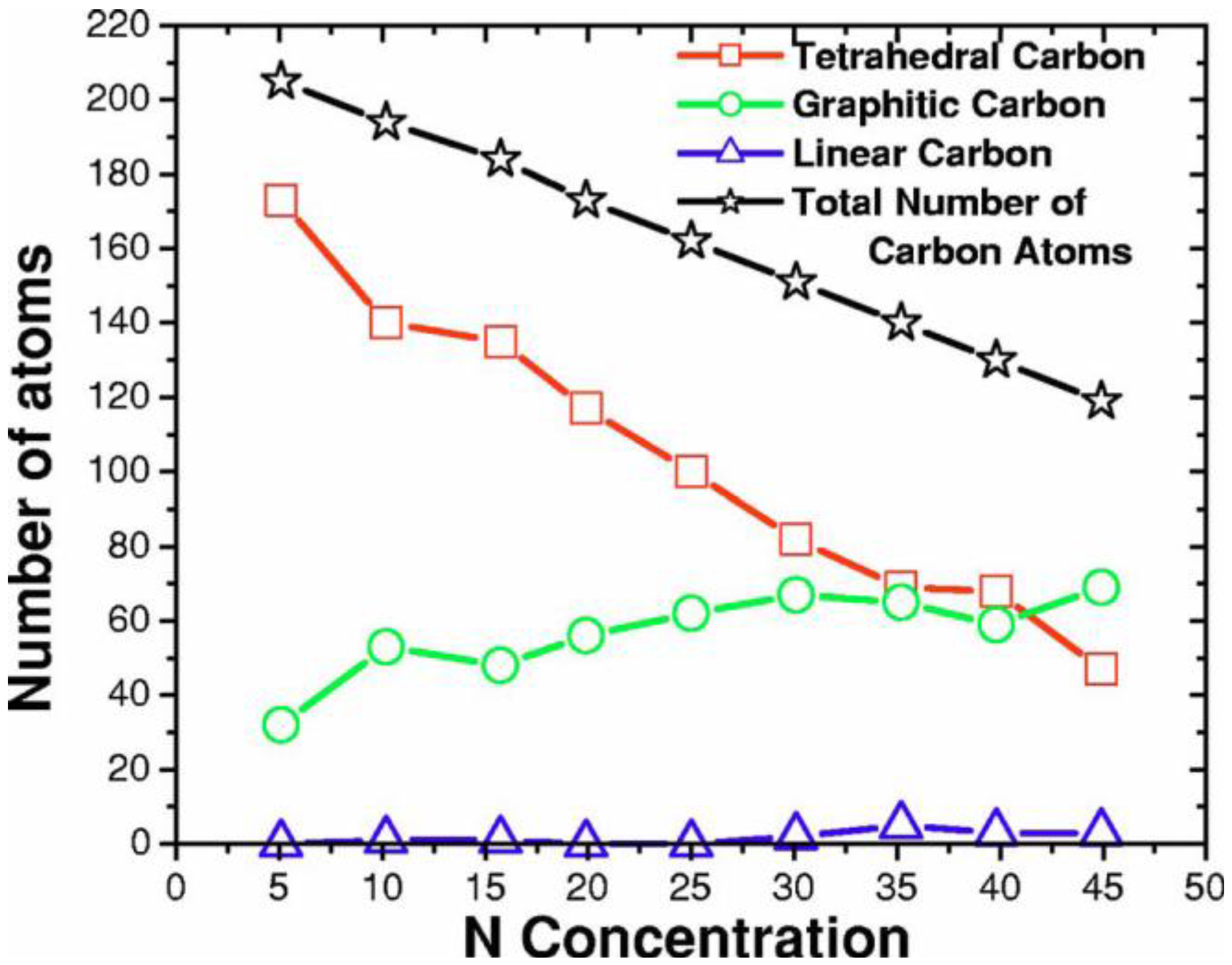

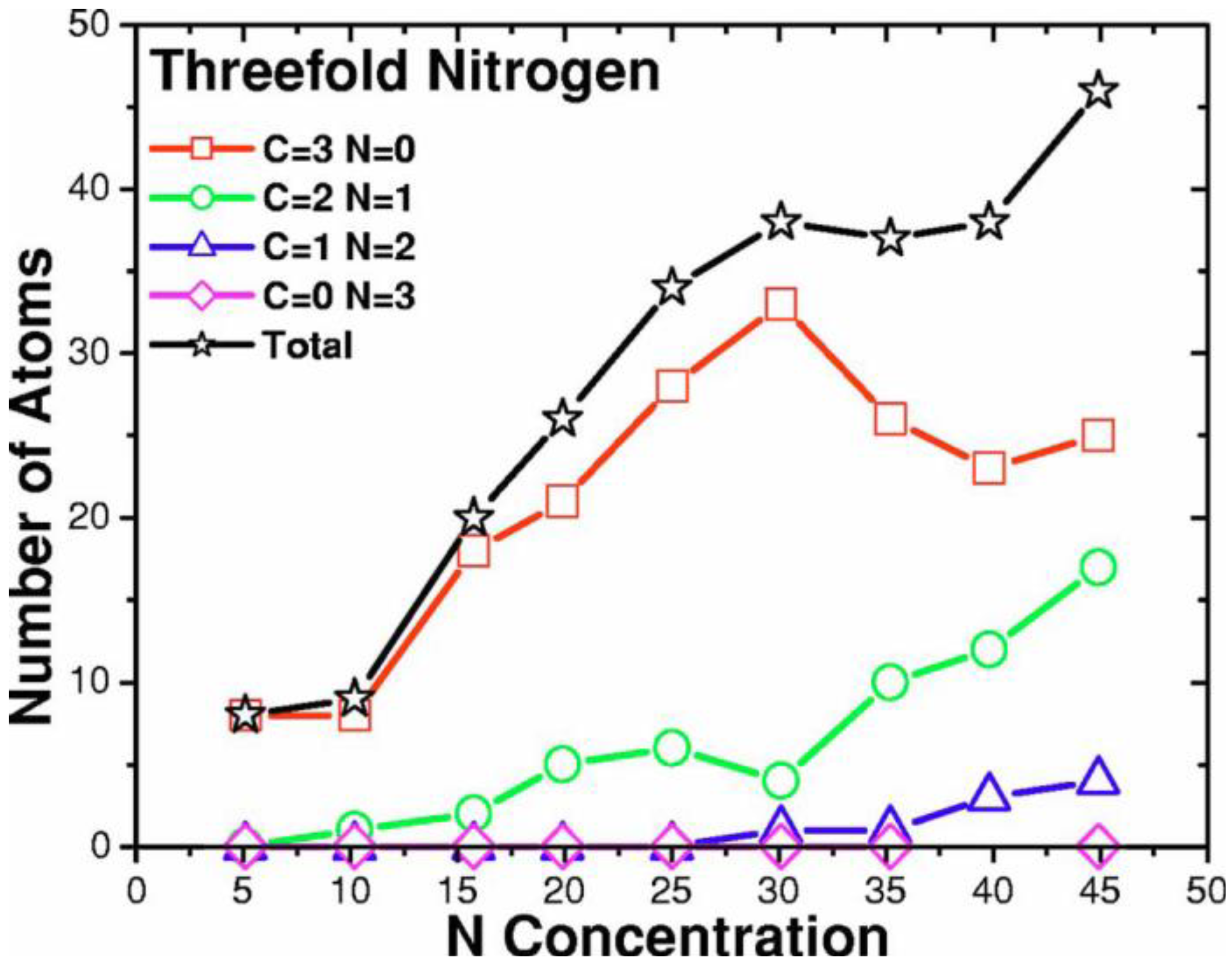
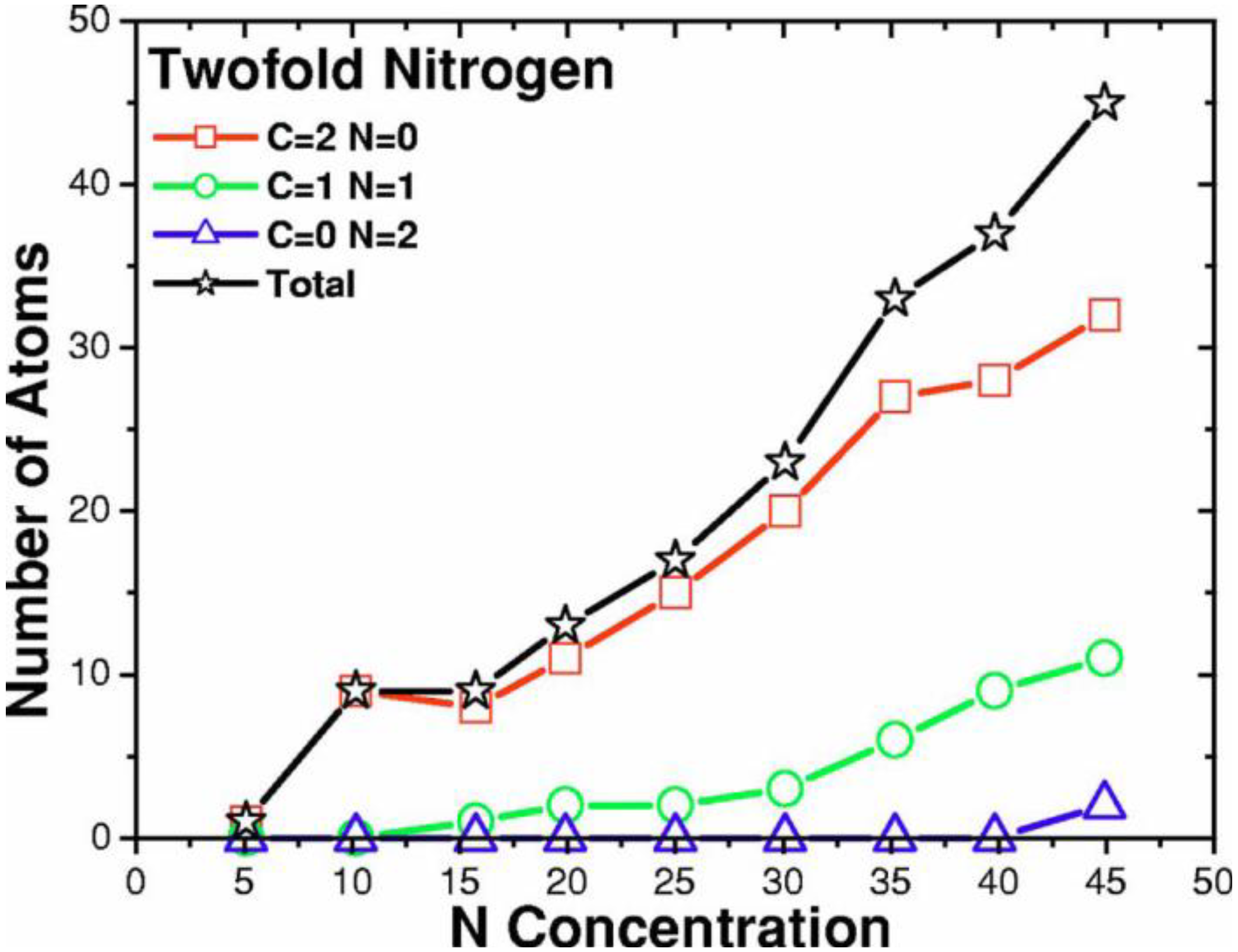
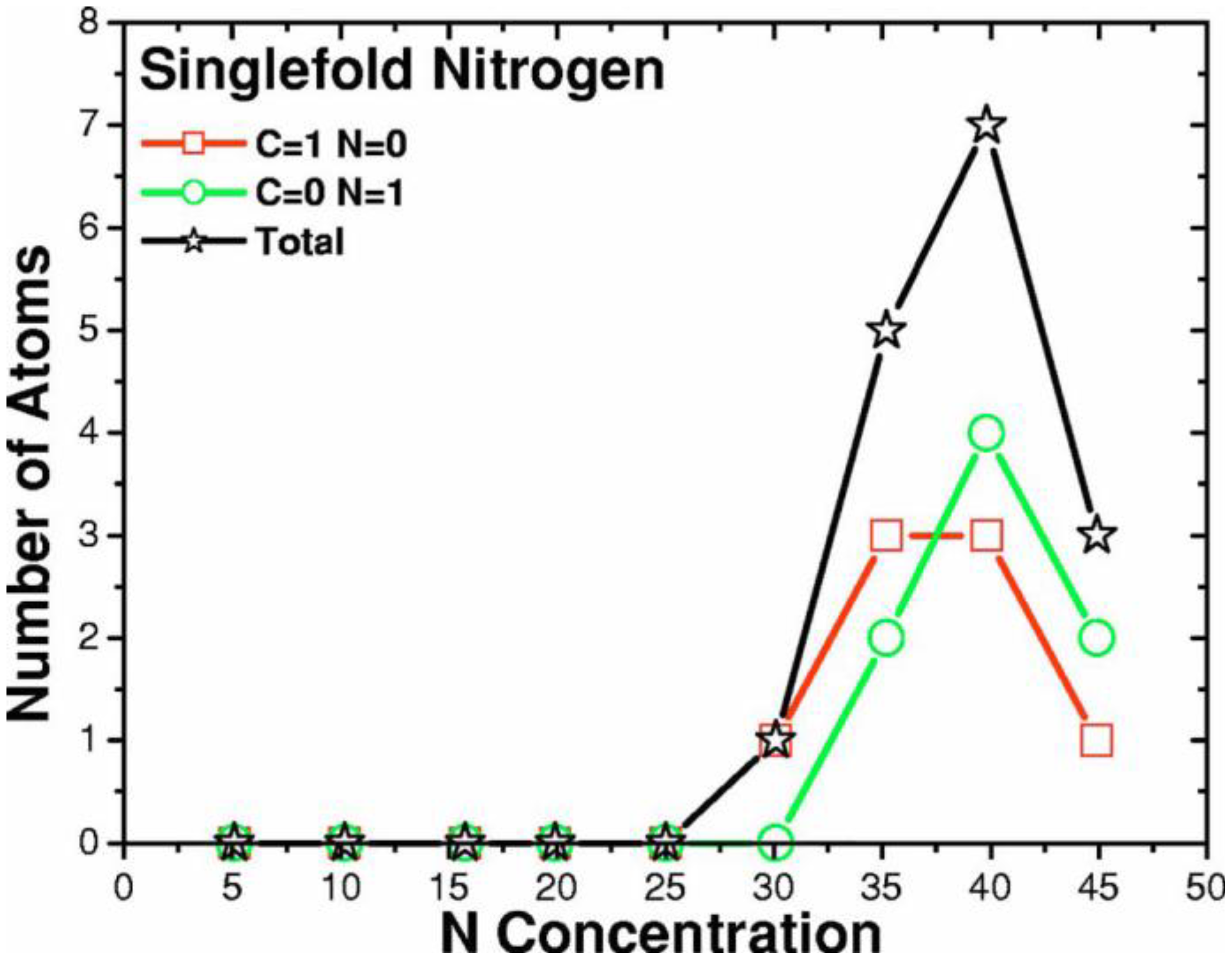
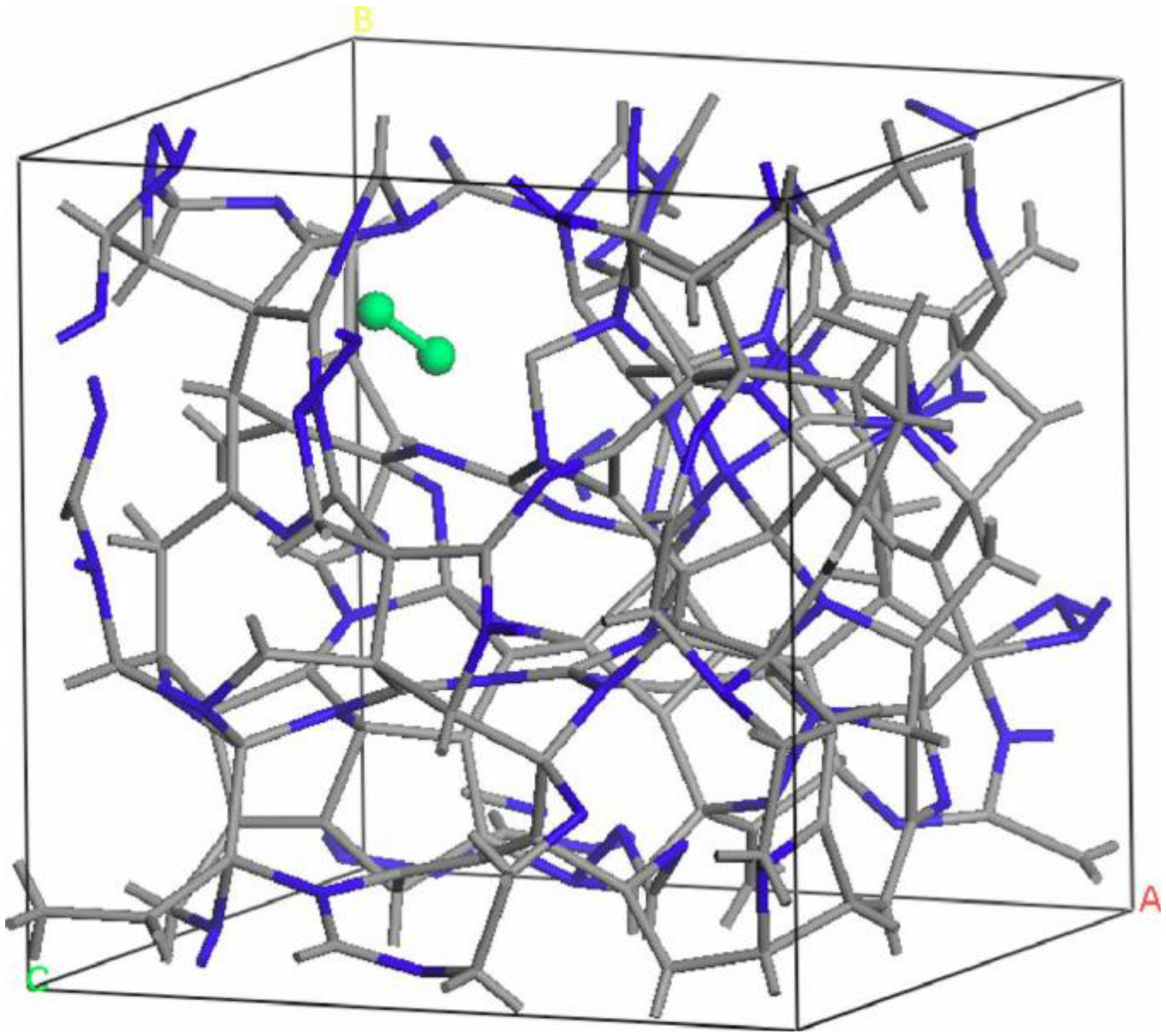
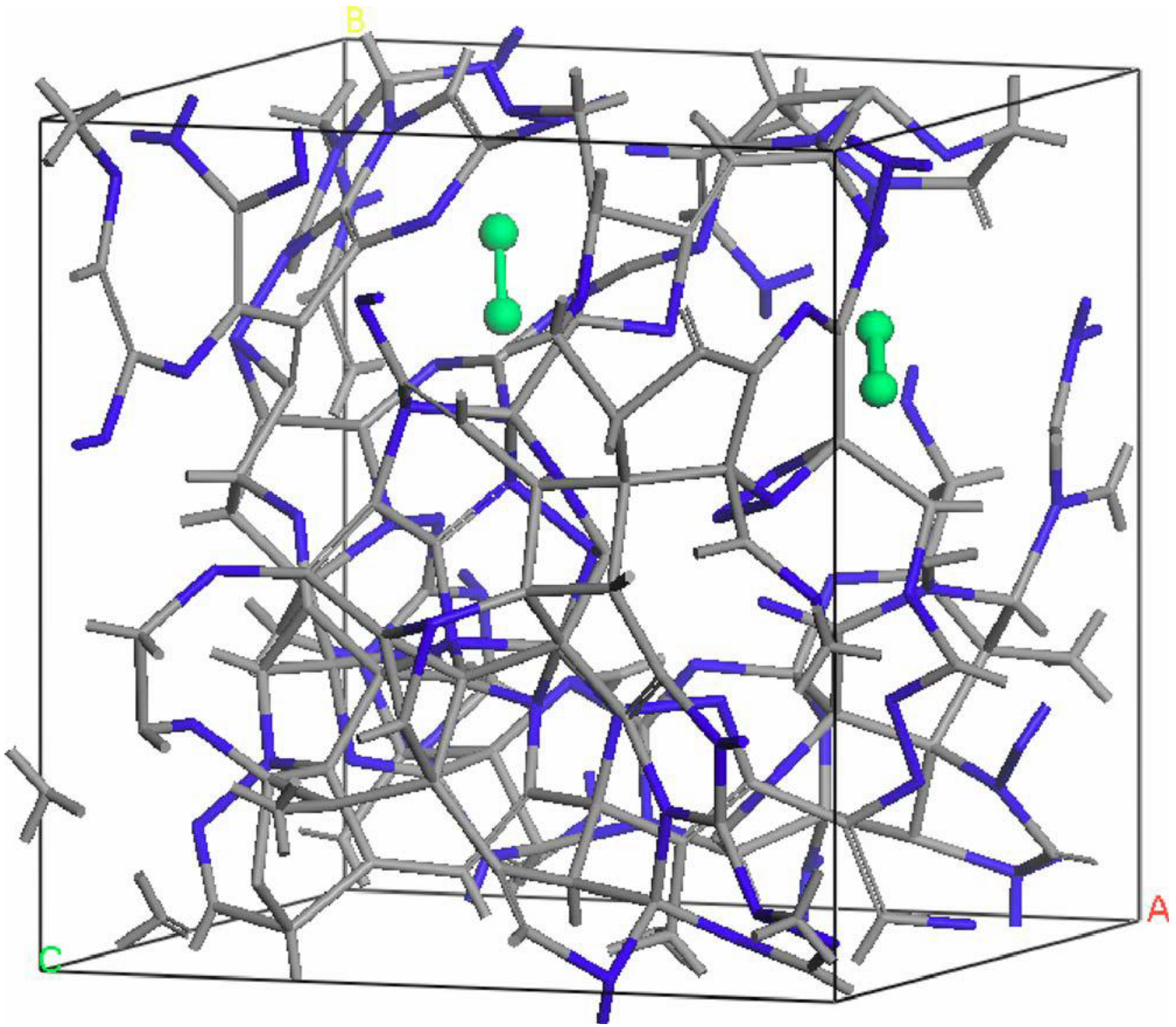
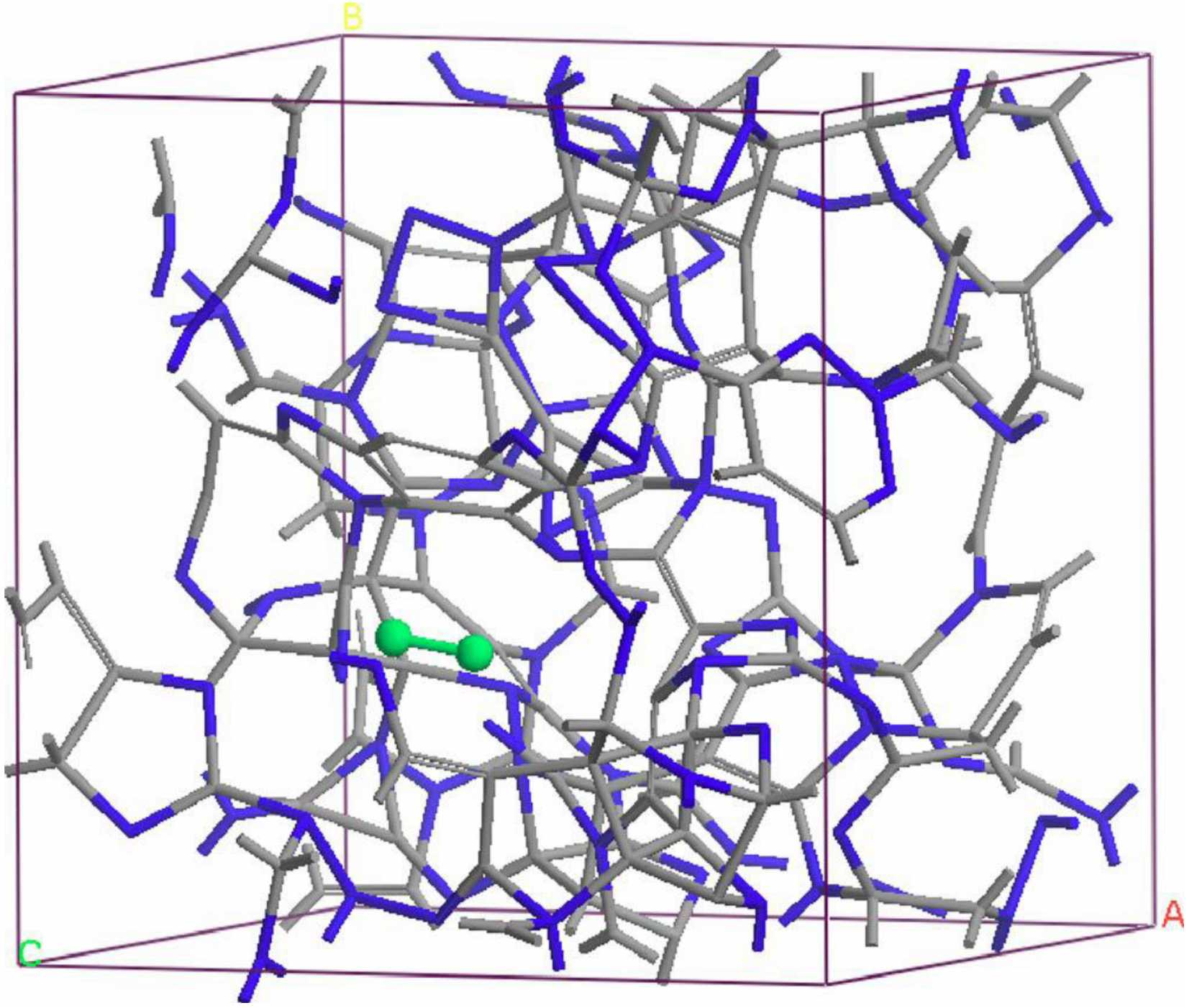
4.3.3. Summary
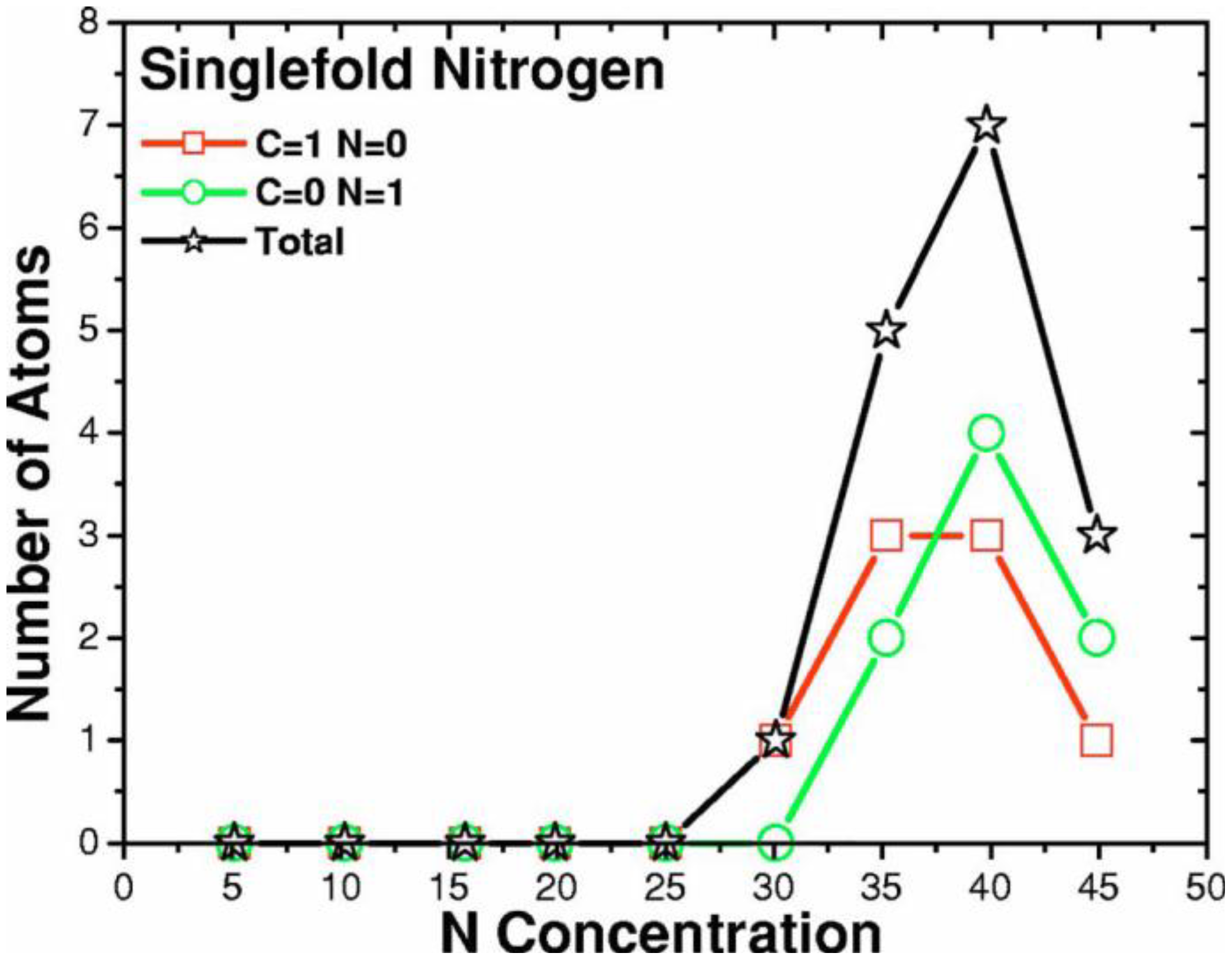
- (i)
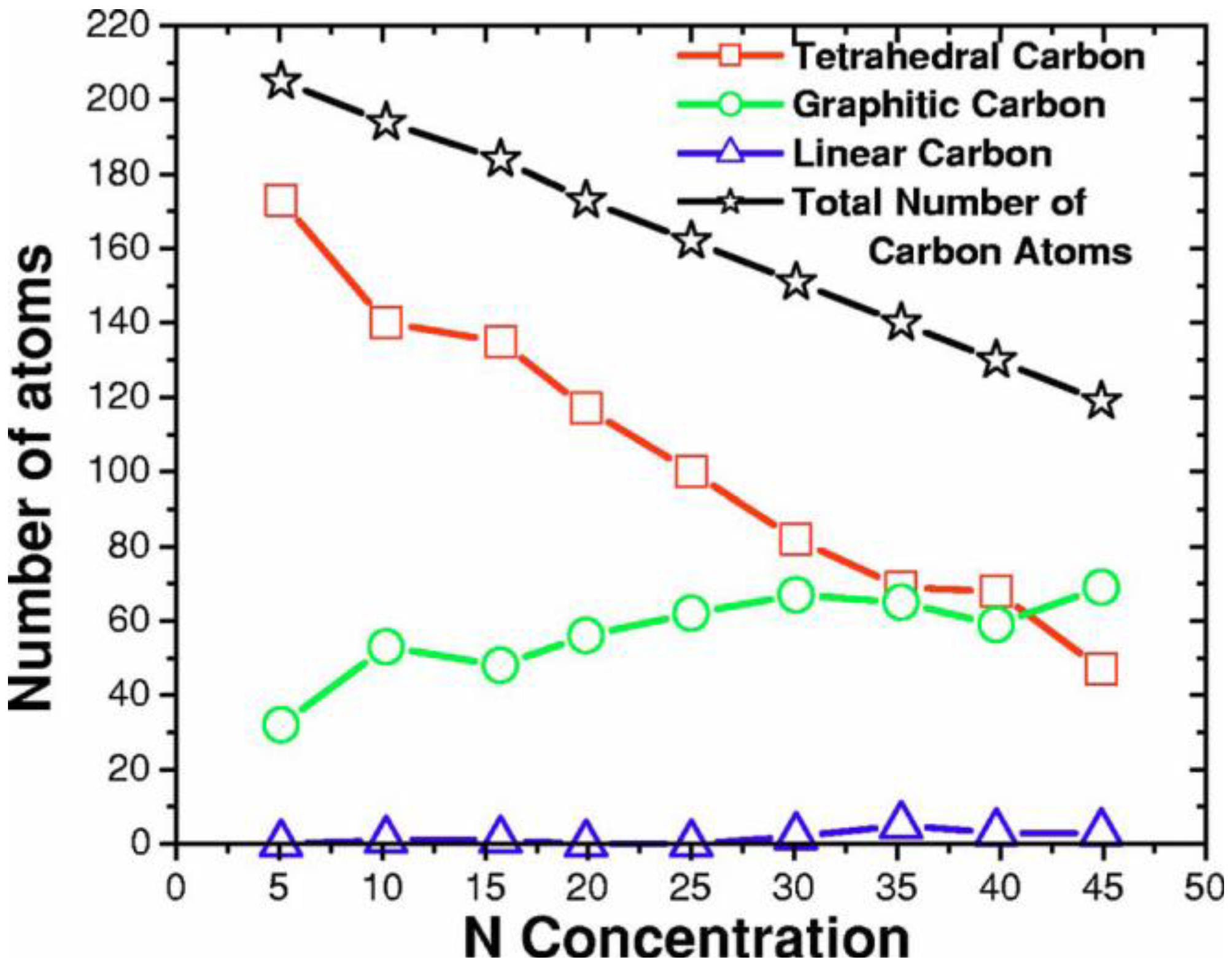
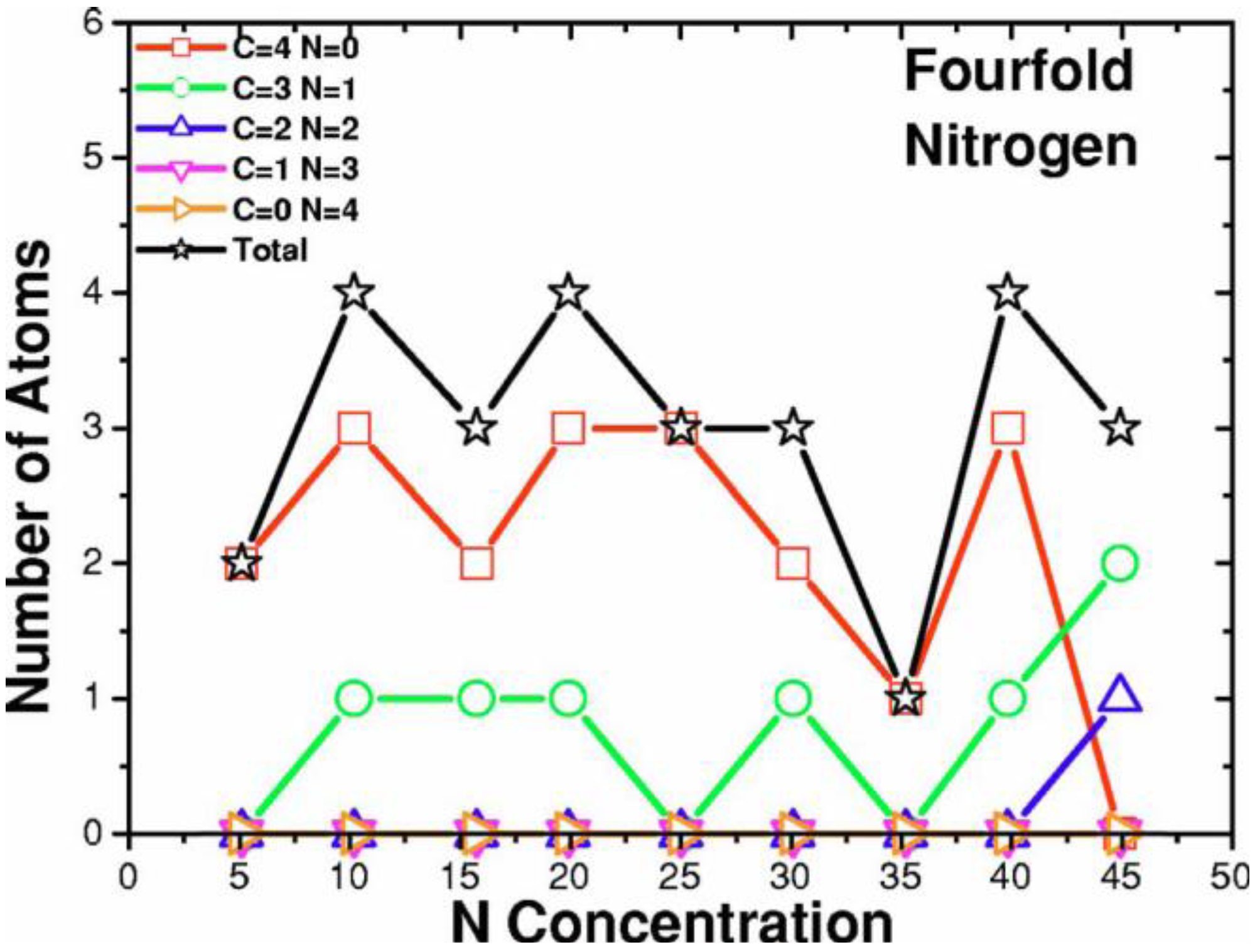
- Tetrahedral nitrogen (ta-N) appears in our ab initio generated CN samples. The amount of ta-N is essentially constant and small (Figure 14) for all concentrations studied and therefore becomes more relevant for concentrations below 5% where the number of other n-fold coordinated nitrogens, with n ≤ 3, is also small. At higher concentrations ta-N exists but its presence is masked by all the other bonding arrangements of nitrogen (Figure 15, Figure 16 and Figure 17). These results are in agreement with the EELS experiments reported in the literature.
- (i)
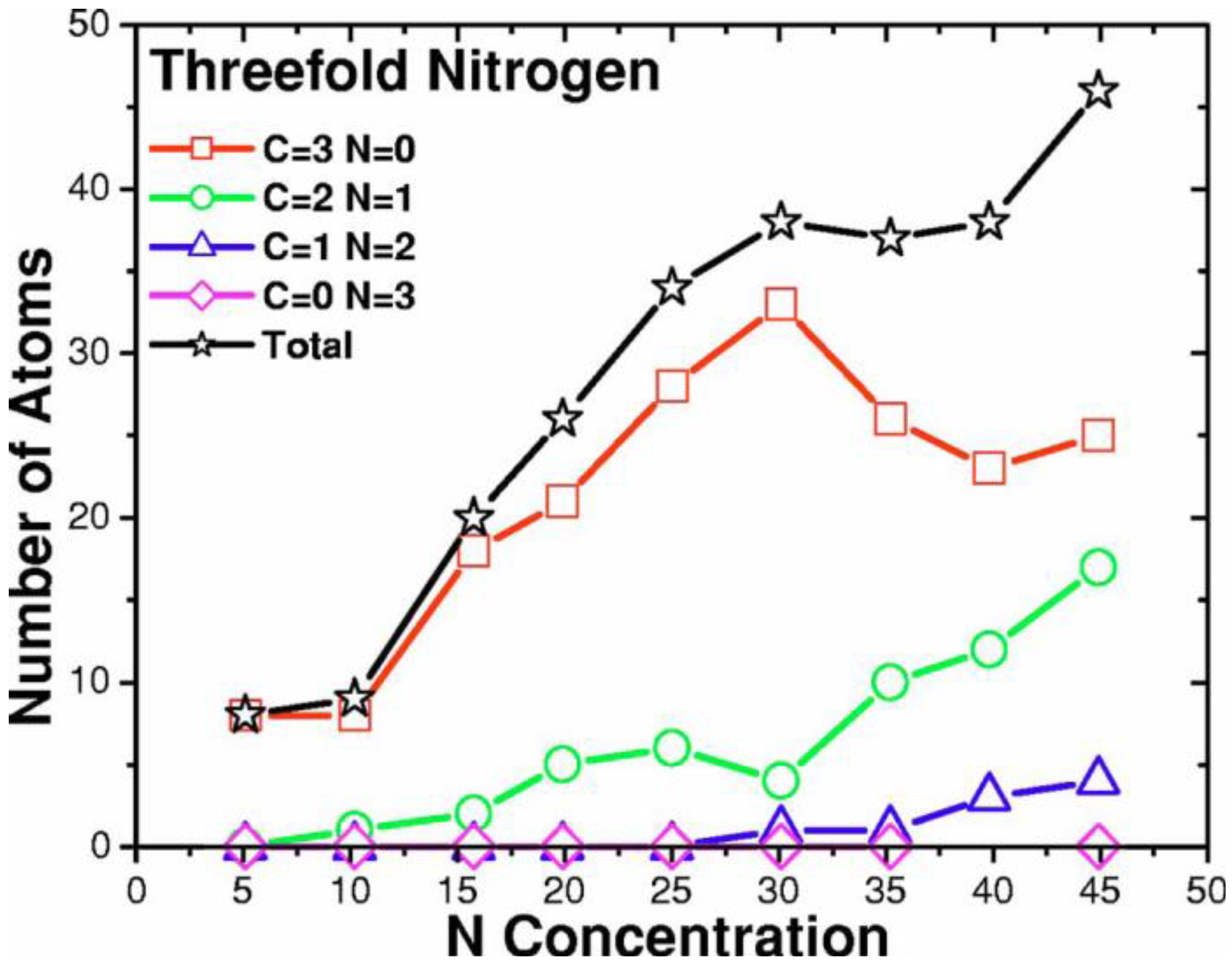
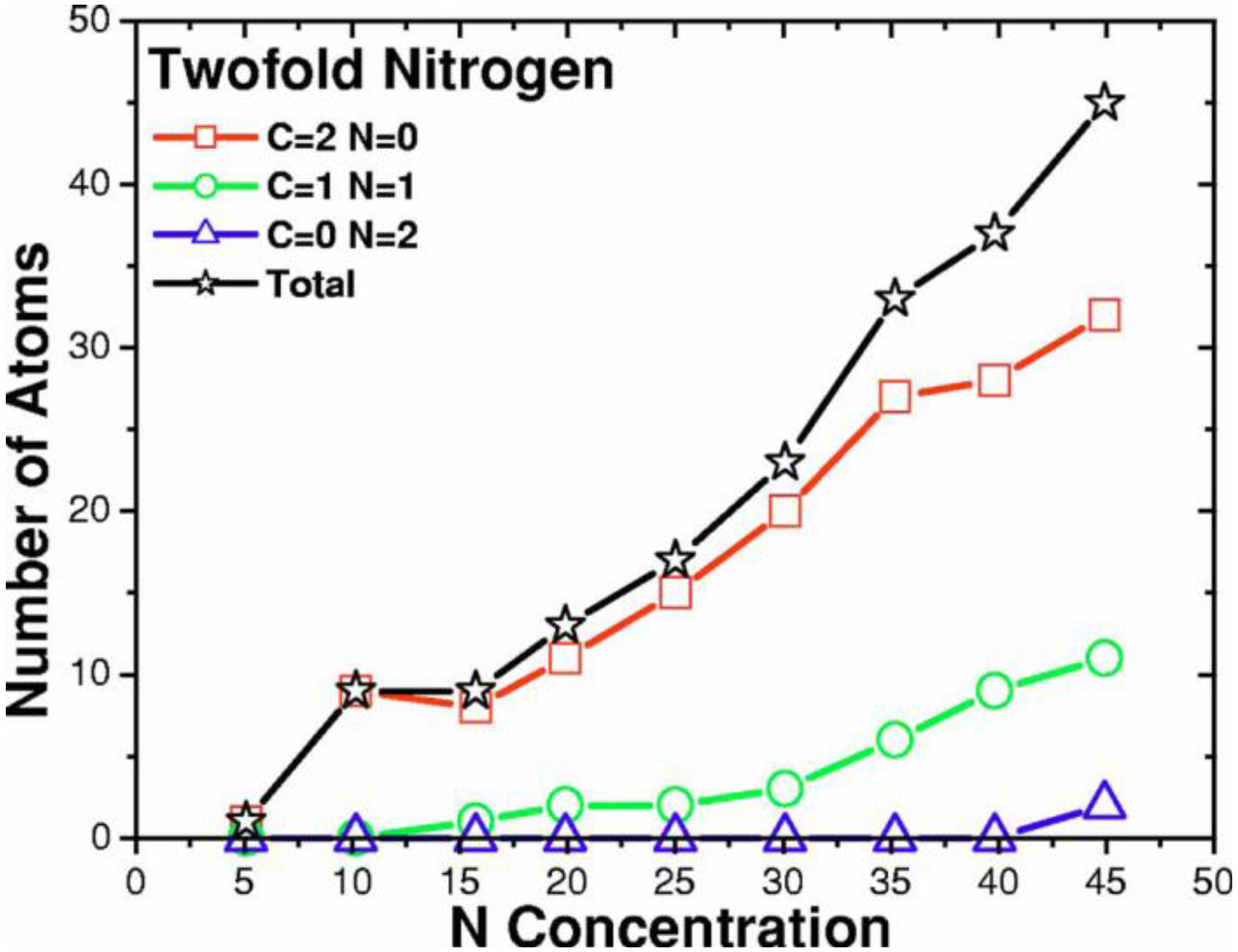
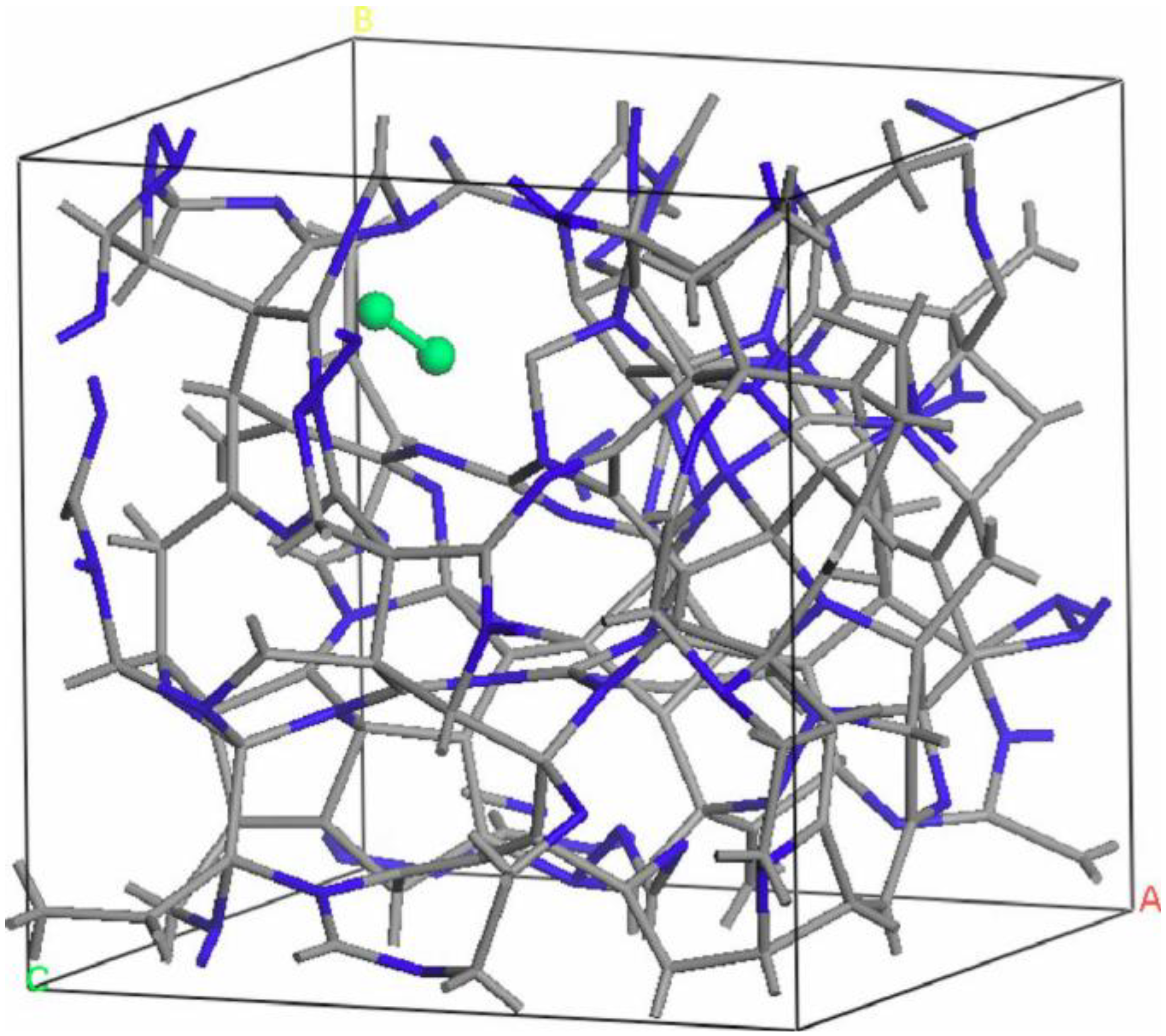
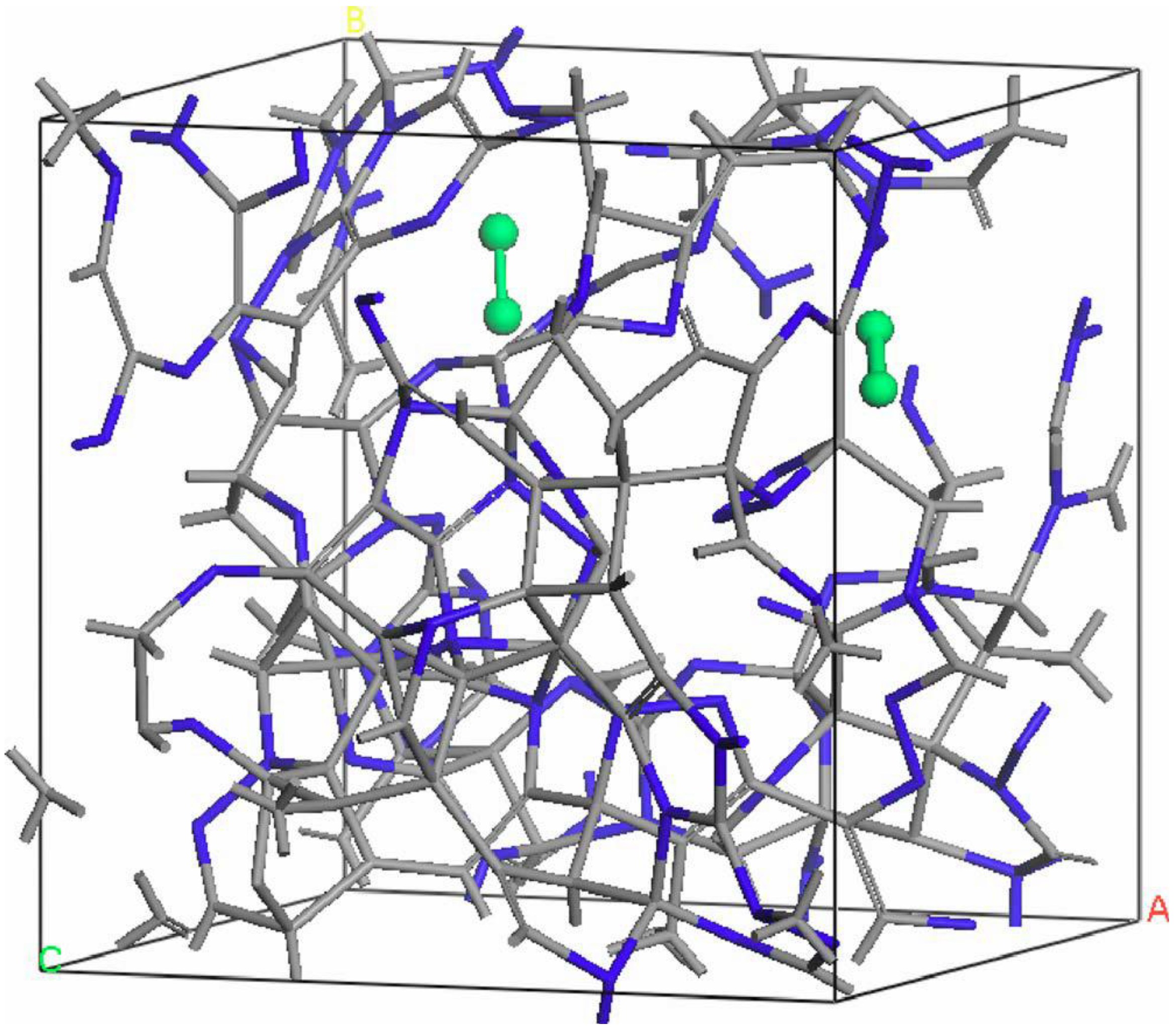
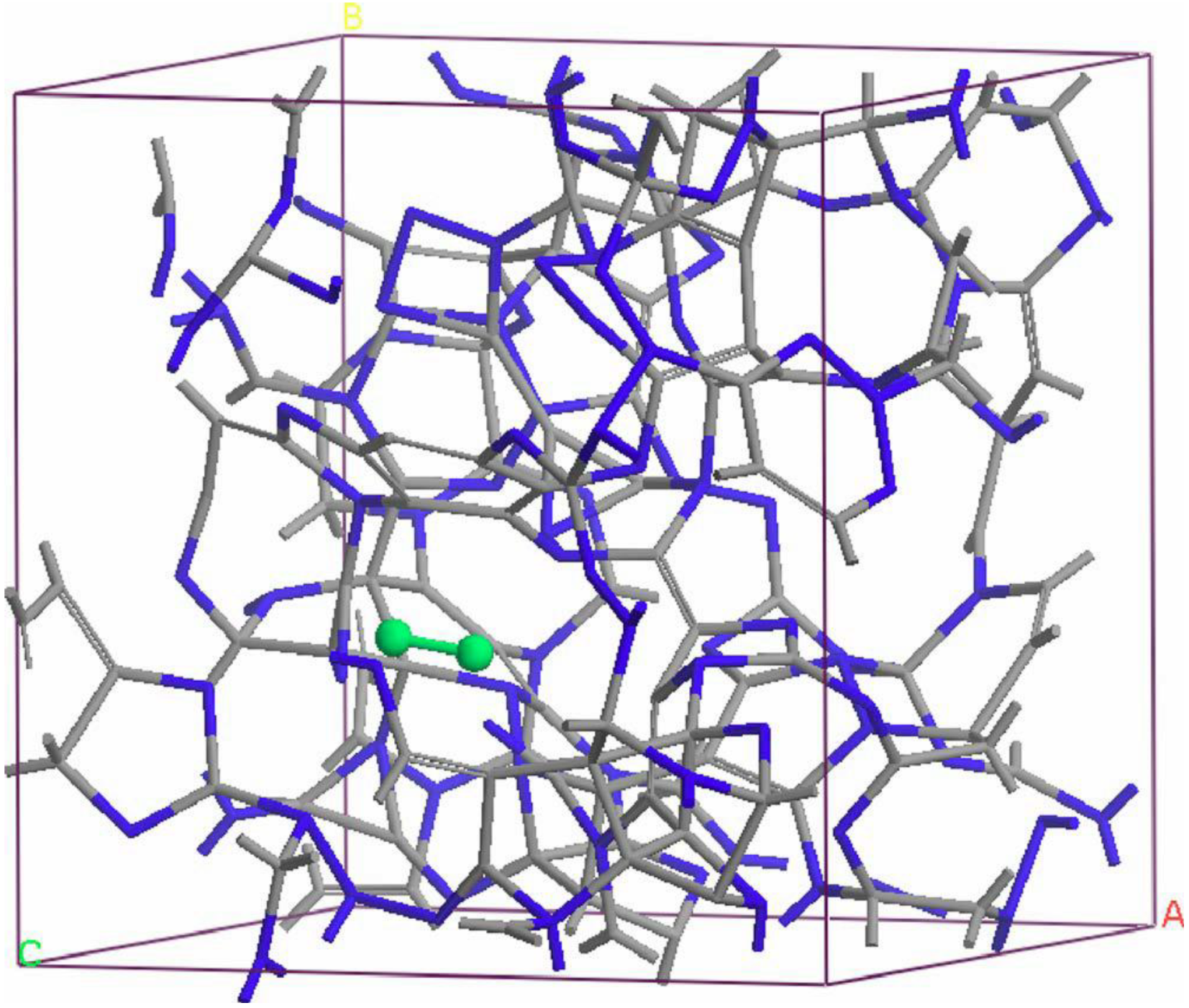
- For concentrations higher that 30%, single-fold nitrogen appears bonded to nitrogen, giving rise to N2 molecules (Figure 18, Figure 19 and Figure 20). We take this as signaling the onset of an upper limit for the incorporation of N in an amorphous carbon matrix, in agreement with experiment and supporting the surmise that the experimental saturation of the nitrogen content within the films is due to the formation of molecular nitrogen either at or below the film surface. Another indication that something drastic happens at 30% is that the three-fold coordination of nitrogen with three carbons starts diminishing abruptly, Figure 15.
4.4. Amorphous Binary Alloys Based on Group IV Elements: The Case of a-Si0.5C0.5 [85,86]
4.4.1. Preamble
4.4.2. Results and Analysis

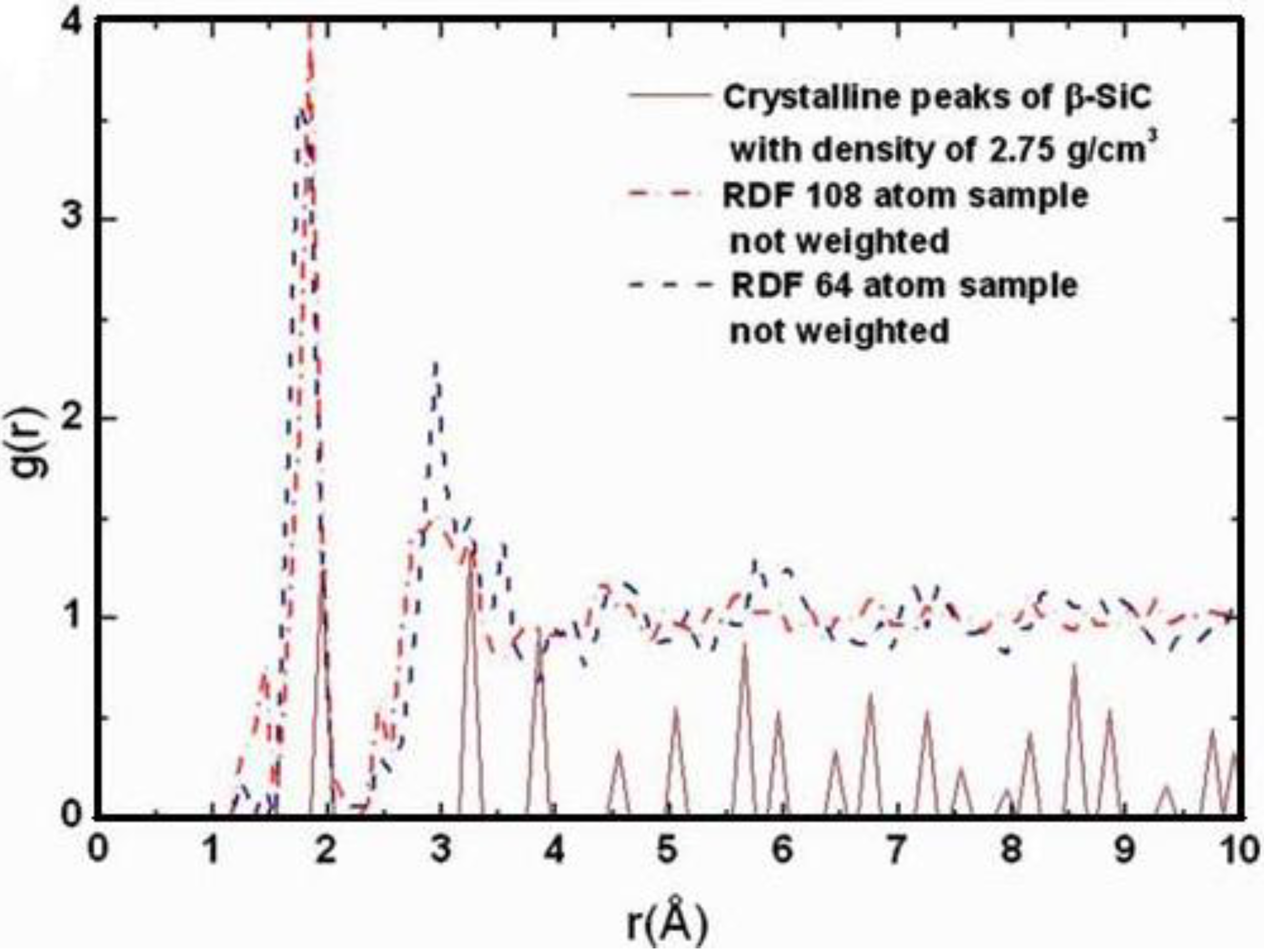
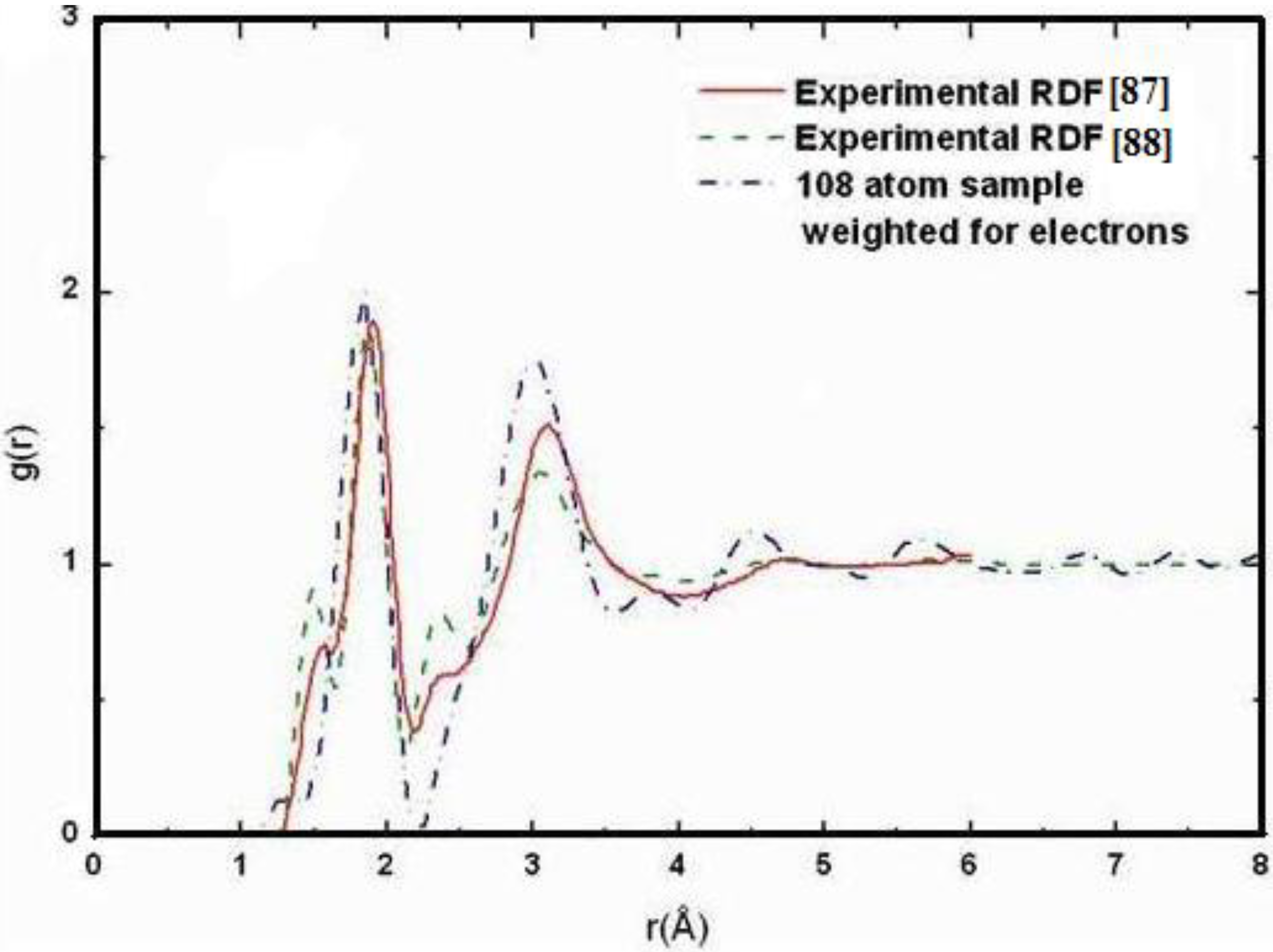
4.4.3. Summary
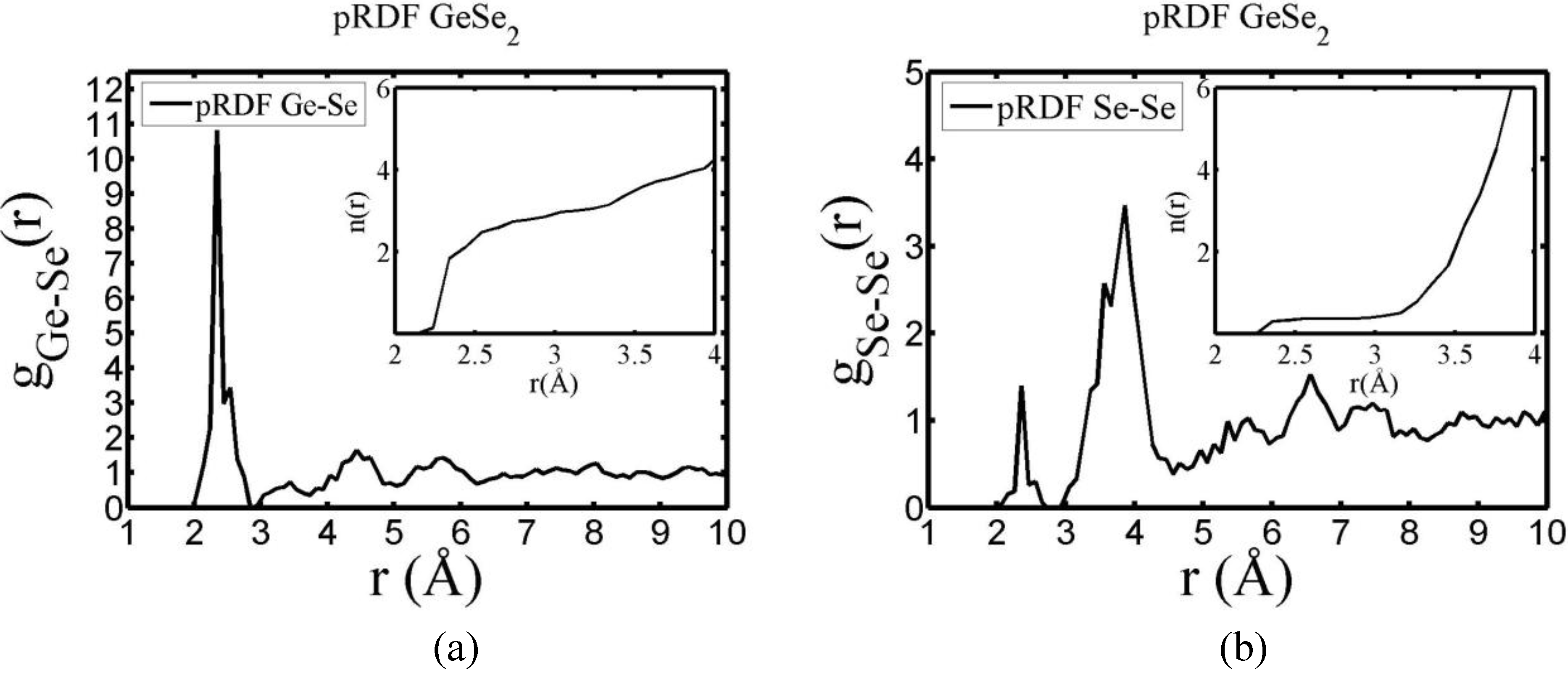
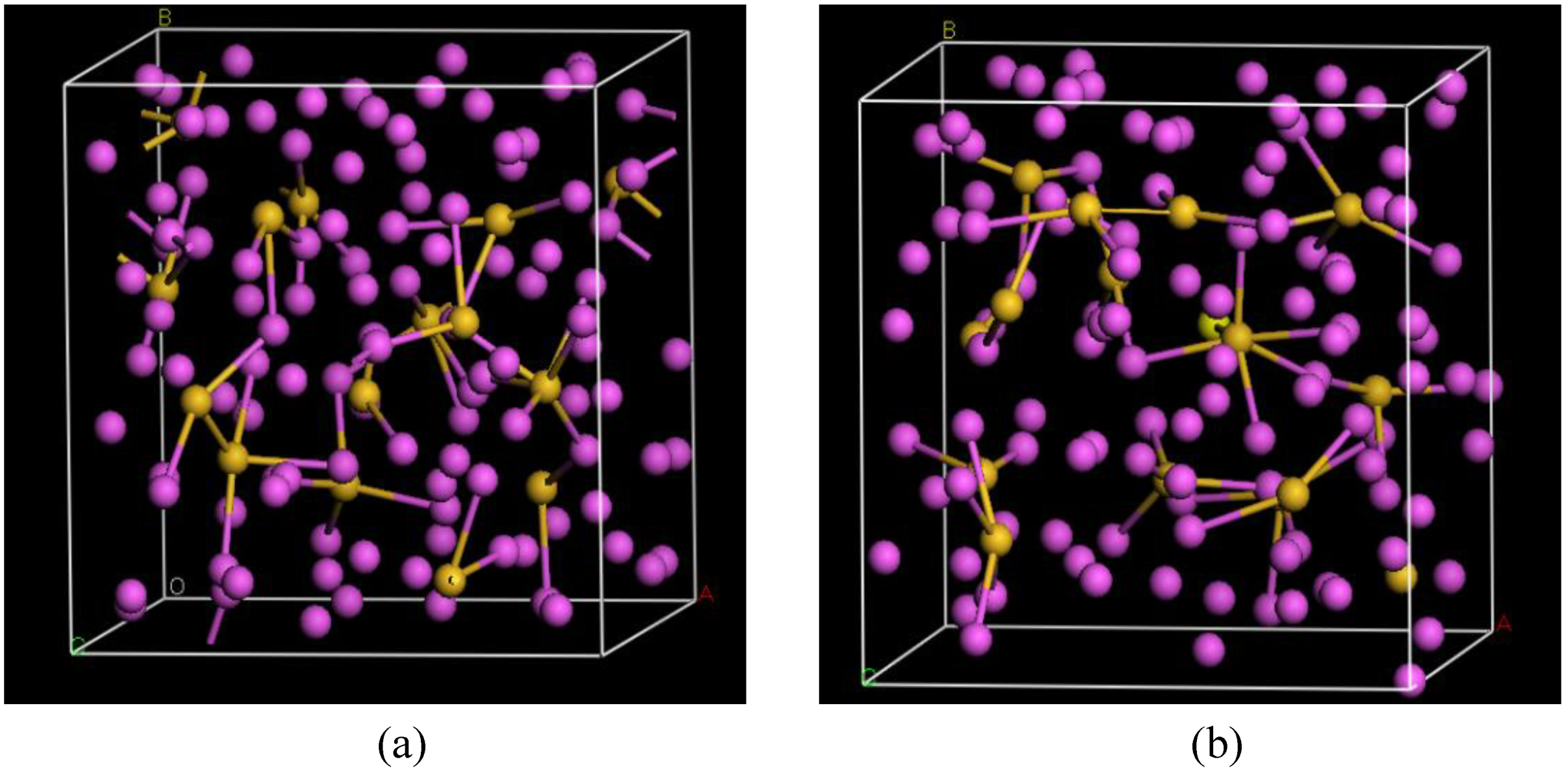
4.5. Amorphous Chalcogenide Alloys: The Case of a-GeSe2 [91,92]
4.5.1. Preamble
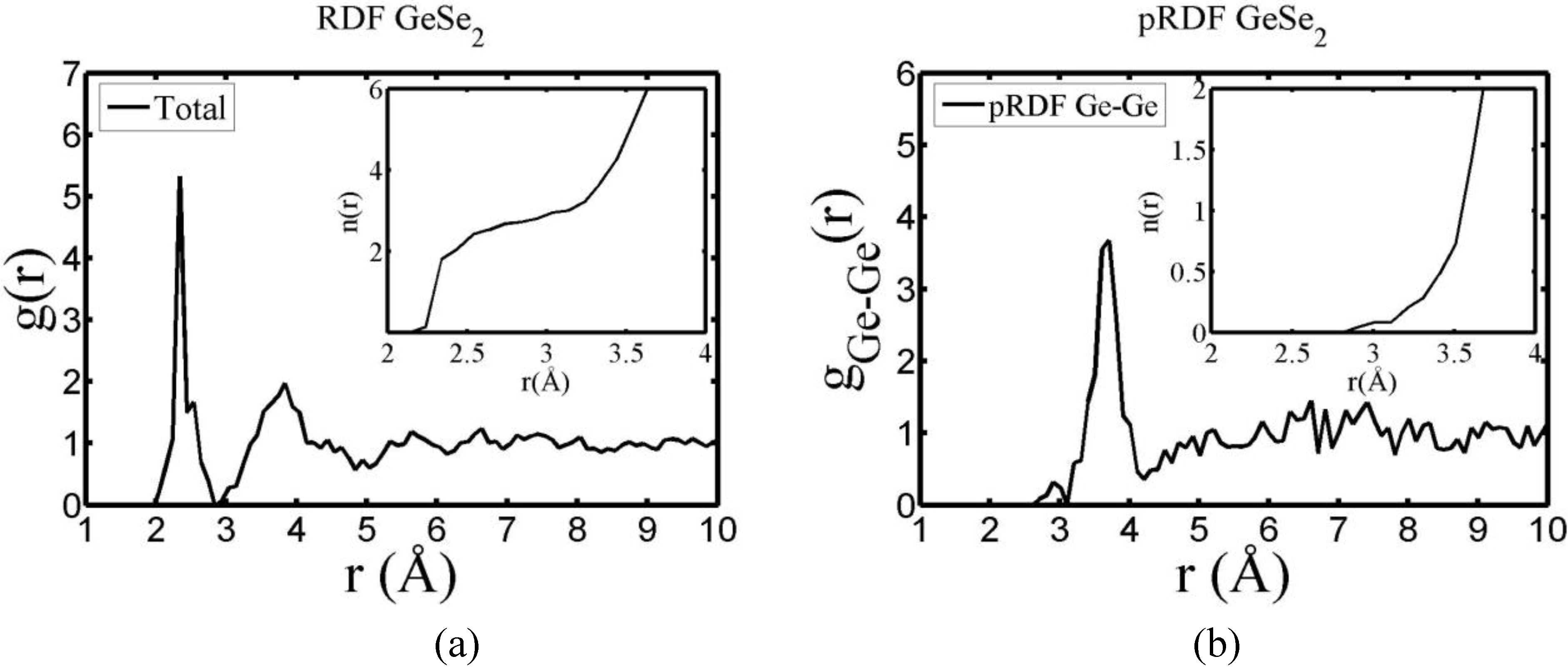
4.5.2. Results and Analysis
| Partial | This work | Massobrio et al. | Cobb et al. | Salmon and Petri |
|---|---|---|---|---|
| gGeGe | 2.91 Å | 2.50 Å | 2.46 Å | 2.42 Å |
| gGeSe | 2.34 Å | 2.37 Å | 2.37 Å | 2.36 Å |
| gSeSe | 2.36 Å | 2.37 Å | 2.40 Å | 2.37 Å |
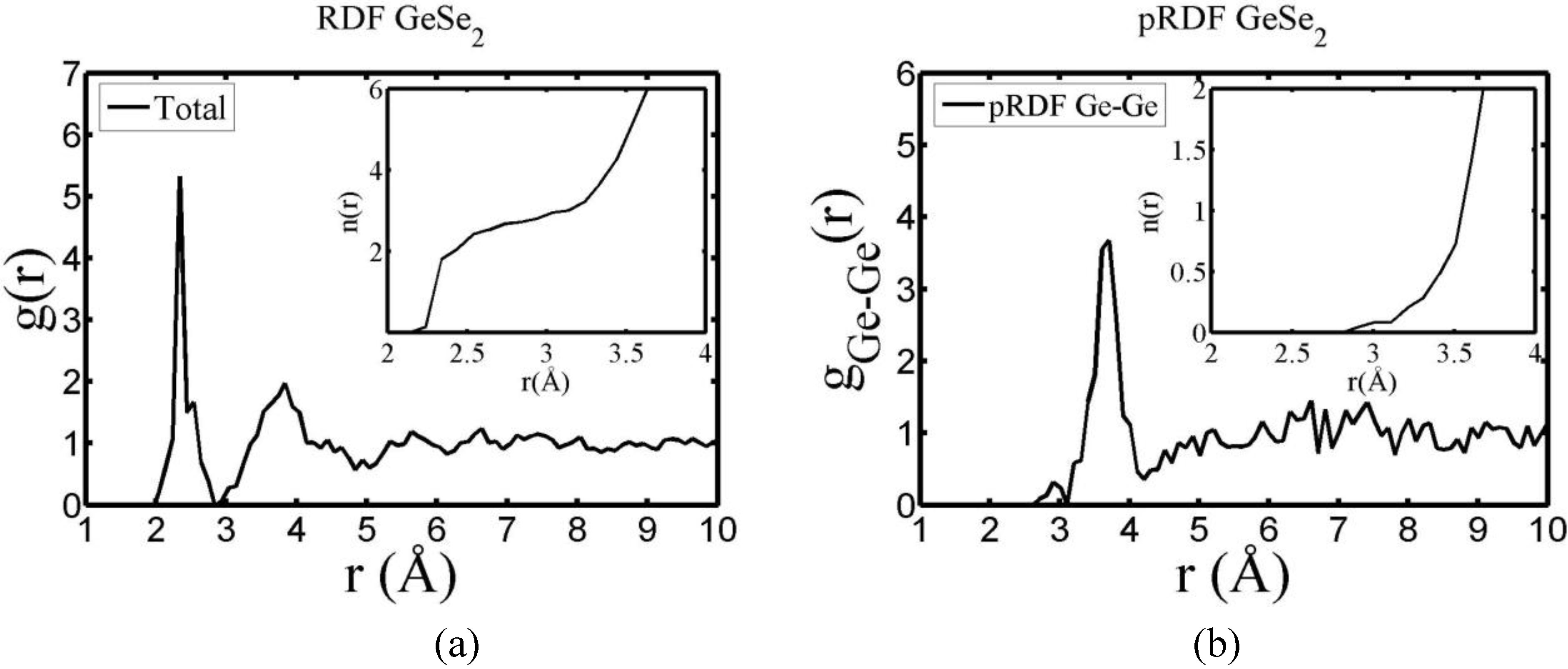
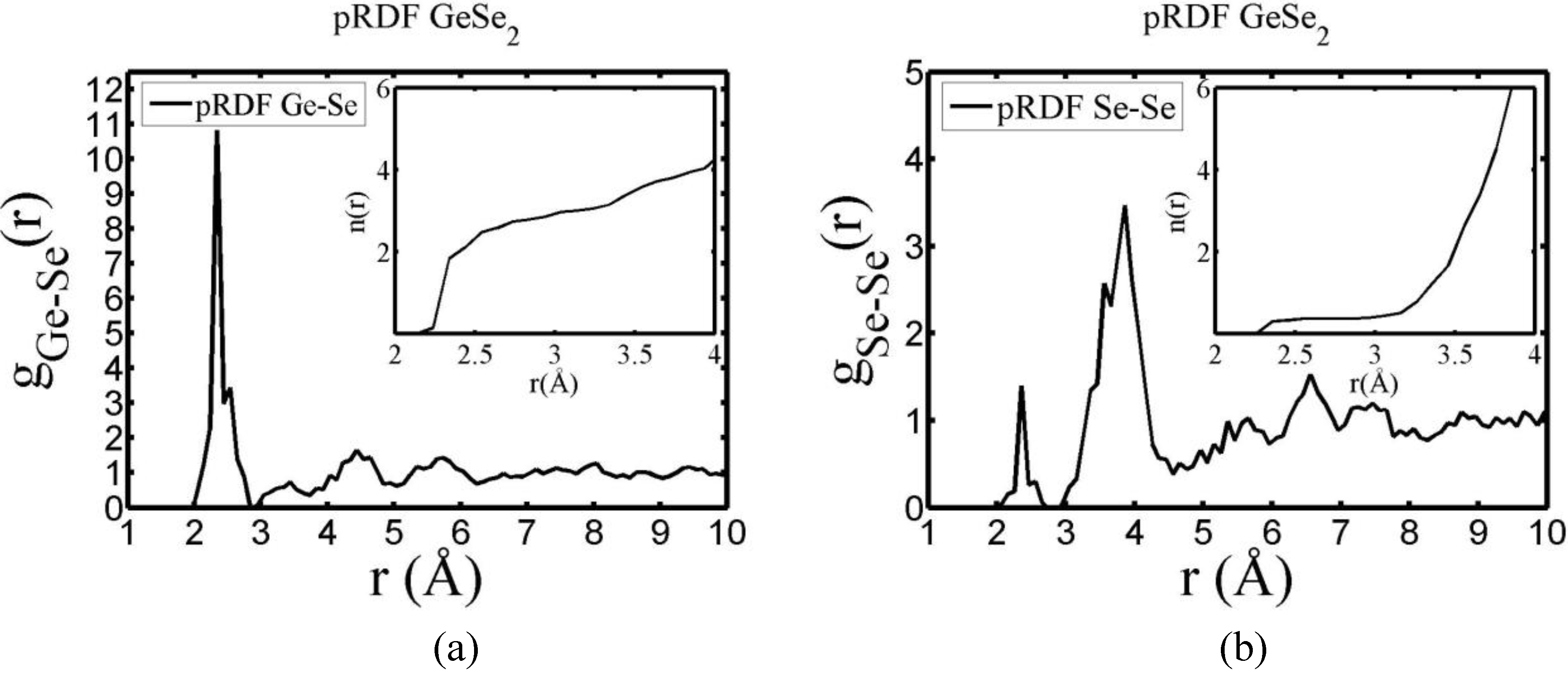
4.5.3. Summary
4.6. Aluminum-based Amorphous Alloys: AlN [96] and Al12at%Si [97,98,99,100]
4.6.1. Preamble
4.6.2. Results and Analysis
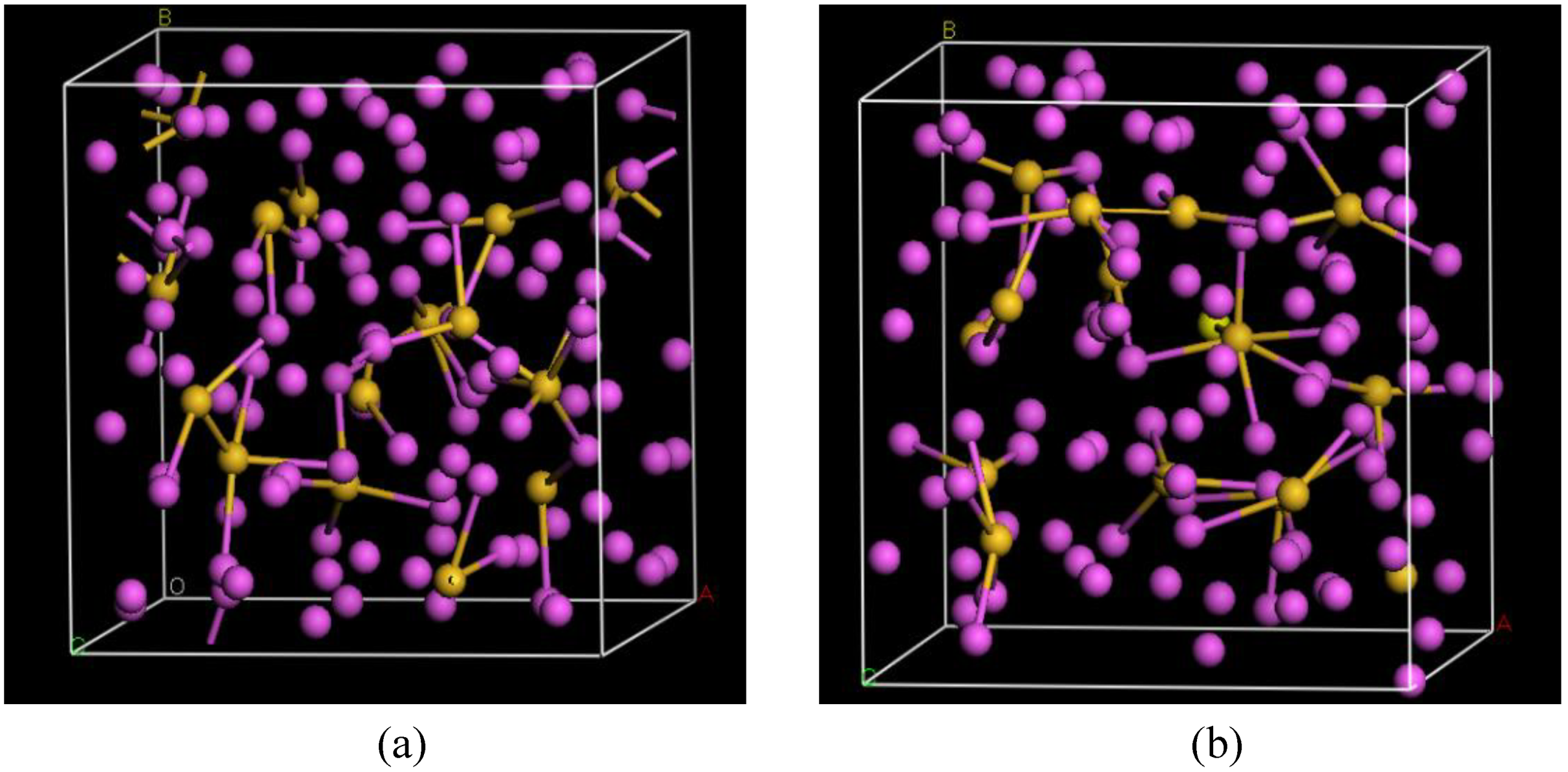
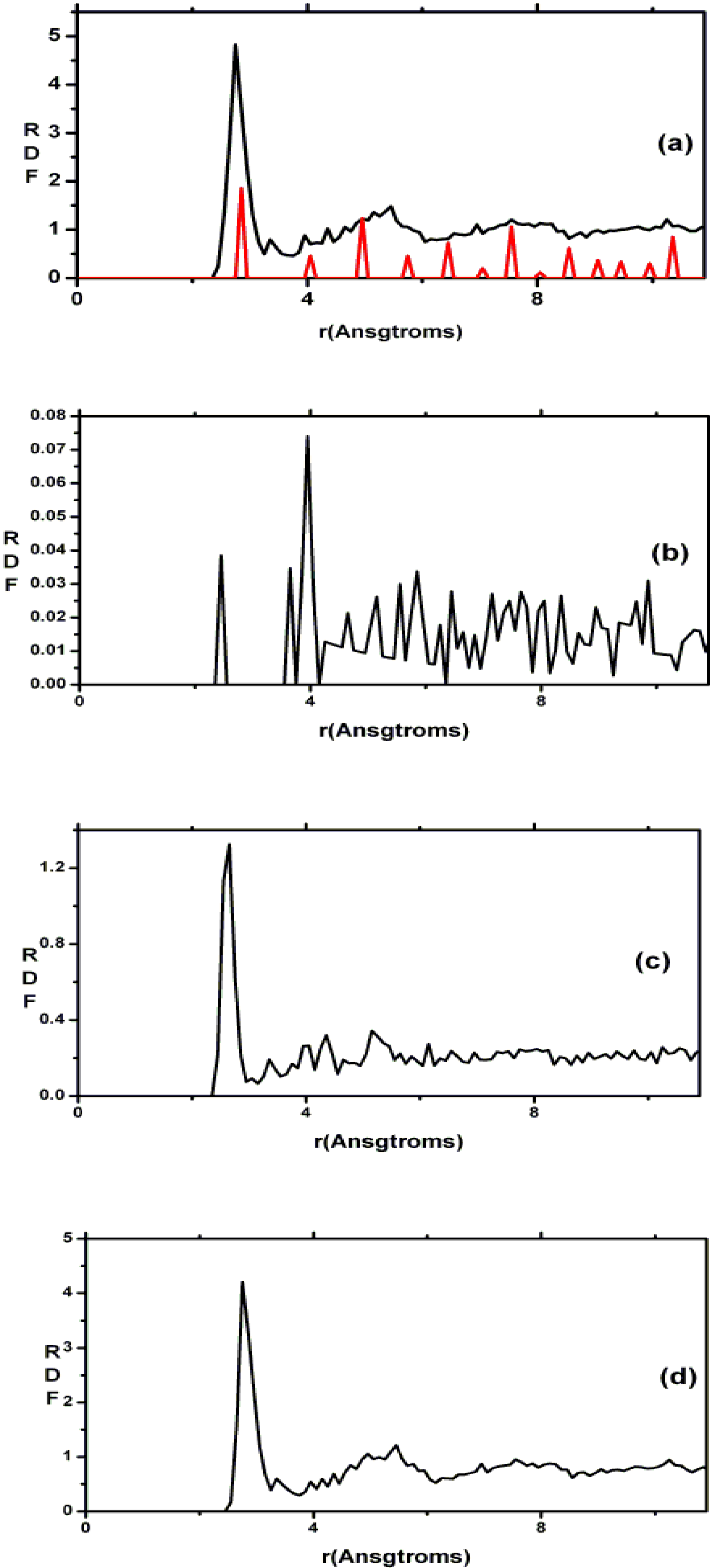
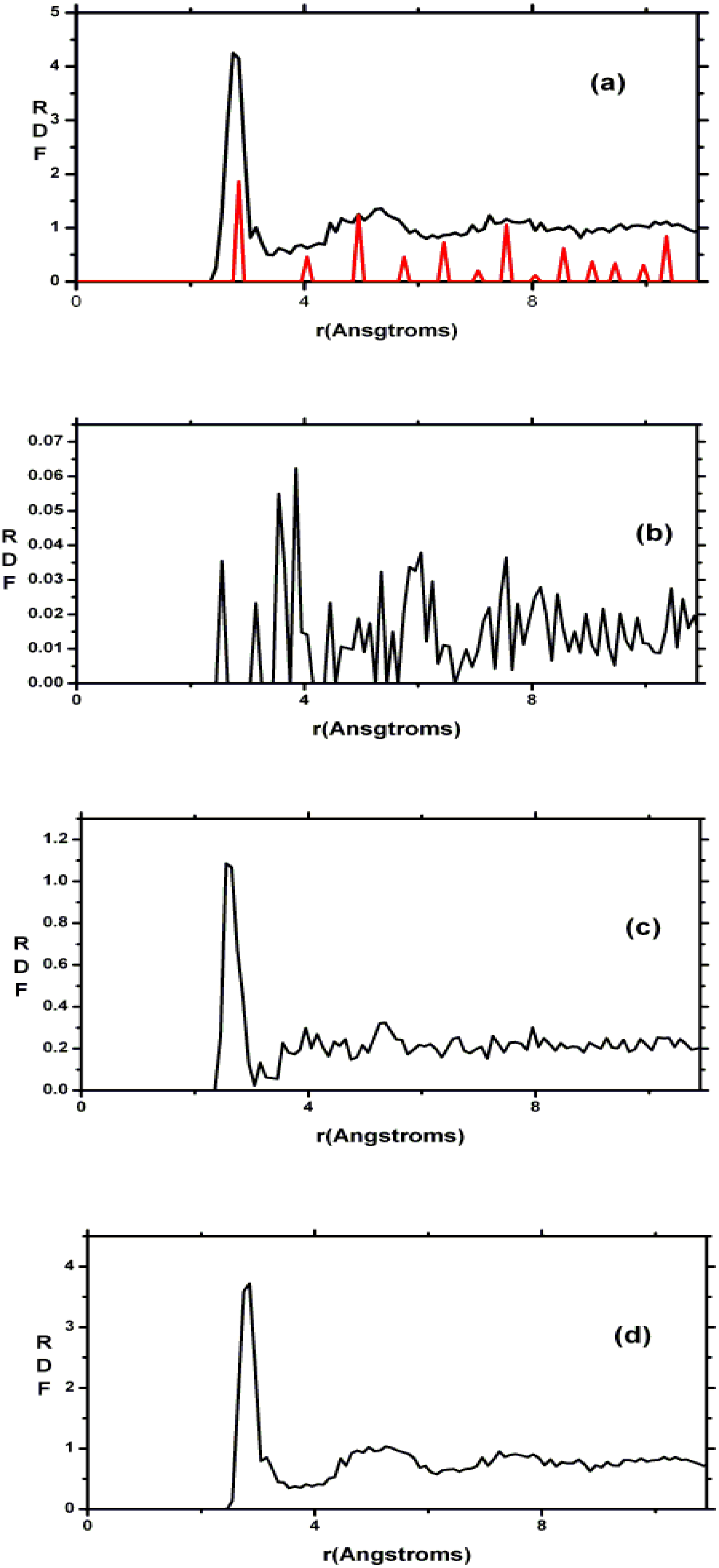
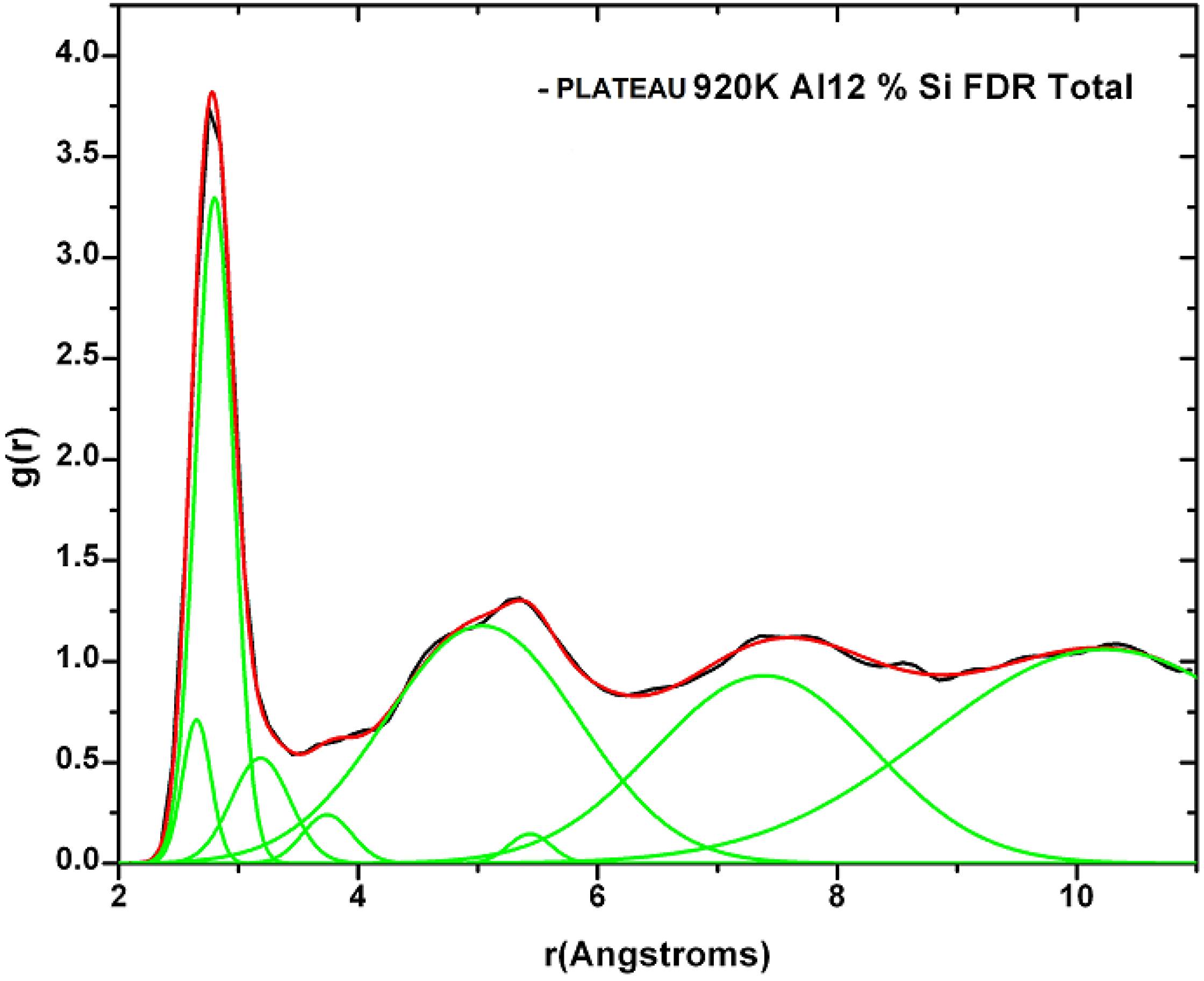
4.6.3. Summary
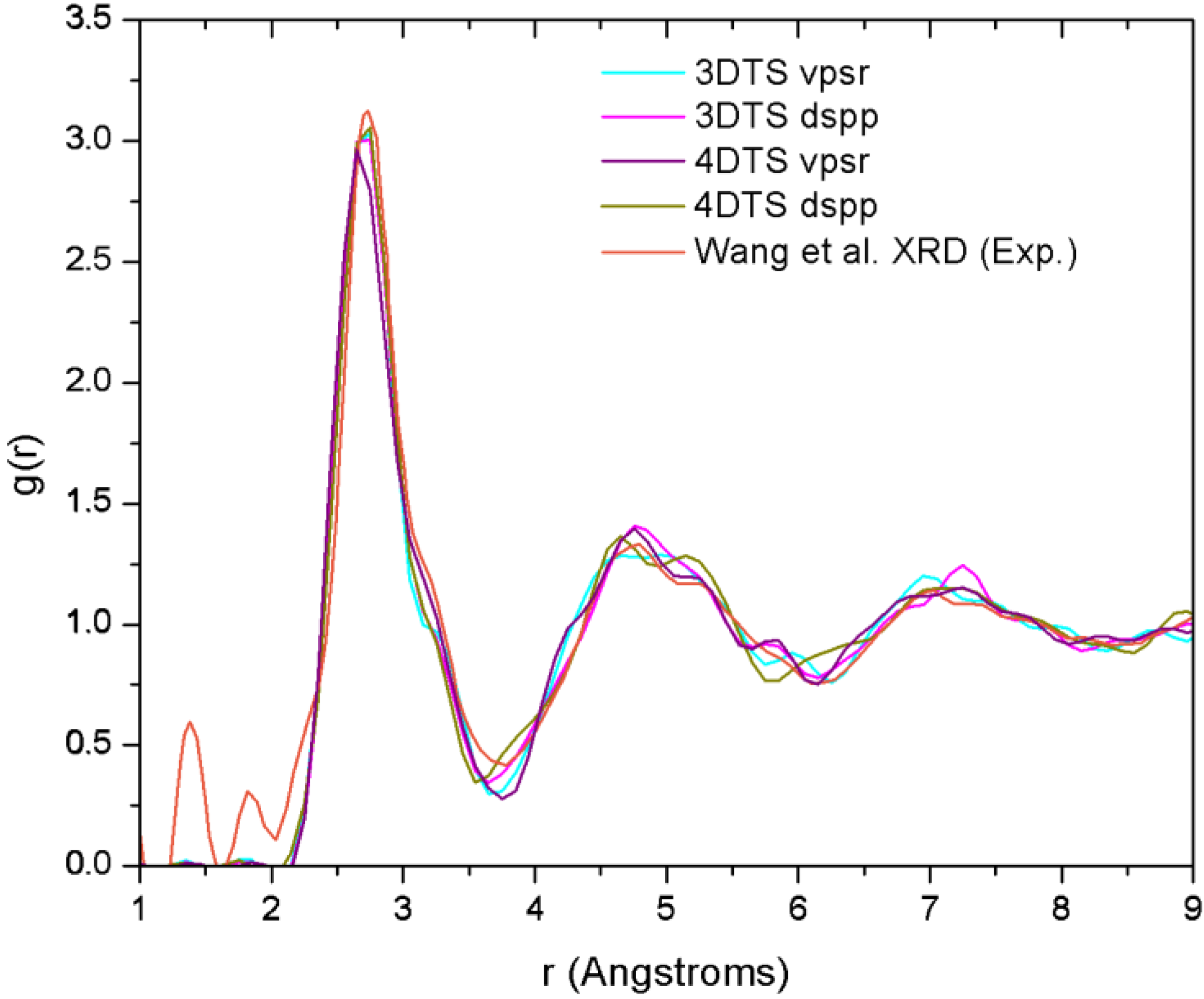
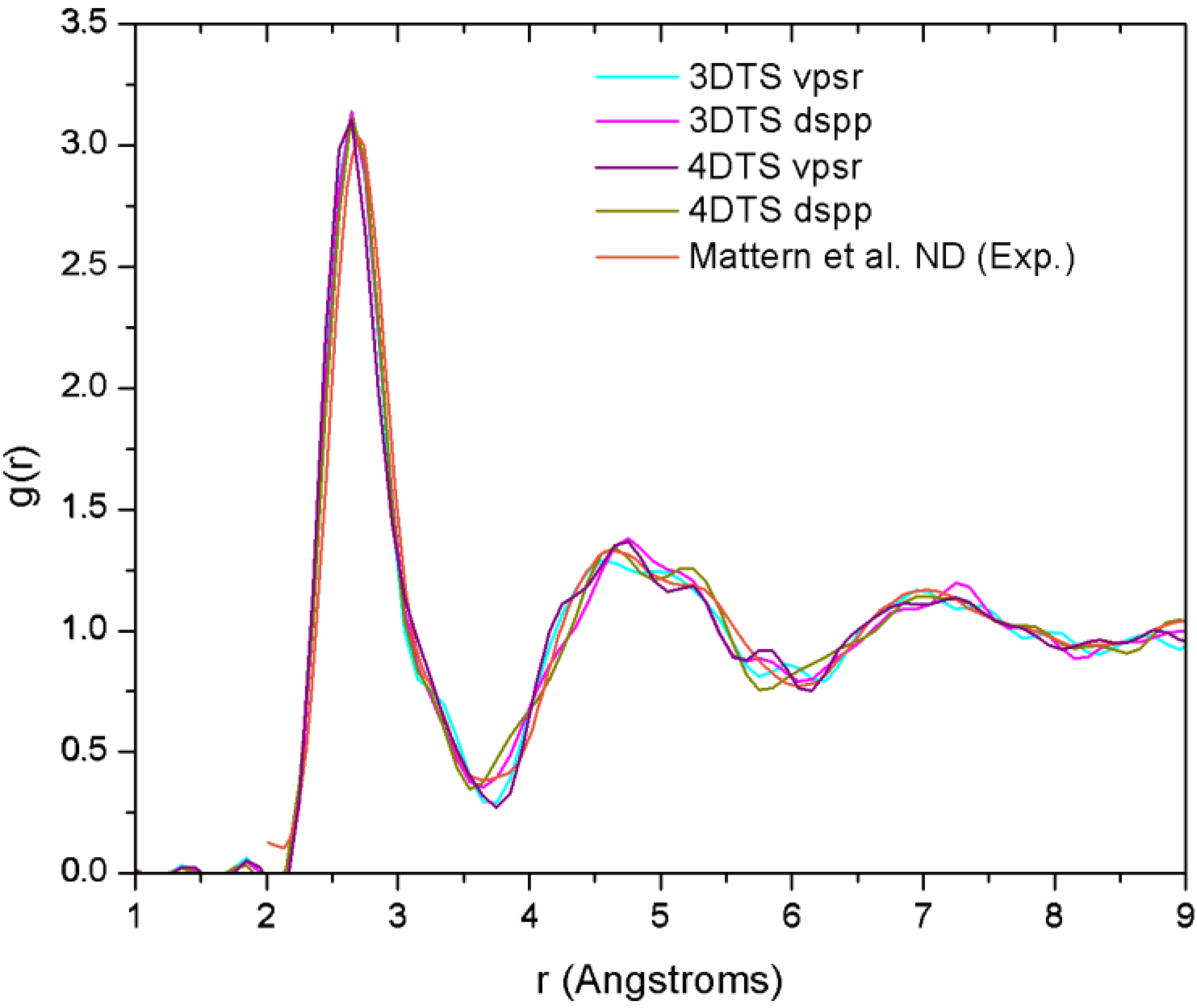
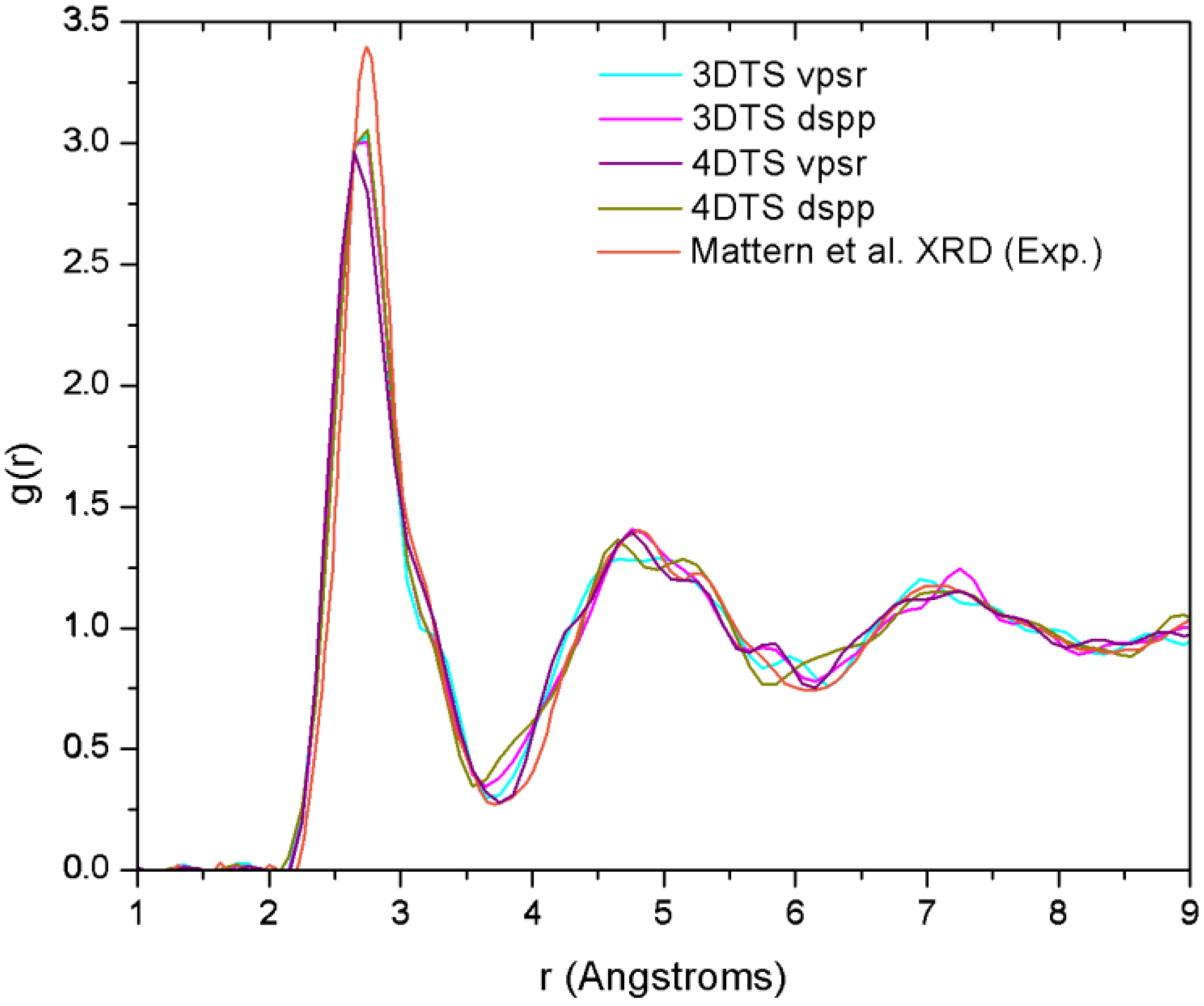
4.7. Amorphous Copper Zirconium Alloys. A Simple BMG-like Material: Cu64 Zr36 [111]
4.7.1. Preamble
4.7.2. Results and Analysis
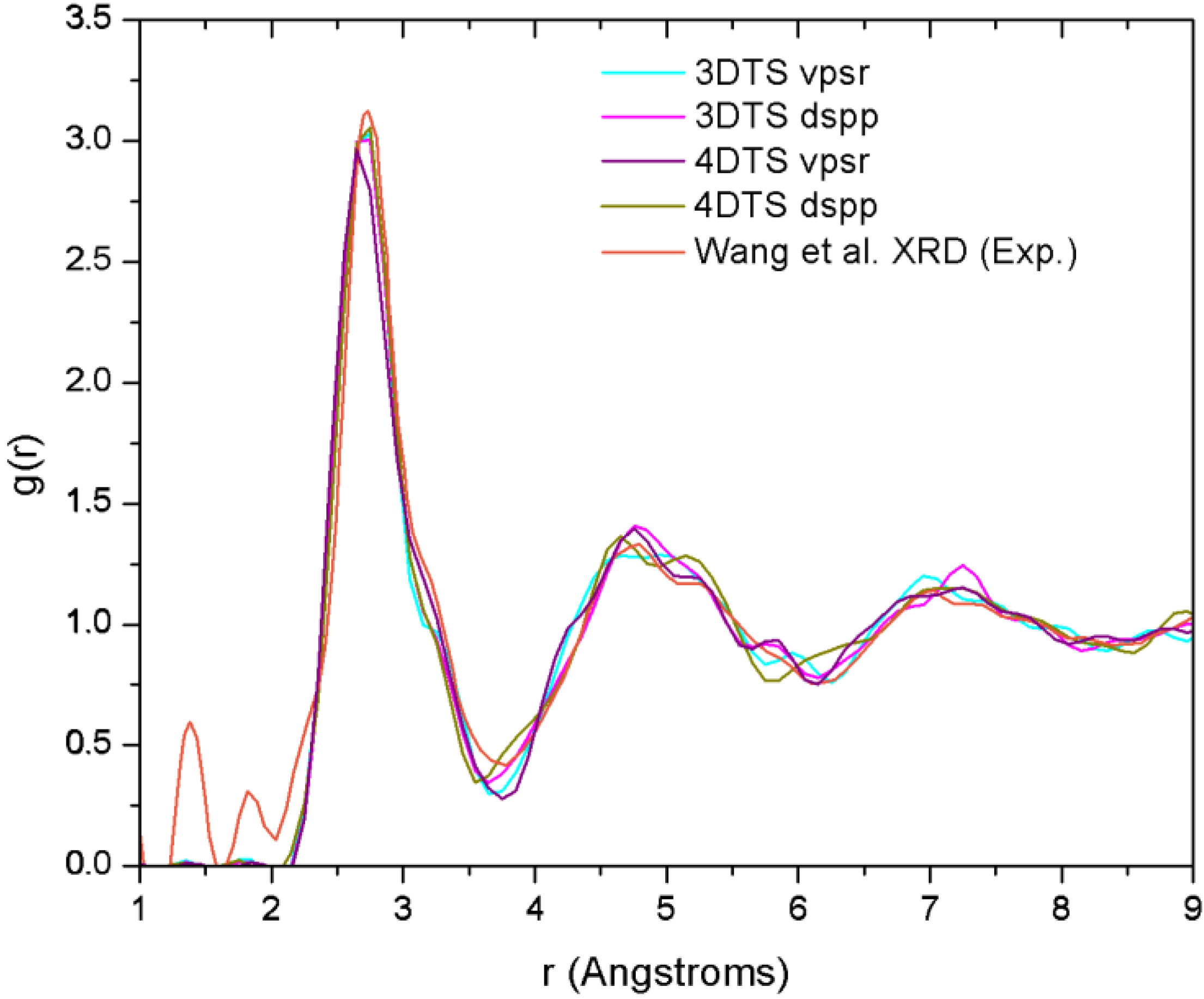
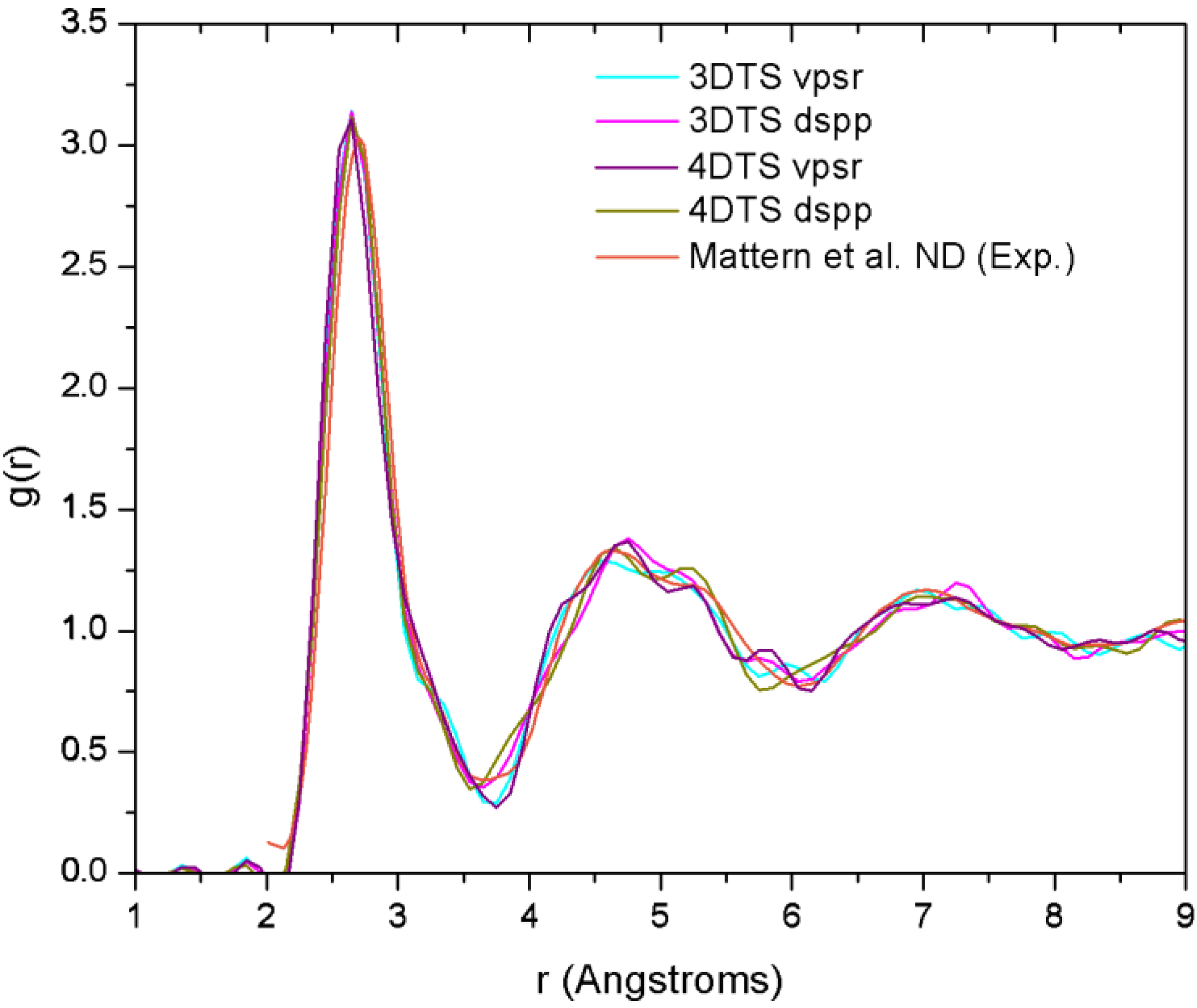
| NCu–Cu | NCu–Zr | NZr–Zr | NZr–Cu | NT | |
|---|---|---|---|---|---|
| Mattern et al.a | 6.7 | 3.9 | 5.9 | 7.6 | 13.2 |
| 3DTS vpsrb | 4.3 | 5.3 | 2.3 | 9.4 | 14.0 |
| 3DTS dsppb | 4.1 | 5.4 | 2.0 | 9.6 | 13.6 |
| 4DTS vpsrb | 3.9 | 5.4 | 2.2 | 9.5 | 13.7 |
| 4DTS dsppb | 3.7 | 5.1 | 2.0 | 9.1 | 13.4 |

4.7.3. Summary
- (a)
- The disparities in the partial coordination numbers with respect to those experimentally reported, are related to the thermal process chosen, i.e., we always stayed under the melting point and therefore the metallic glass was never obtained. Moreover, from the AIMD studies and the reported experimental values, only Jakse and Pasturel [32] establish with precision the method they used to compute the coordination numbers.
- (b)
- The energies of the samples let us know that a time step of 10.71 fs, along with the vpsr pp, lead to a more metastable amorphous structure. Therefore the use of the dspp pp is indicated.
- (c)
- The amorphous alloy shares structural characteristics with the metallic glass, namely, the RDFs are very similar and the total coordination values are also very similar. Nevertheless differences are to be noted.
5. Amorphous Alloys: Their Properties [1]
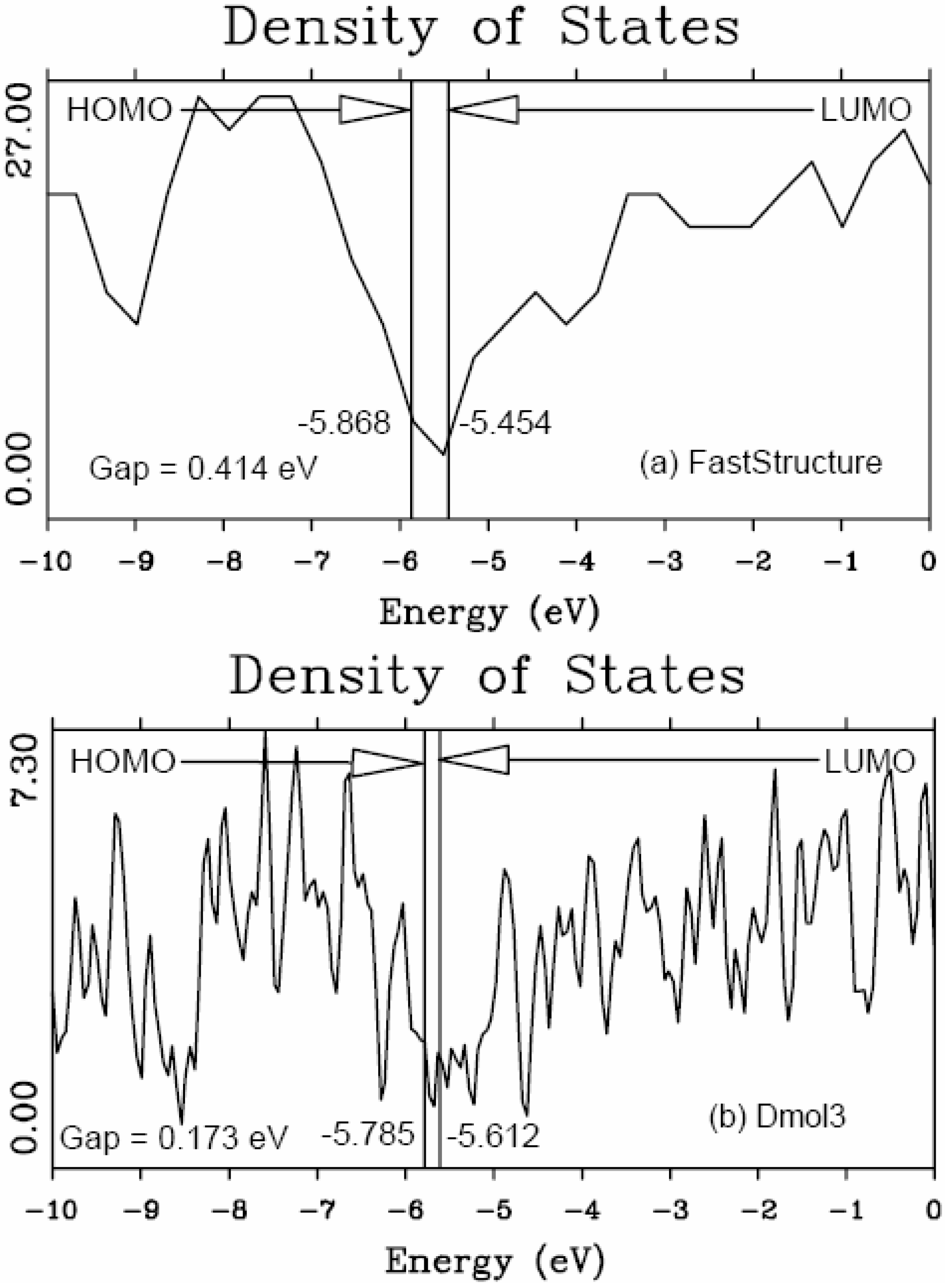
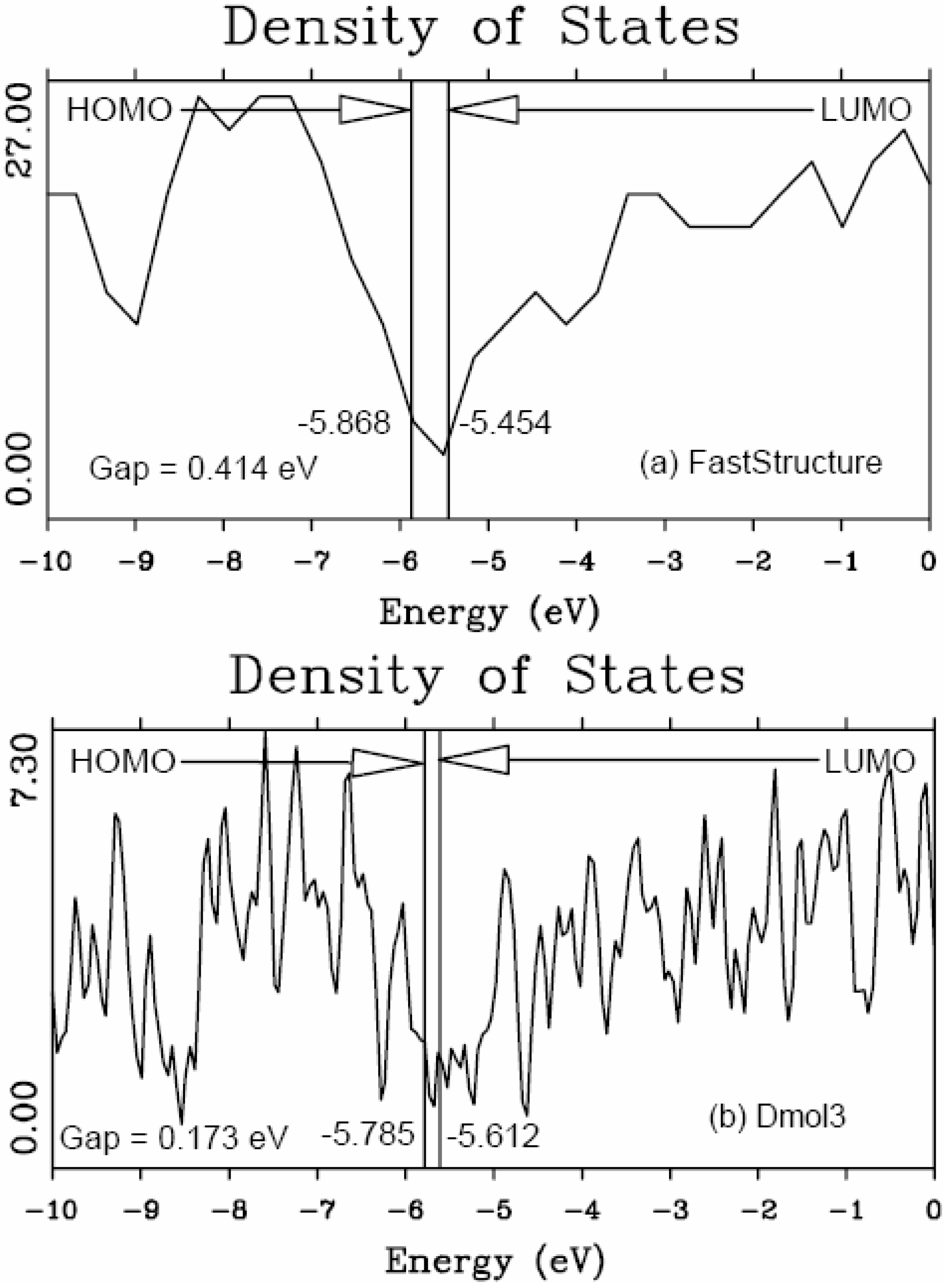
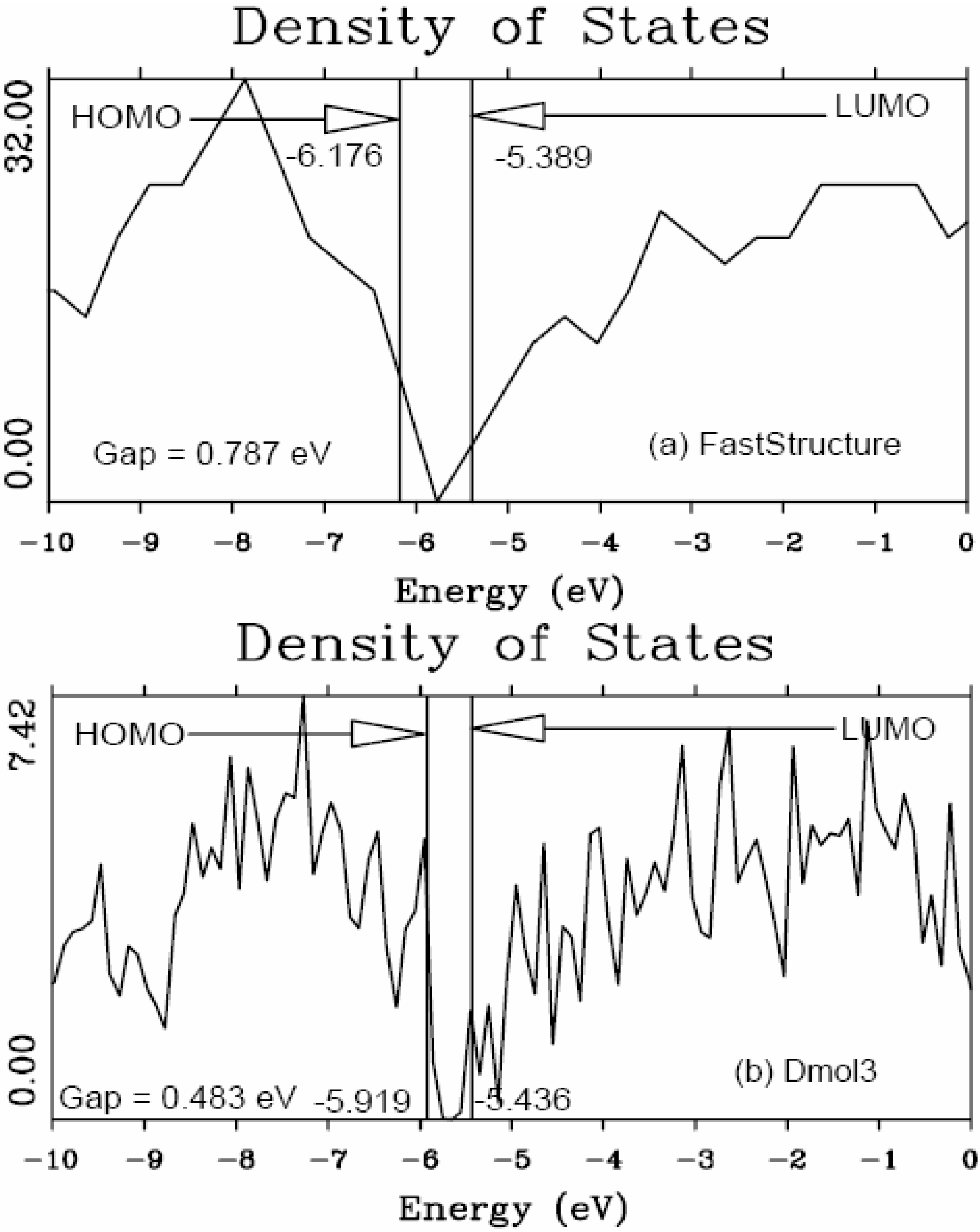
5.1. Electronic Energy Gaps of Amorphous Hydrogenated Silicon [48]


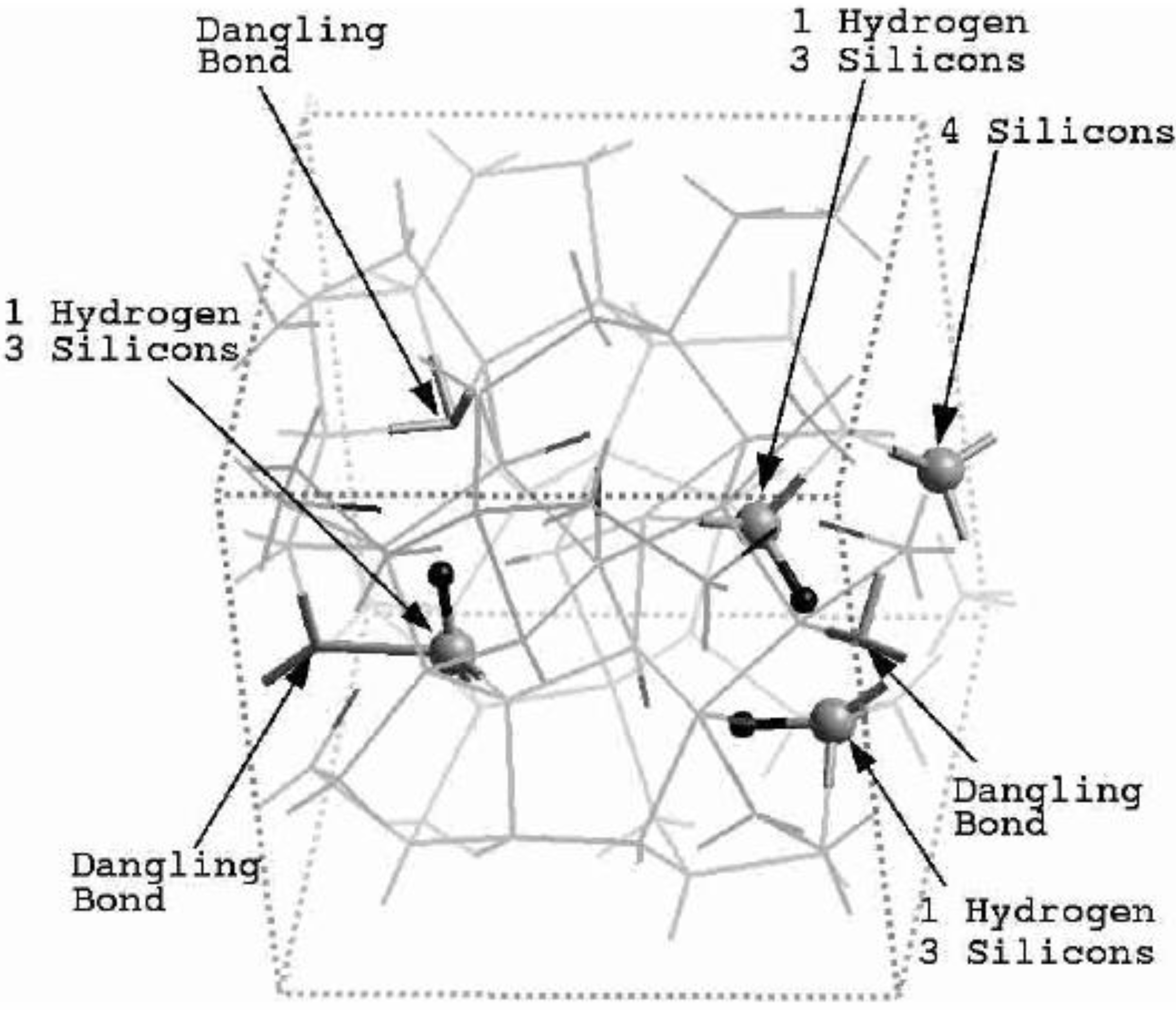
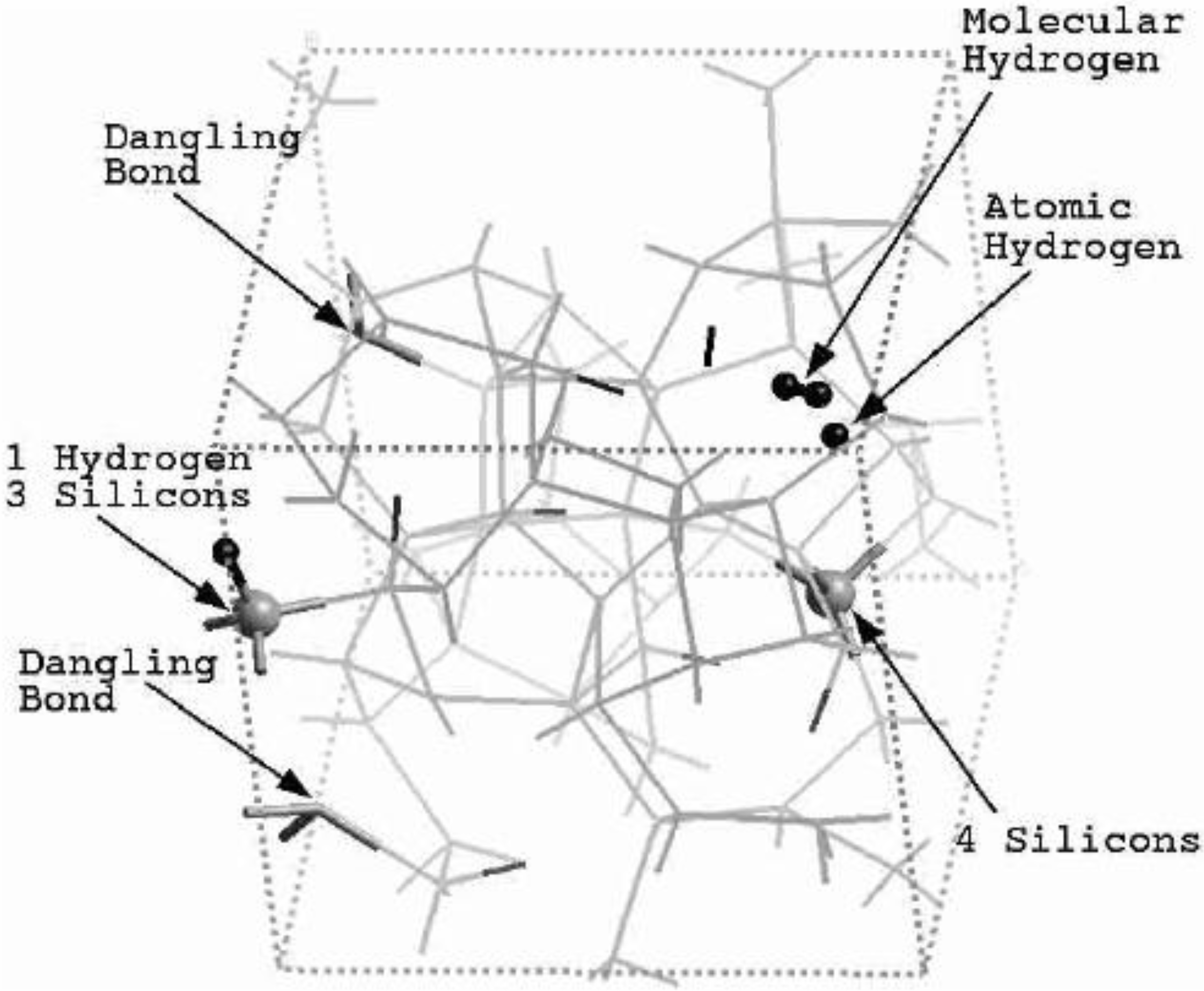
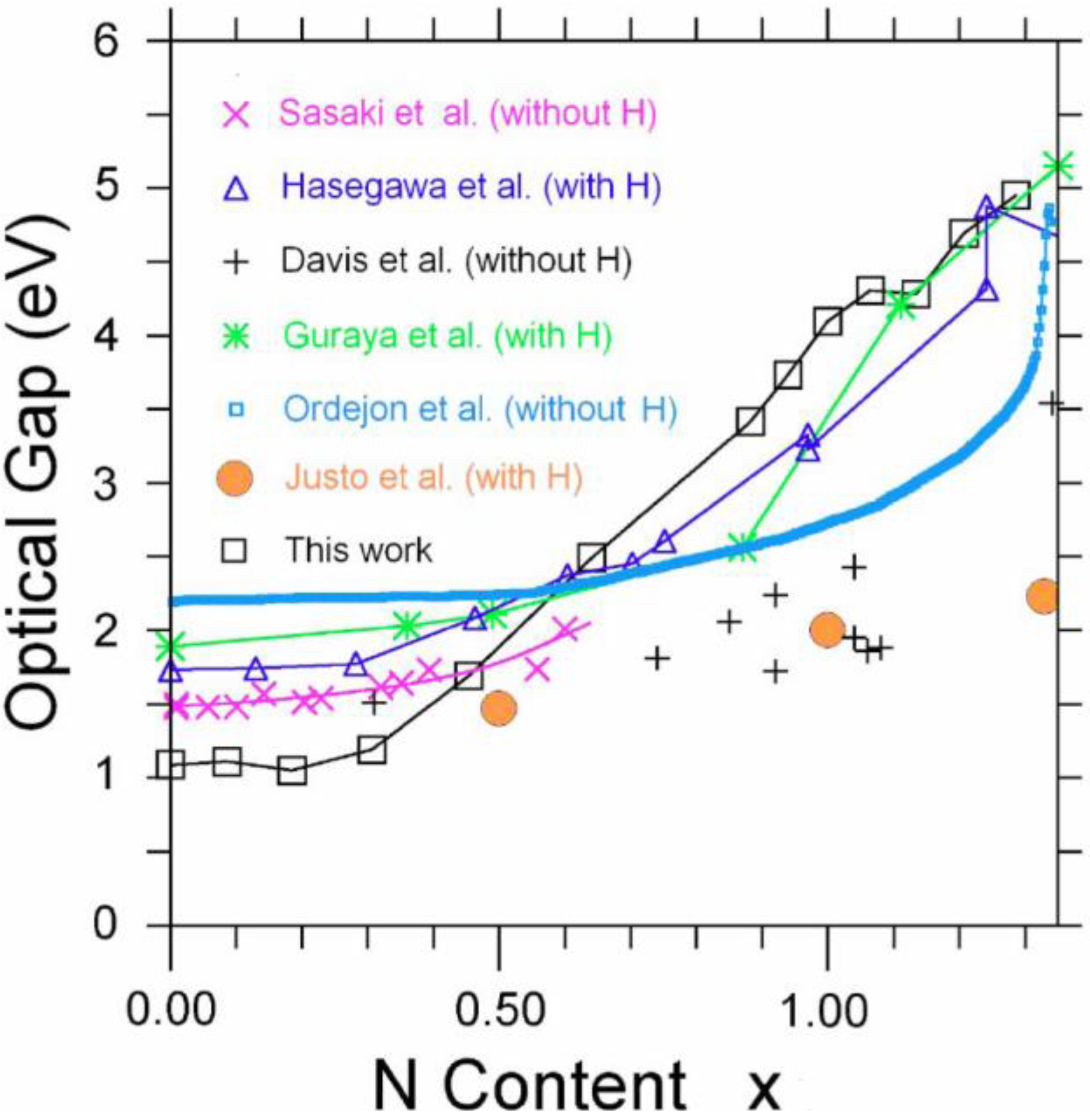
5.2. Optical Gaps of Amorphous Materials a la Tauc. A Case Study: Amorphous Silicon-Nitrogen Alloys [1,52,53,54,55]
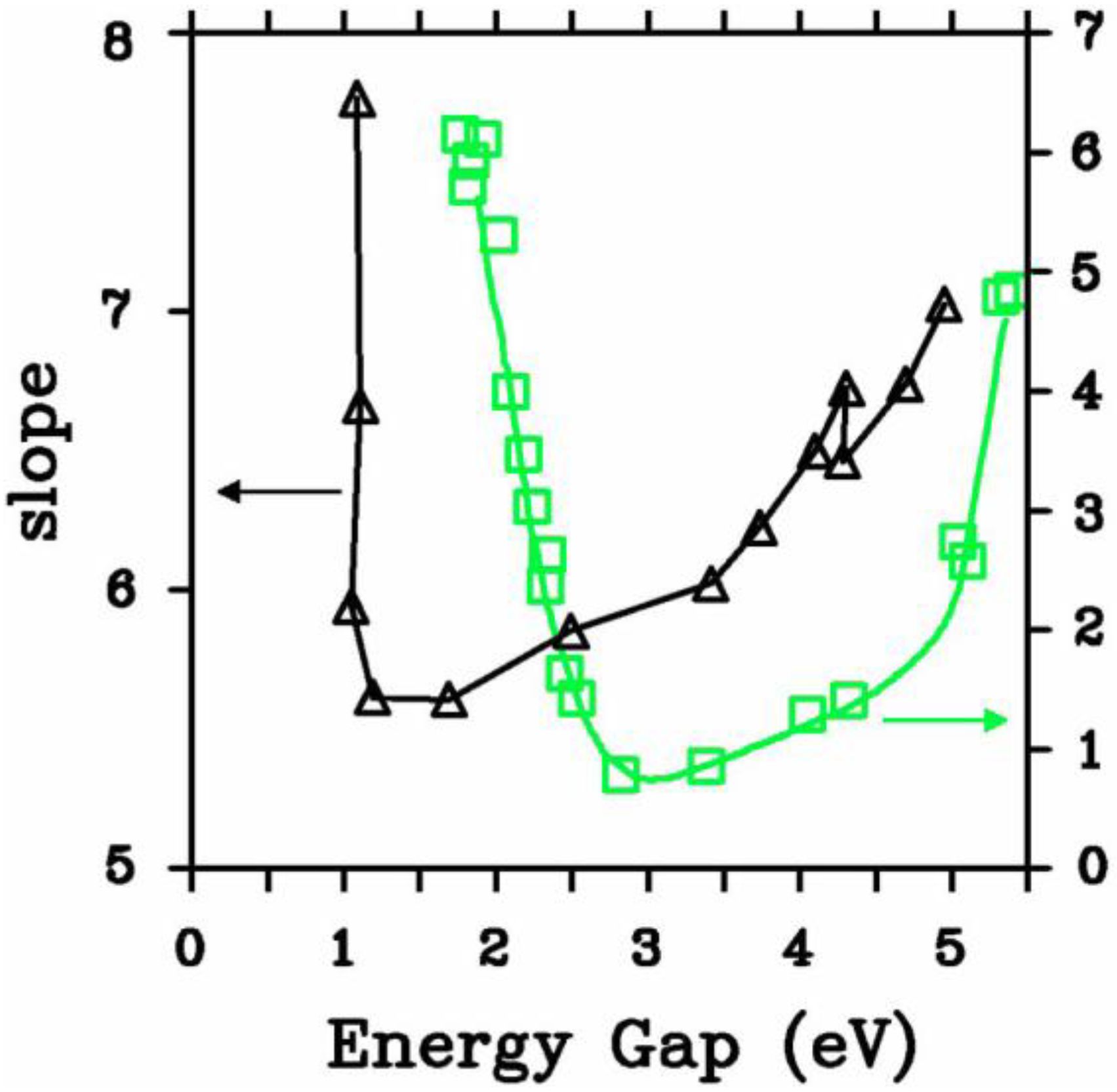
5.2.1. Optical Gaps of Amorphous Materials a la Tauc
- (a)
- The matrix elements for the electronic transitions are constant over the range of photon energies of interest and given by D = π (a/Ω)½.
- (b)
- The k-conservation selection rule is relaxed. As in Mott and Davis we take the matrix element to be the same whether or not either the initial or final states, or both, are localized.
5.2.2. A Case Study: Amorphous Silicon-Nitrogen Alloys
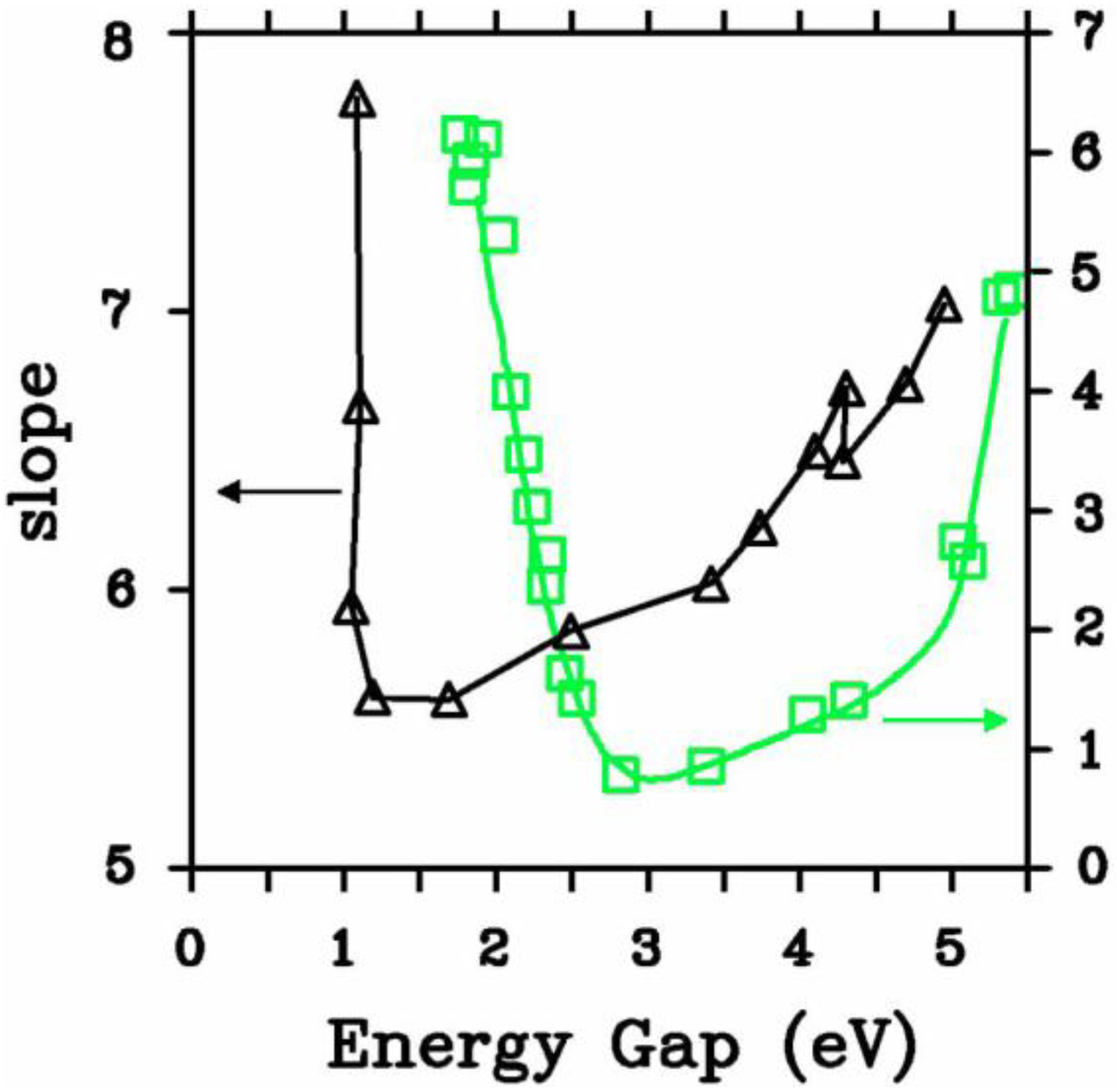
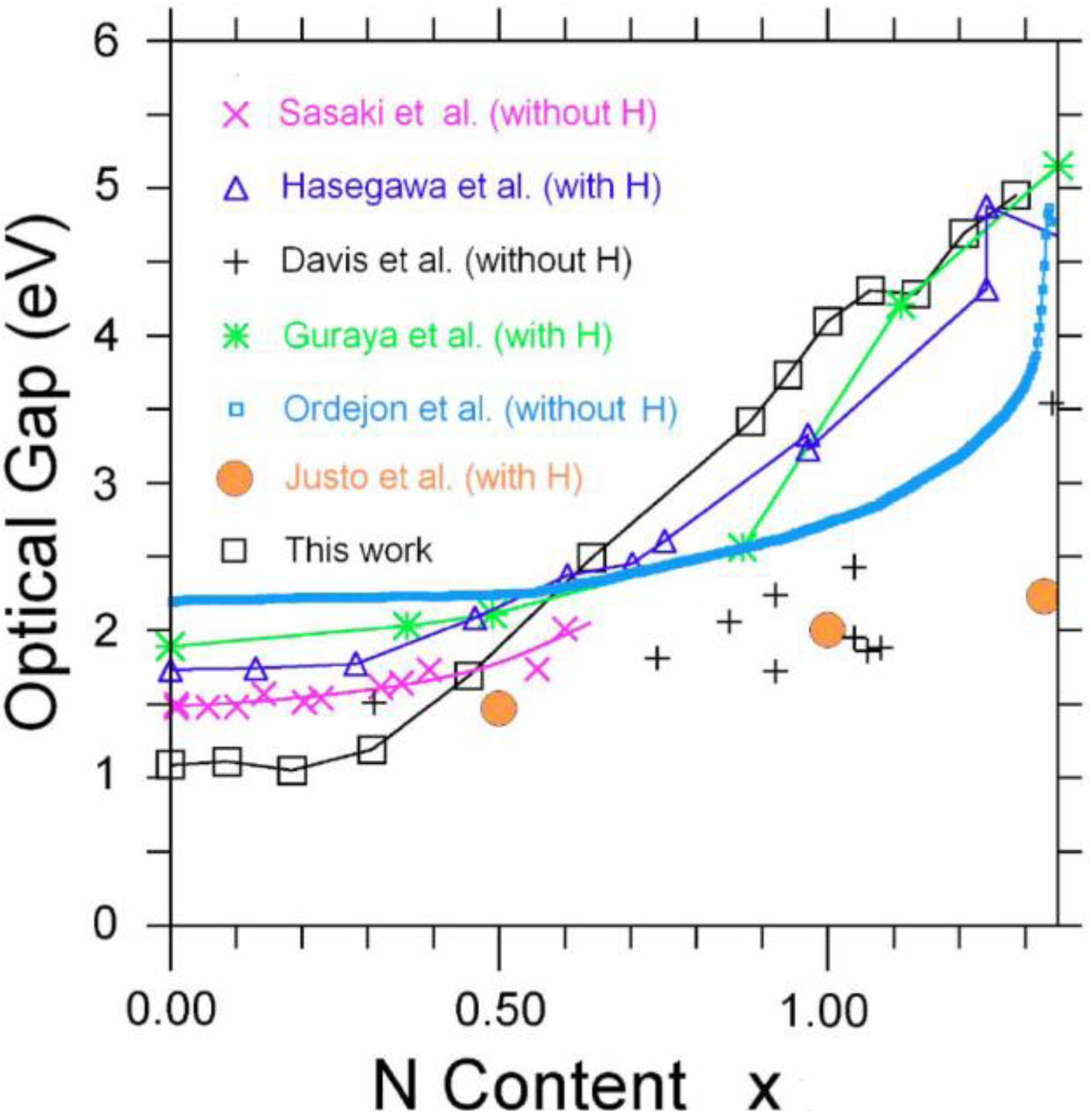
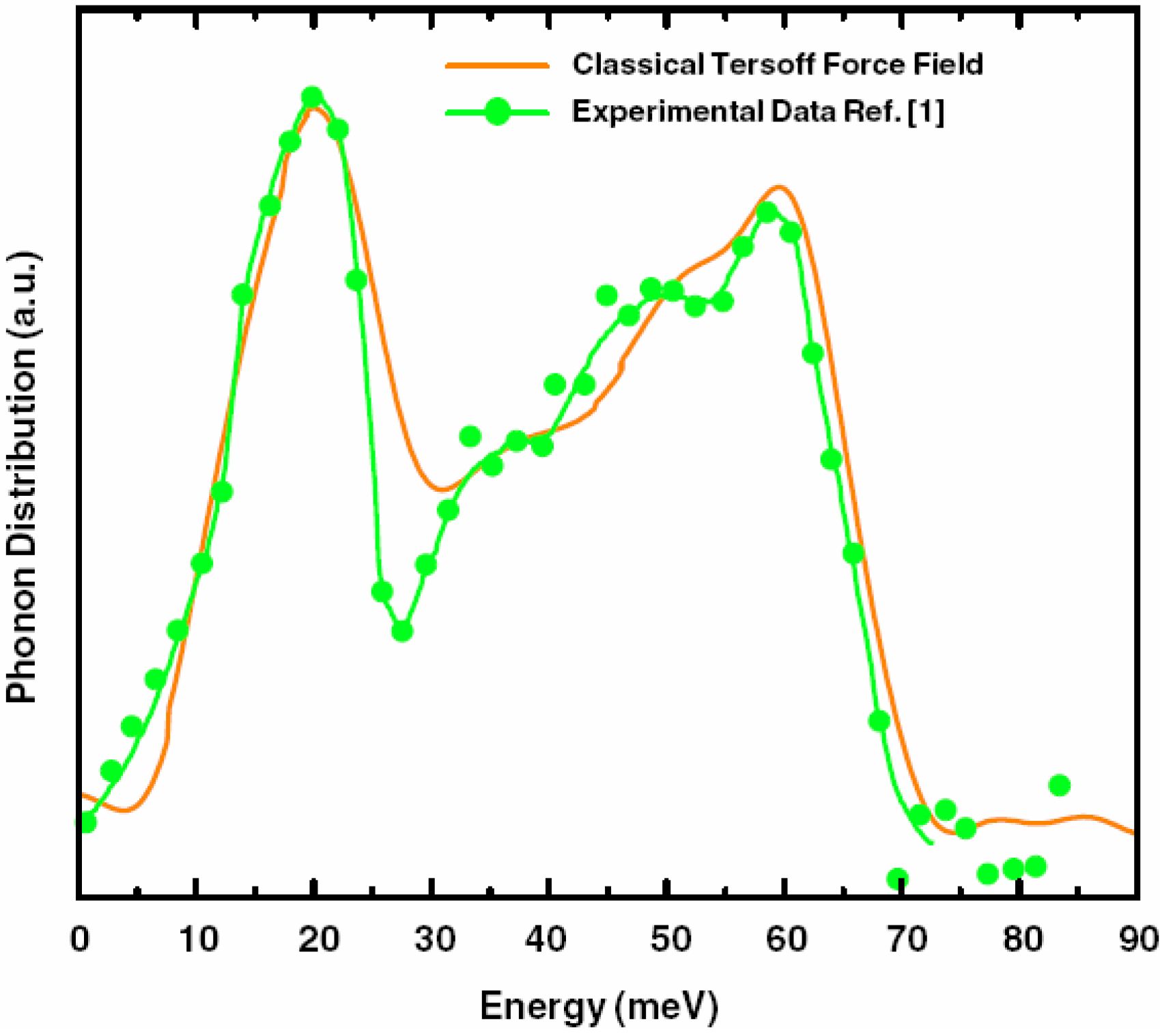
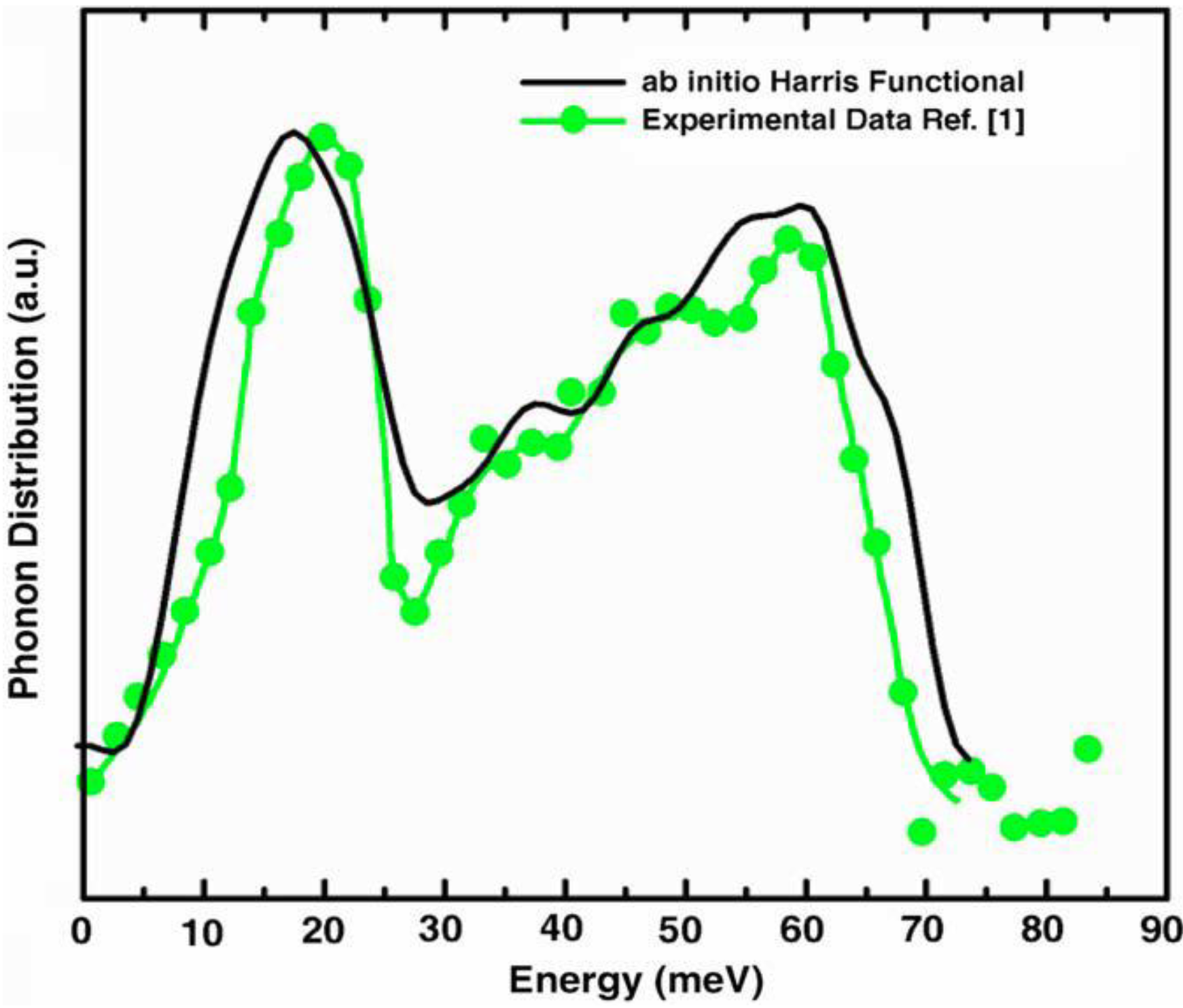
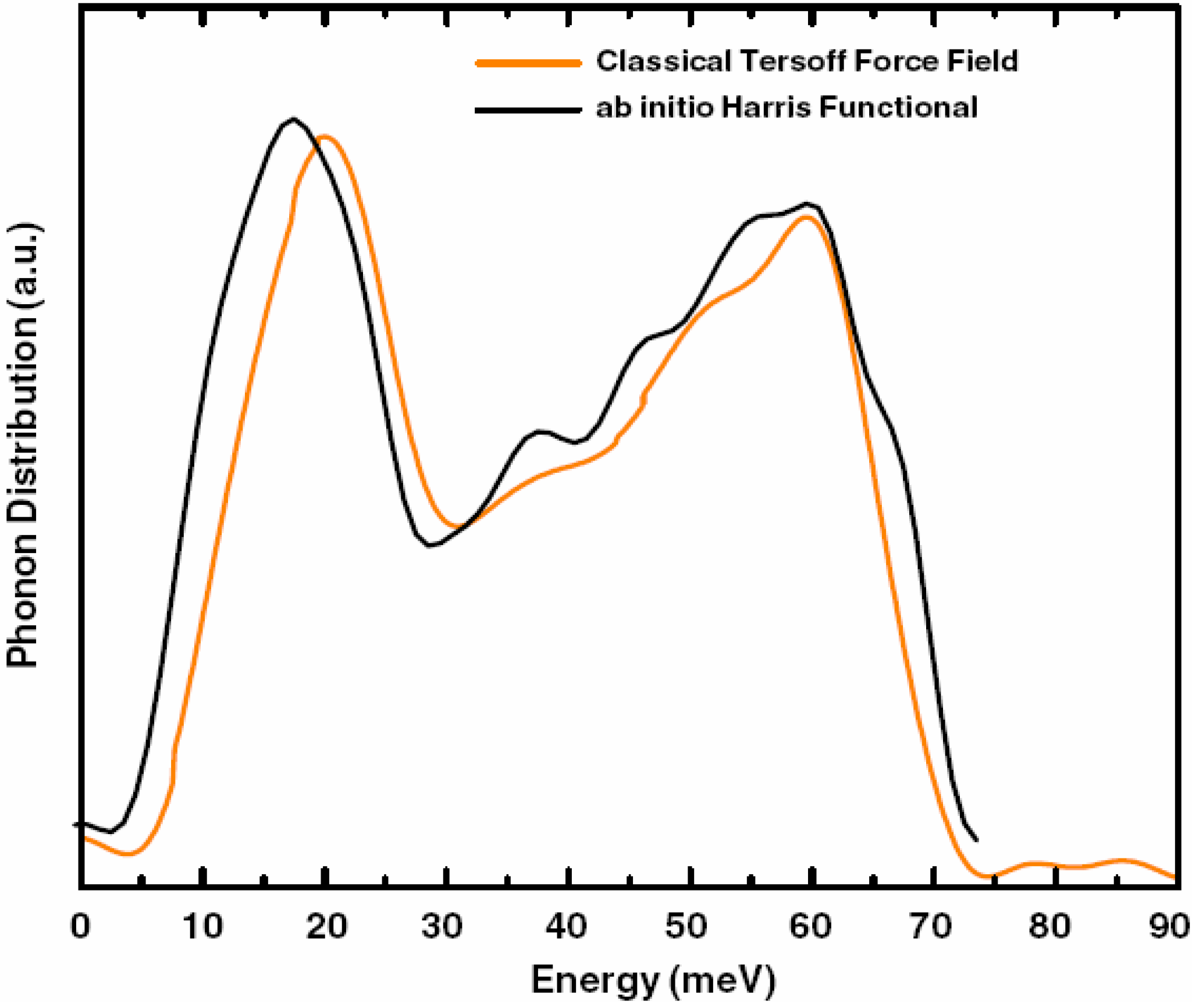
5.3. Vibrational Properties in the Harmonic Approximation [118]
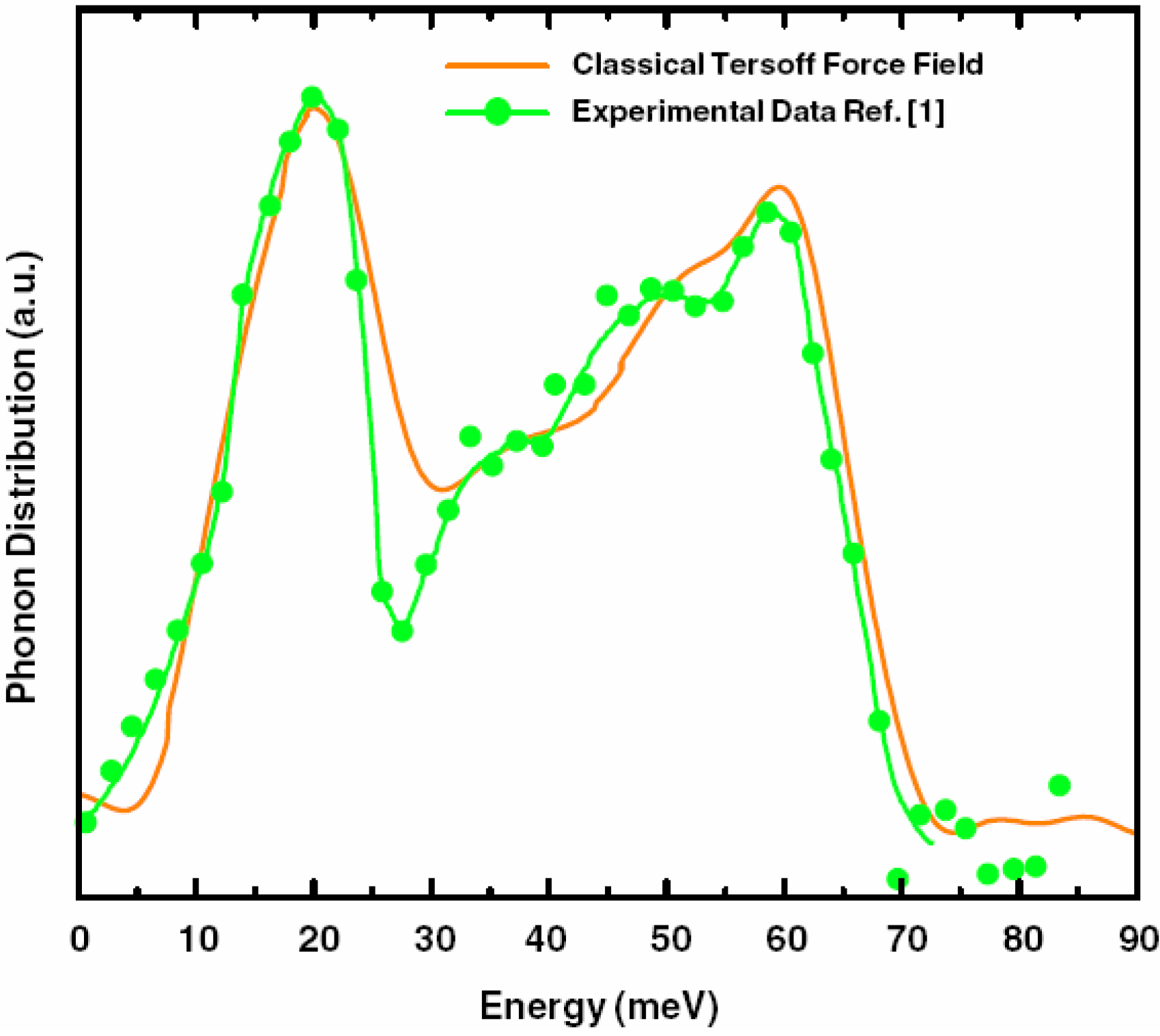
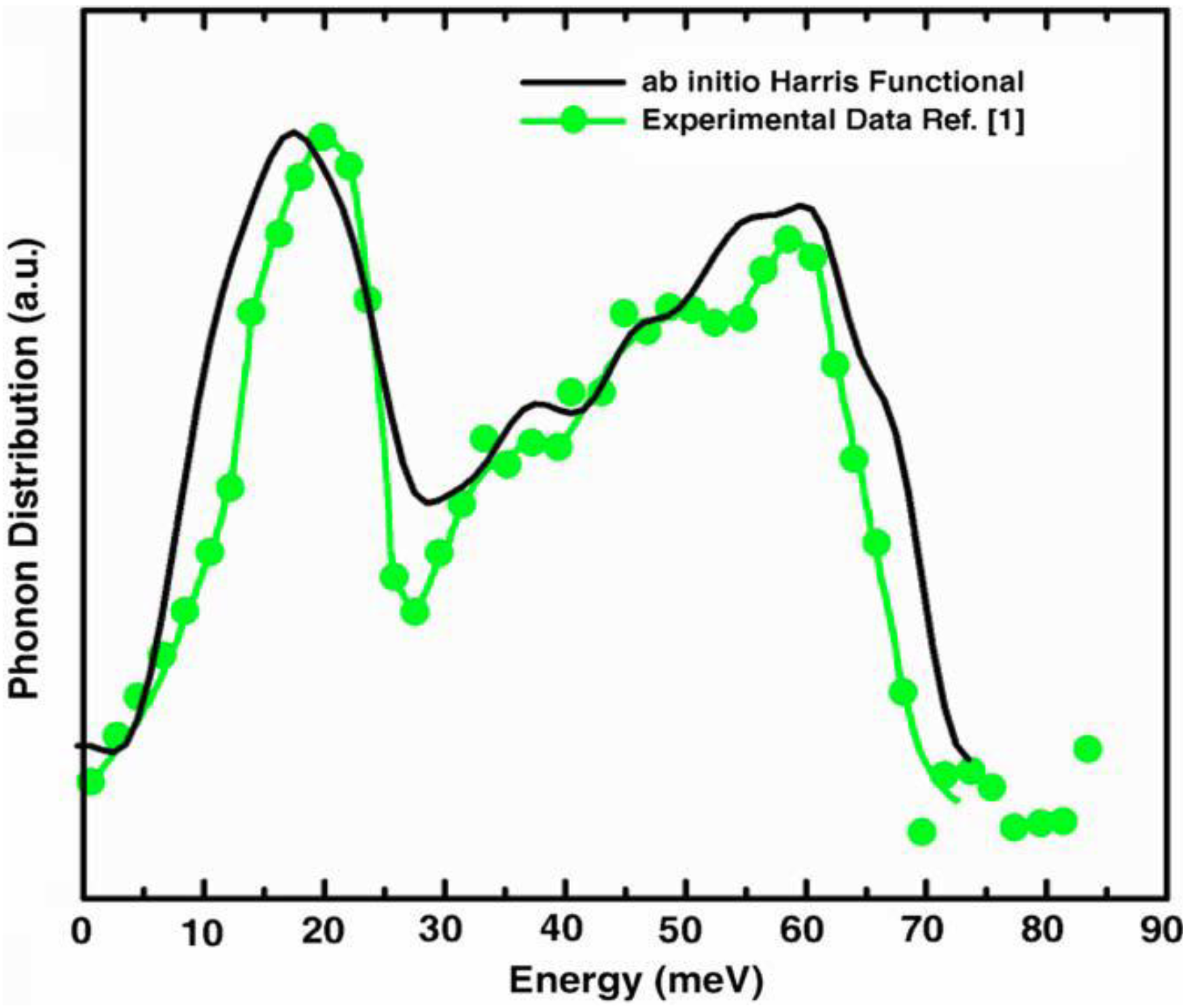

- Our frequency calculations are based on the 216-atom random structure that we ab initio generated using the undermelt-quench approach + the code Fast.
- The relative heights of the two prominent peaks of the vDOS from both experiment and simulations essentially coincide and so do the overall shapes.
- The positions of the two prominent peaks are essentially the same for the simulations and experiment. The first peak of the Harris calculation is somewhat displaced towards lower energies.
- In the region 30–50 meV the Harris simulation agrees better with experiment than Tersoff’s.
6. Conclusions
Acknowledgments
References
- Valladares, A.A. A new approach to the ab initio generation of amorphous semiconducting structures. Electronic and vibrational studies. In Glass Materials Research Progress; Wolf, J.C., Lange, L., Eds.; Nova Science Publishers Inc.: New York, NY, USA, 2008; Chapter 3; pp. 61–123. [Google Scholar]
- Romero, C.; Noyola, J.C.; Santiago, U.; Valladares, R.M.; Valladares, A.; Valladares, A.A. A new approach to the computer modeling of amorphous nanoporous structures of semiconducting and metallic materials: A review. Materials 2010, 3, 467–502. [Google Scholar] [CrossRef]
- Bulk Metallic Glasses—An Overview; Miller, M.; Liaw, P. (Eds.) Springer Science & Business Media, LLC: New York, NY, USA, 2008; pp. xi–xii.
- Aga, R.S.; Morris, J.R. Modeling: The Role of Atomistic Simulations. In Bulk Metallic Glasses. An Overview; Miller, M., Liaw, P., Eds.; Springer Science & Business Media, LLC: New York, NY, USA, 2008; Chapter 3; pp. 57–85. [Google Scholar]
- Hui, L. Shoulder-peak formation in the process of quenching. Phys. Rev. B 2003, 68, 024210:1–024210:5. [Google Scholar]
- Car, R.; Parrinello, M. Structural, dynamical, and electronic properties of amorphous silicon: An ab initio molecular-dynamics study. Phys. Rev. Lett. 1988, 60, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Stich, I.; Car, R.; Parrinello, M. Bonding and disorder in liquid silicon. Phys. Rev. Lett. 1989, 63, 2240–2243. [Google Scholar] [CrossRef] [PubMed]
- Stich, I.; Car, R.; Parrinello, M. Structural, bonding, dynamical, and electronic properties of liquid silicon: An ab initio molecular-dynamics study. Phys. Rev. B 1991, 44, 4262–4274. [Google Scholar] [CrossRef]
- Stich, I.; Car, R.; Parrinello, M. Amorphous silicon studied by ab initio molecular dynamics: Preparation, structure, and properties. Phys. Rev. B 1991, 44, 11092–11104. [Google Scholar] [CrossRef]
- Drabold, D.A.; Fedders, P.A.; Sankey, O.F.; Dow, J.D. Molecular-dynamics simulations of amorphous Si. Phys. Rev. B 1990, 42, 5135–5141. [Google Scholar] [CrossRef]
- Fedders, P.A.; Drabold, D.A.; Klemm, S. Defects, tight binding, and first-principles molecular-dynamics simulations on a-Si. Phys. Rev. B 1992, 45, 4048–4055. [Google Scholar] [CrossRef]
- Lee, I.; Chang, K.J. Atomic and electronic structure of amorphous Si from first-principles molecular-dynamics simulations. Phys. Rev. B 1994, 50, 18083–18089. [Google Scholar] [CrossRef]
- Buda, F.; Chiarotti, G.L.; Car, R.; Parrinello, M. Structure of hydrogenated amorphous silicon from ab initio molecular dynamics. Phys. Rev. B 1991, 44, 5908–5911. [Google Scholar] [CrossRef]
- Fedders, P.A.; Drabold, D.A. Hydrogen and defects in first-principles molecular-dynamics-modeled a-Si:H. Phys. Rev. B 1993, 47, 13277–13282. [Google Scholar] [CrossRef]
- Tuttle, B.; Adams, J.B. Structure of a-Si:H from Harris-functional molecular dynamics. Phys. Rev. B 1996, 53, 16265–16271. [Google Scholar] [CrossRef]
- Merchant, A.R.; McKenzie, D.R.; McCulloch, D.G. Ab initio simulations of amorphous carbon nitrides. Phys. Rev. B 2001, 65, 24208:1–24208:9. [Google Scholar] [CrossRef]
- McKenzie, D.R.; Gerstner, E.G.; Merchant, A.R.; McCulloch, D.G.; Goa, P.E.; Cooper, N.C.; Goringe, C.M. The electronic structure and memory device applications of tetrahedral amorphous carbon. Int. J. Mod. Phys. B 2000, 14, 230–241. [Google Scholar] [CrossRef]
- Walters, J.K.; Kuhn, M.; Spaeth, C.; Fischer, H.; Richter, F.; Newport, R.J. Neutron-diffraction studies of amorphous CNx materials. Phys. Rev. B 1997, 56, 14315–14321. [Google Scholar] [CrossRef]
- Finocchi, F.; Galli, G.; Parrinello, M.; Bertoni, C.M. Microscopic structure of amorphous covalent alloys probed by ab initio molecular dynamics: SiC. Phys. Rev. Lett. 1992, 68, 3044–3047. [Google Scholar] [CrossRef] [PubMed]
- Finocchi, F.; Galli, G.; Parrinello, M.; Bertoni, C.M. Chemical order in amorphous covalent alloys: A theoretical study of a-SiC. Physica B 1993, 185, 379–383. [Google Scholar] [CrossRef]
- Finocchi, F.; Galli, G. Ab initio study of the hydrogenation effects in amorphous silicon carbide. Phys. Rev. B 1994, 50, 7393–7397. [Google Scholar] [CrossRef]
- Massobrio, C.; Pasquarello, A. Short and intermediate range order in amorphous GeSe2. Phys. Rev. B 2008, 77, 144207:1–144207:10. [Google Scholar] [CrossRef]
- Massobrio, C.; Pasquarello, A. Structural properties of amorphous GeSe2. J. Phys. Condens. Mat. 2007, 19, 415111:1–415111:9. [Google Scholar] [CrossRef]
- Sankey, O.F.; Niklewsky, D.J. Ab initio multicenter tight-binding model for molecular-dynamics simulations and other applications in covalent systems. Phys. Rev. B 1989, 40, 3979–3995. [Google Scholar] [CrossRef]
- Cappelletti, R.L.; Cobb, M.; Drabold, D.A.; Kamitakahara, W.A. Neutron-scattering and ab initio molecular-dynamics study of vibrations in glassy GeSe2. Phys. Rev. B 1995, 52, 9133–9136. [Google Scholar] [CrossRef]
- Cobb, M.; Drabold, D.A.; Cappelletti, R.L. Ab initio molecular-dynamics study of the structural, vibrational, and electronic properties of glassy GeSe2. Phys Rev. B 1996, 54, 12162–12171. [Google Scholar] [CrossRef]
- Kohary, K.; Burlakov, V.M.; Pettifor, D.G.; Nguyen-Manh, D. Modeling In-Se amorphous alloys. Phys. Rev. B 2005, 71, 184203:1–184203:7. [Google Scholar]
- Alemany, M.M.G.; Gallego, L.J.; González, D.J. Kohn-Sham ab initio molecular dynamics study of liquid Al near melting. Phys. Rev. B 2004, 70, 134206:1–134206:6. [Google Scholar] [CrossRef]
- Wang, X.D.; Yin, S.; Cao, Q.P.; Jiang, J.Z.; Franz, H.; Jin, Z.H. Atomic structure of binary Cu64.5Zr35.5 bulk metallic glass. App. Phys. Lett. 2008, 92, 011902:1–011902:3. [Google Scholar]
- Mattern, N.; Jóvari, P.; Kaban, I.; Gruner, S.; Elsner, A.; Kokotin, V.; Franz, H.; Beuneu, B.; Eckert, J. Short-range order of Cu–Zr metallic glass. J. Alloy Compd. 2009, 485, 163–169. [Google Scholar] [CrossRef]
- Jakse, N.; Pasturel, A. Glass forming ability and short-range order in a binary bulk metallic glass by ab initio molecular dynamics. App. Phys. Lett. 2008, 93, 113104:1–113104:3. [Google Scholar] [CrossRef]
- Jakse, N.; Pasturel, A. Local order and dynamic properties of liquid and undercooled CuxZr1−x alloys by ab initio molecular dynamics. Phys. Rev. B 2008, 78, 214204:1–214204:9. [Google Scholar] [CrossRef]
- Sun, Y.L.; Shen, J.; Valladares, A.A. Atomic structure and diffusion in Cu60Zr40 metallic liquid and glass: Molecular dynamics simulations. J. Appl. Phys. 2009, 106, 073520:1–073520:8. [Google Scholar]
- Fast Structure Simulated Annealing—User Guide. Release 4.0.0.; Molecular Simulations Inc.: San Diego, CA, USA, 1996.
- Li, X.; Andzelm, J.; Harris, J.; Chaka, A.M. A fast density-functional method for chemistry. In Chemical Applications of Density-Functional Theory; Laird, B.B., Ross, R.B., Ziegler, T., Eds.; American Chemical Society: Washington, DC, USA, 1996; Chapter 26. [Google Scholar]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Harris, J. Simplified method for calculating the energy of weakly interacting fragments. Phys. Rev. B 1985, 31, 1770–1779. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B. Analytic energy derivatives in the numerical local-density-functional approach. J. Chem. Phys. 1991, 94, 7245–7250. [Google Scholar] [CrossRef]
- Lin, Z.J.; Harris, J. A localized-basis scheme for molecular dynamics. J. Phys. Condens. Mat. 1993, 5, 1055–1080. [Google Scholar] [CrossRef]
- Romero, C.; Mata, Z.; Lozano, M.; Barrón, H.; Valladares, R.M.; Álvarez, F.; Valladares, A.A. Amorphizing non-cubic structures of carbon. The case of rhombohedral and hexagonal crystalline supercells. J. Non-Cryst. Solids 2004, 338–340, 513–516. [Google Scholar] [CrossRef]
- Barrón-Escobar, H. Amorfización de estructuras cúbicas de carbón. M.Sc. Thesis, PCeIM, UNAM, Mexico, 2009. [Google Scholar]
- Yang, S.H. Ab initio local-orbital density-functional method for transition metals and semiconductors. Phys. Rev. B 1998, 58, 1832–1838. [Google Scholar] [CrossRef]
- Quantum Chemistry, DMol3 User Guide; Cerius2-3.5; Molecular Simulations, Inc.: San Diego, CA, USA, 1996.
- Kugler, S.; Kohary, K.; Kadas, K.; Pusztai, L. Unusual atomic arrangements in amorphous silicon. Solid State Commun. 2003, 127, 305–309. [Google Scholar] [CrossRef]
- Valladares, A.A.; Alvarez, F.; Liu, Z.; Sticht, J.; Harris, J. Ab initio studies of the atomic and electronic structure of pure and hydrogenated a-Si. Eur. Phys. J. B 2001, 22, 443–453. [Google Scholar] [CrossRef]
- Bellissent, R.; Chenevas-Paule, A.; Chieux, P.; Menelle, A. a-Si:H short range order by neutron scattering. J. Non-Cryst. Solids 1985, 77–78, 213–216. [Google Scholar] [CrossRef]
- Nakamura, K.G.; Ishioka, K.; Kitajima, M.; Murakami, K. Ab initio calculation of the hydrogen molecule in silicon. Solid State Comm. 1997, 101, 735–738. [Google Scholar] [CrossRef]
- Álvarez, F.; Díaz, C.C.; Valladares, A.A.; Valladares, R.M. Radial distribution functions of ab initio generated amorphous covalent networks. Phys. Rev. B 2002, 65, 113108:1–113108:4. [Google Scholar]
- Álvarez, F.; Valladares, A.A. Optical gaps of ab initio generated random networks for a-SiNx alloys. Appl. Phys. Lett. 2002, 80, 58–60. [Google Scholar] [CrossRef]
- Álvarez, F.; Valladares, A.A. The atomic and electronic structure of amorphous silicon nitride. Rev. Mex. Fís. 2002, 48, 528–533. [Google Scholar]
- Álvarez, F.; Valladares, A.A. Atomic topology and radial distribution functions of a-SiNx alloys: Ab initio simulations. Solid State Commun. 2003, 127, 483–487. [Google Scholar] [CrossRef]
- Álvarez, F.; Valladares, A.A. First-principles simulations of atomic networks and optical properties of amorphous SiNx alloys. Phys. Rev. B 2003, 68, 205203:1–205203:10. [Google Scholar]
- Justo, J.F.; de Brito Mota, F.; Fazzio, A. First-principles investigation of a-SiNx:H. Phys. Rev. B 2002, 65, 073202:1–073202:4. [Google Scholar] [CrossRef]
- Hasegawa, S.; Matuura, M.; Kurata, Y. Amorphous SiNx:H dielectrics with low density of defects. Appl. Phys. Lett. 1986, 49, 1272–1274. [Google Scholar] [CrossRef]
- Davis, E.A.; Piggins, N.; Bayliss, S.C. Optical properties of amorphous SiNx(:H) films. J. Phys. Condens. Mat. 1987, 20, 4415–4427. [Google Scholar]
- Guraya, M.M.; Ascolani, H.; Zampieri, G.; Cisneros, J.I.; Dias da Silva, J.H.; Cantaõ, M.P. Bond densities and electronic structure of amorphous SiNx:H. Phys. Rev. B 1990, 42, 5677–5684. [Google Scholar] [CrossRef]
- Santana, G.; Morales-Acevedo, A. Optimization of PECVD SiN:H films for silicon solar cells. Sol. Energy Mater. Sol. Cells 2000, 60, 135–142. [Google Scholar] [CrossRef]
- Aiyama, T.; Fukunaga, T.; Niihara, K.; Hirai, T.; Suzuki, K. X-ray diffraction study of the amorphous structure of chemically vapor-deposited silicon nitride. J. Non-Cryst. Solids 1979, 33, 131–139. [Google Scholar] [CrossRef]
- Misawa, M.; Fukunaga, T.; Niihara, K.; Hirai, T.; Suzuki, K. Structure characterization of CVD amorphous Si3N4 by pulsed neutron total scattering. J. Non-Cryst. Solids 1979, 34, 313–321. [Google Scholar] [CrossRef]
- Fukunaga, T.; Goto, T.; Misawa, M.; Hirai, T.; Suzuki, K. Atomic-scale structure of CVD amorphous Si3N4-BN composite. J. Non-Cryst. Solids 1987, 95–96, 1119–1126. [Google Scholar] [CrossRef]
- De Brito Mota, F.; Justo, J.F.; Fazzio, A. Structural properties of amorphous silicon nitride. Phys. Rev. B 1998, 58, 8323–8328. [Google Scholar] [CrossRef]
- De Brito Mota, F.; Justo, J.F.; Fazzio, A. Structural and electronic properties of silicon nitride materials. Int. J. Quantum Chem. 1998, 70, 973–980. [Google Scholar] [CrossRef]
- De Brito Mota, F.; Justo, J.F.; Fazzio, A. Hydrogen role on the properties of amorphous silicon nitride. J. Appl. Phys. 1999, 86, 1843–1847. [Google Scholar] [CrossRef]
- Justo, J.F.; de Brito Mota, F.; Fazzio, A. Hydrogenated amorphous silicon nitride: structural and electronic properties. Mater. Res. Soc. Symp. Proc. 1999, 538, 555–560. [Google Scholar] [CrossRef]
- Valladares, A.A.; Álvarez-Ramírez, F. Bonding in amorphous carbon-nitrogen alloys: A first principles study. Phys. Rev. B 2006, 73, 024206:1–024206:7. [Google Scholar] [CrossRef]
- McKenzie, D.R. Tetrahedral bonding in amorphous carbon. Rep. Prog. Phys. 1996, 59, 1611–1664. [Google Scholar] [CrossRef]
- Robertson, J. Hard amorphous (diamond-like) carbons. Prog. Solid State Chem. 1991, 21, 199–333. [Google Scholar] [CrossRef]
- Robertson, J. Diamond-like amorphous carbon. Mater. Sci. Eng. R. 2002, 37, 129–281. [Google Scholar] [CrossRef]
- Kroke, E.; Schwarz, M. Novel group 14 nitrides. Coord. Chem. Rev. 2004, 248, 493–532. [Google Scholar] [CrossRef]
- Rodil, S. Preparation and characterization of carbon nitride thin films. Ph.D. Thesis, University of Cambridge, Cambridge, UK, November 2000. [Google Scholar]
- Davis, C.A.; McKenzie, D.R.; Yin, Y.; Kravtchinskaia, E.; Amaratunga, G.A.J.; Veerasamy, V.S. Substitutional nitrogen doping of tetrahedral amorphous carbon. Philos. Mag. B 1994, 69, 1133–1140. [Google Scholar] [CrossRef]
- Shi, X.; Fu, H.; Shi, J.R.; Cheah, L.X.; Tay, B.K.; Hui, P. Electronic transport properties of nitrogen doped amorphous carbon films deposited by the filtered cathodic vacuum arc technique. J. Phys. Condens. Mat. 1998, 10, 9293–9302. [Google Scholar] [CrossRef]
- Stanishevsky, A.; Khriachtchev, L.; Akula, I. Deposition of carbon films containing nitrogen by filtered pulsed cathodic arc discharge method. Diamond Relat. Mater. 1998, 7, 1190–1195. [Google Scholar] [CrossRef]
- Chhowalla, M.; Alexandrou, I.; Kiely, C.; Amaratunga, G.A.J.; Aharonov, R.; Fontana, R.F. Investigation of carbon nitride films by cathodic arc evaporation. Thin Solid Films 1996, 290–291, 103–106. [Google Scholar] [CrossRef]
- Marton, D.; Boyd, K.J.; Al-Bayati, A.H.; Todorov, S.S.; Rabalais, J.W. Carbon nitride deposited using energetic species: A two-phase system. Phys. Rev. Lett. 1994, 73, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Marton, D.; Boyd, K.J.; Rabalais, J.W. Synthesis of carbon nitride. Int. J. Mod. Phys. B 1995, 9, 3527–3558. [Google Scholar] [CrossRef]
- Aarao Reis, F.D.A.; Franceschini, D.F. Two species model for deposition and erosion of carbon-nitrogen films. Appl. Phys. Lett. 1999, 74, 209–211. [Google Scholar] [CrossRef]
- Silva, S.R.P.; Robertson, J.; Amaratunga, G.A.J.; Rafferty, B.; Brown, L.M.; Schwan, J.; Franceschini, D.F.; Mariotto, G. Nitrogen modification of hydrogenated amorphous carbon films. J. Appl. Phys. 1997, 81, 2626–2634. [Google Scholar] [CrossRef] [Green Version]
- Veerasamy, V.S.; Amaratunga, G.A.J.; Davis, C.A.; Timbs, A.E.; Milne, W.I.; McKenzie, D.R. N-type doping of highly tetrahedral diamond-like amorphous carbon. J. Phys. Condens. Mat. 1993, 5, L169–L174. [Google Scholar] [CrossRef]
- Álvarez, F.; Díaz, C.C.; Valladares, R.M.; Valladares, A.A. Ab initio generation of amorphous carbon structures. Diam. Relat. Mater. 2002, 11, 1015–1018. [Google Scholar] [CrossRef]
- Valladares, A.A.; Valladares, A.; Valladares, R.M.; Mc Nelis, M.A. Ab initio cluster simulation of N doped tetrahedral amorphous carbon. J. Non-Cryst. Solids 1998, 231, 209–221. [Google Scholar] [CrossRef]
- Mejía-Mendoza, L.M. Estudio computacional de aleaciones amorfas basadas en silicio-carbono y silicio-germanio. Ph.D. Thesis, PCeIM, UNAM, Mexico, 2011. [Google Scholar]
- Mejía Mendoza, L.M.; Valladares, R.M.; Valladares, A.A. Simulating the structure of amorphous Si0.5C0.5 using Lin-Harris molecular dynamics. Mol. Simulat. 2008, 34, 989–995. [Google Scholar] [CrossRef]
- The goodfellow web site information about Nicalon (Si05C0.5) physical properties. Available online: http://www.goodfellow.com/E/Silicon-Carbide'.html (accessed on 7 April 2011).
- Tersoff, J. Chemical order in amorphous silicon carbide. Phys. Rev. B 1994, 49, 16349–16352. [Google Scholar] [CrossRef]
- Ishimaru, M.; Bae, I.T.; Hirotsu, Y.; Matsumura, S.; Sickafus, K.E. Structural relaxation of amorphous silicon carbide. Phys. Rev. Lett. 2002, 89, 055502:1–055502:4. [Google Scholar] [CrossRef]
- Ishimaru, M. Electron-beam radial distribution analysis of irradiation-induced amorphous SiC. Nucl. Instr. Meth. B 2006, 250, 309–314. [Google Scholar] [CrossRef]
- Reyes-Retana, J.A. Simulaciones computacionales de los calcogenuros amorfos. Ph.D. Thesis, PCeIM, UNAM, Mexico, 2011. [Google Scholar]
- Reyes-Retana, J.A.; Valladares, A.A. Structural properties of amorphous selenium: An ab initio molecular-dynamics simulation. Comput. Mater. Sci. 2010, 47, 934–939. [Google Scholar] [CrossRef]
- Salmon, P.S. Structure of liquids and glasses in the Ge–Se binary system. J. Non-Cryst. Solids 2007, 353, 2959–2974. [Google Scholar] [CrossRef]
- Azoulay, R.; Thibierge, H.; Brenac, A. Devitrification characteristics of GexSe1−x glasses. J. Non-Cryst. Solids 1975, 18, 33–53. [Google Scholar] [CrossRef]
- Salmon, P.S.; Petri, I. Structure of glassy and liquid GeSe2. J. Phys. Condens. Mat. 2003, 15, S1509–S1528. [Google Scholar] [CrossRef]
- Valladares, A.A.; Valladares, A.; Valladares, R.M.; Calles, A. Structural properties of amorphous aluminum and aluminum-nitrogen alloys. Computer simulations. Mater. Res. Soc. Symp. Proc. 2005, 848, 463–477. [Google Scholar]
- Díaz-Celaya, J.A.; Valladares, R.M.; Valladares, A.A. Computational generation of disordered structures of Al-12%Si. An ab initio approach. Mater. Res. Soc. Symp. Proc. 2007, 1048, Z08-20:1–Z08-20:6. [Google Scholar] [CrossRef]
- Díaz-Celaya, J.A.; Valladares, A.A.; Valladares, R.M. An ab initio molecular dynamics calculation of the density of the liquid metallic alloy Al–Si 12 at% as a function of temperature. Intermetallics 2010, 18, 1818–1820. [Google Scholar] [CrossRef]
- Díaz-Celaya, J.A.; Valladares, A.A.; Valladares, R.M. Atomic Coordination Number in Eutectic Aluminum-Silicon as a function of temperature in the liquid phase: An ab initio study. In Proceedings of Liquid and Amorphous Metals XIV, Rome, Italy, 11–15 July 2010.
- Díaz-Celaya, J.A. Estudio del sistema aluminio-silicio líquido y amorfo. Ph.D. Thesis, PCeIM, UNAM, Mexico, 2011. [Google Scholar]
- Valladares, A.A. Generating amorphous and liquid aluminum: A new approach. J. Non-Cryst. Solids 2007, 353, 3540–3544. [Google Scholar] [CrossRef]
- Daw, M.S.; Baskes, M.I. Embedded-atom method-Derivation and application to impurities, surfaces, and other defects in metals. Phys. Rev. B 1984, 29, 6443–6453. [Google Scholar] [CrossRef]
- Sutton, A.P.; Chen, J. Long-range Finnis Sinclair potentials. Philos. Mag. Lett. 1990, 61, 139–146. [Google Scholar] [CrossRef]
- Ducastelle, F. Mudules élastiques des métaux de transition. J. Phys. France 1970, 31, 1055–1062. [Google Scholar] [CrossRef]
- Murray, J.L.; McAlister, A.J. The Aluminum-Silicon system. Bull. Alloy Phase Dia. 1984, 5, 74–84. [Google Scholar] [CrossRef]
- Napolitano, R.E.; Meco, H.; Jung, C. Faceted solidification morphologies in low-growth-rate Al-Si eutectics. JOM 2004, 56, 16–21. [Google Scholar] [CrossRef]
- Bian, X.F.; Wang, W.M.; Qin, J.Y. Liquid structure Al-12.5%Si alloy modified by antimony. Mater. Charact. 2001, 46, 25–29. [Google Scholar] [CrossRef]
- Stepanyuk, V.S.; Katsnelson, A.A.; Szasz, A.; Trushin, O.S.; Müller, H.; Watson, L.M.; Kirchmayr, H. The microstructure of liquid and amorphous aluminum. J. Non-Cryst. Solids 1992, 151, 169–174. [Google Scholar] [CrossRef]
- Finney, J.L. Modeling of liquids and amorphous solids. In Amorphous Solids and the Liquid State; March, N.H., Street, R.A., Tosi, M., Eds.; Plenum: New York, NY, USA, 1985; pp. 31–51. [Google Scholar]
- Waseda, Y. The Structure of Non-Crystalline Materials, Liquids and Amorphous Solids, 1st ed.; McGraw-Hill Inc: Columbus, OH, USA, 1980; p. 90. [Google Scholar]
- Galván-Colín, J. Método computational ab initio para la amorfización de una aleación Cu-Zr (Cu64Zr36). M.Sc. Thesis, PCeIM, UNAM, Mexico, 2011. [Google Scholar]
- Peker, A.; Johnson, W.L. A highly processable metallic-glass Zr41.2Ti13.8Cu12.5Ni10.0Be22.5. Appl. Phys. Lett. 1993, 63, 2342–2344. [Google Scholar] [CrossRef]
- Xu, D.; Lohwongwatana, B.; Duan, G.; Johnson, W.L.; Garland, C. Bulk metallic glass formation in binary Cu-rich alloy series—Cu100−xZrx (x = 34, 36, 38.2, 40 at%) and mechanical properties of bulk Cu64Zr36 glass. Acta Mater. 2004, 52, 2621–2624. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Subramanian, P.R.; Chakrabarti, D.J.; Laughlin, D.E. Phase Diagrams of Binary Copper Alloys; Materials Information Society: Novelty, Ohio, USA, 1994. [Google Scholar]
- Mott, N.F.; Davis, E.A. Electronic Processes in Non-crystalline Materials; Oxford University Press: Oxford, UK, 1971; p. 238. [Google Scholar]
- Tauc, J. Optical Properties of Solids; Abelès, F., Ed.; North Holland: Amsterdam, The Netherlands, 1970; p. 277. [Google Scholar]
- Valladares, A.; Valladares, R.M.; Alvarez-Ramírez, F.; Valladares, A.A. Studies of the phonon density of states in ab initio generated amorphous structures of pure silicon. J. Non-Cryst. Solids 2006, 352, 1032–1036. [Google Scholar] [CrossRef]
- Tersoff, J. Modeling solid-state chemistry—interatomic potentials for multicomponent systems. Phys. Rev. B 1989, 39, 5566–5568. [Google Scholar] [CrossRef]
- Kamitakahara, W.A.; Shanks, H.R.; McClelland, J.F.; Buchenau, U.; Gompf, F.; Pintschovius, L. Measurement of phonon densities of states for pure and hydrogenated amorphous-silicon. Phys. Rev. Lett. 1984, 52, 644–647. [Google Scholar] [CrossRef]
- Kamitakahara, W.A.; Soukoulis, C.M.; Shanks, H.R.; Buchenau, U.; Grest, G.S. Vibrational spectrum of amorphous silicon: Experiment and computer simulation. Phys. Rev. B 1987, 36, 6539–6542. [Google Scholar] [CrossRef]
- Oxford Order N Package (OXON); Developed at the Materials Modeling Laboratory; Department of Materials, the University of Oxford: Oxford, UK, December 2000.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Valladares, A.A.; Díaz-Celaya, J.A.; Galván-Colín, J.; Mejía-Mendoza, L.M.; Reyes-Retana, J.A.; Valladares, R.M.; Valladares, A.; Alvarez-Ramirez, F.; Qu, D.; Shen, J. New Approaches to the Computer Simulation of Amorphous Alloys: A Review. Materials 2011, 4, 716-781. https://doi.org/10.3390/ma4040716
Valladares AA, Díaz-Celaya JA, Galván-Colín J, Mejía-Mendoza LM, Reyes-Retana JA, Valladares RM, Valladares A, Alvarez-Ramirez F, Qu D, Shen J. New Approaches to the Computer Simulation of Amorphous Alloys: A Review. Materials. 2011; 4(4):716-781. https://doi.org/10.3390/ma4040716
Chicago/Turabian StyleValladares, Ariel A., Juan A. Díaz-Celaya, Jonathan Galván-Colín, Luis M. Mejía-Mendoza, José A. Reyes-Retana, Renela M. Valladares, Alexander Valladares, Fernando Alvarez-Ramirez, Dongdong Qu, and Jun Shen. 2011. "New Approaches to the Computer Simulation of Amorphous Alloys: A Review" Materials 4, no. 4: 716-781. https://doi.org/10.3390/ma4040716
APA StyleValladares, A. A., Díaz-Celaya, J. A., Galván-Colín, J., Mejía-Mendoza, L. M., Reyes-Retana, J. A., Valladares, R. M., Valladares, A., Alvarez-Ramirez, F., Qu, D., & Shen, J. (2011). New Approaches to the Computer Simulation of Amorphous Alloys: A Review. Materials, 4(4), 716-781. https://doi.org/10.3390/ma4040716