Organic Zeolite Analogues Based on Multi-Component Liquid Crystals: Recognition and Transformation of Molecules within Constrained Environments

Abstract
: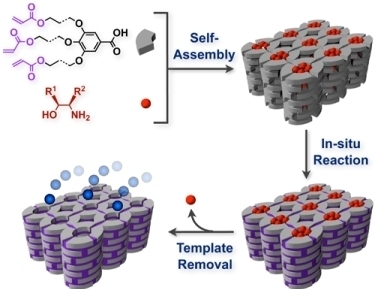
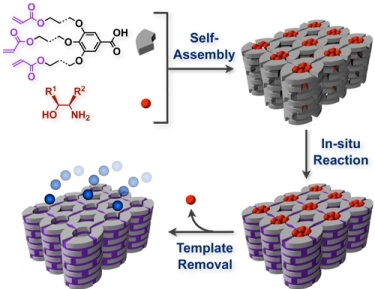
1. Introduction
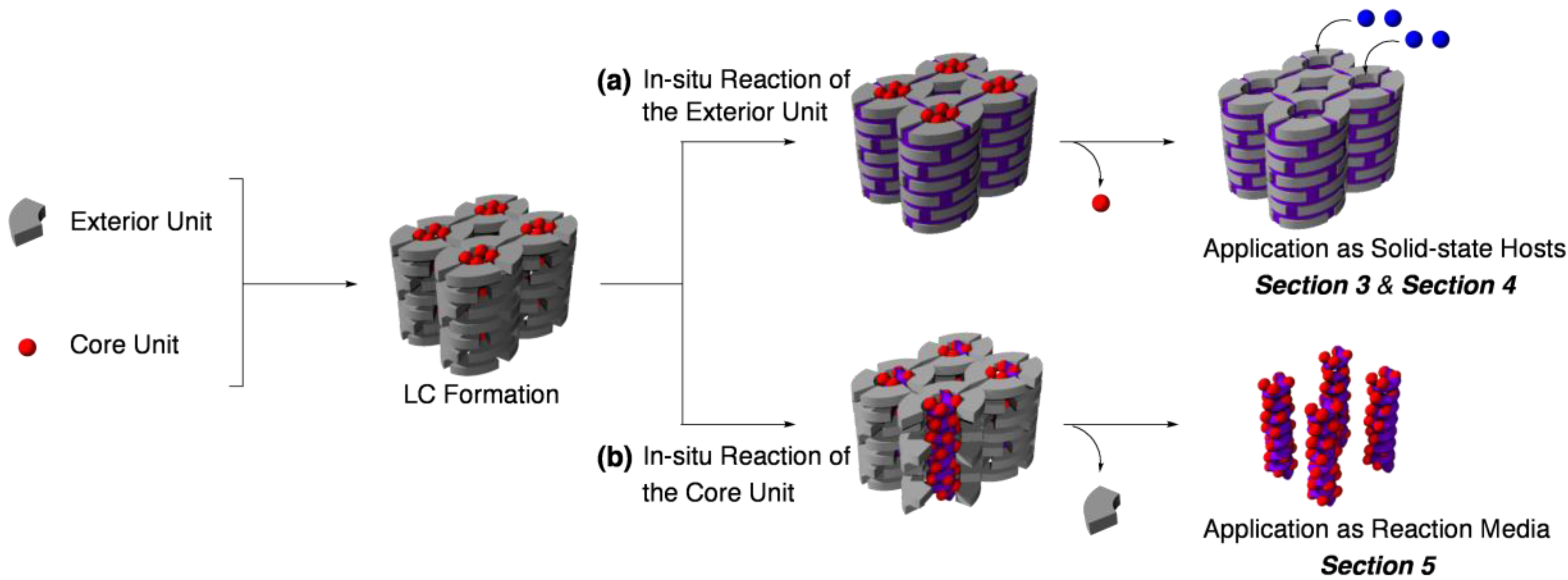
2. Classification of In Situ Reactions in Liquid Crystals
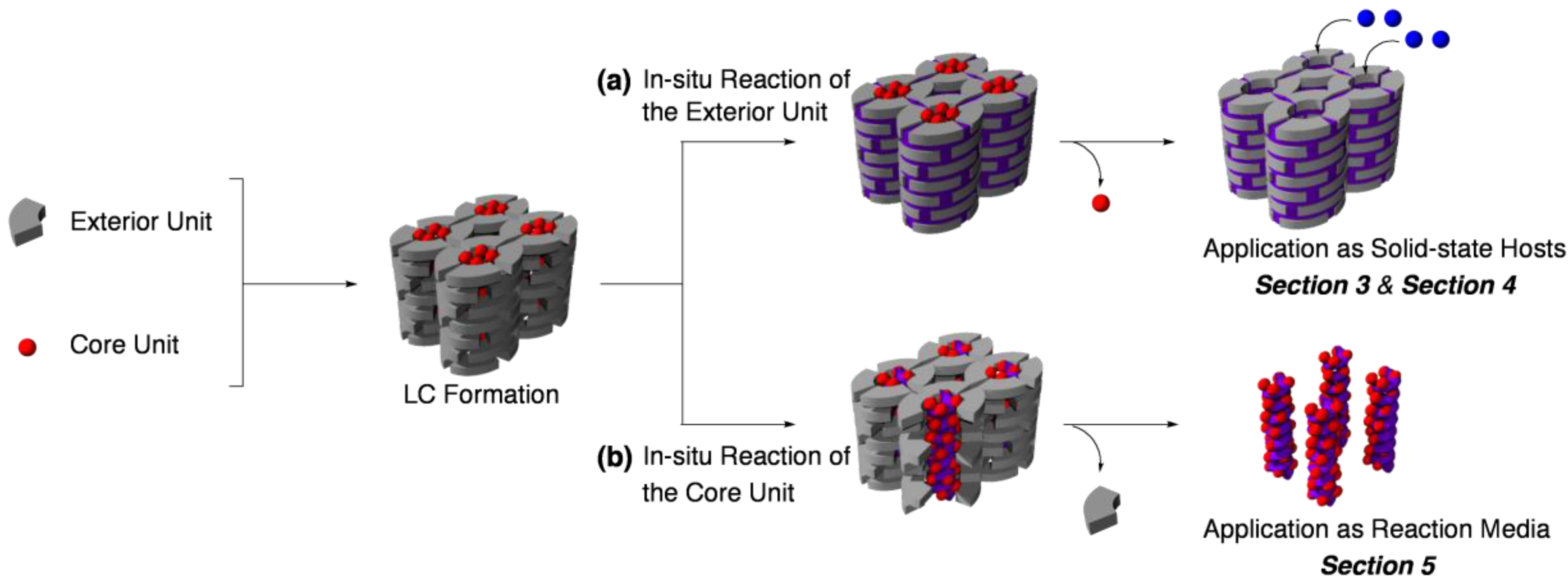
3. Guest-Selective Molecular Sieves
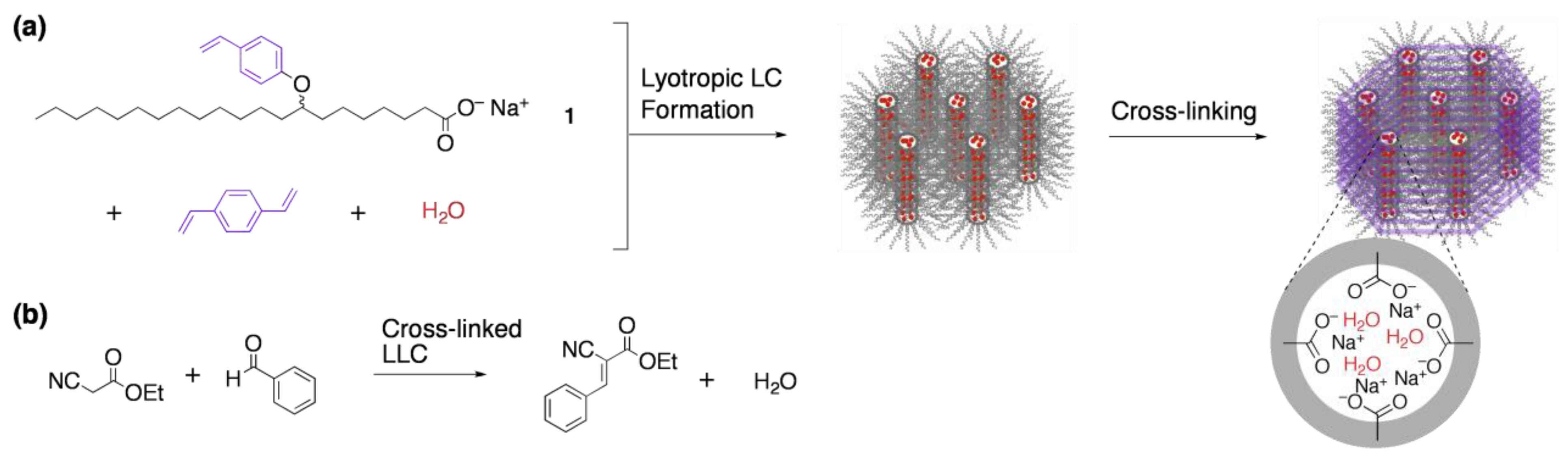
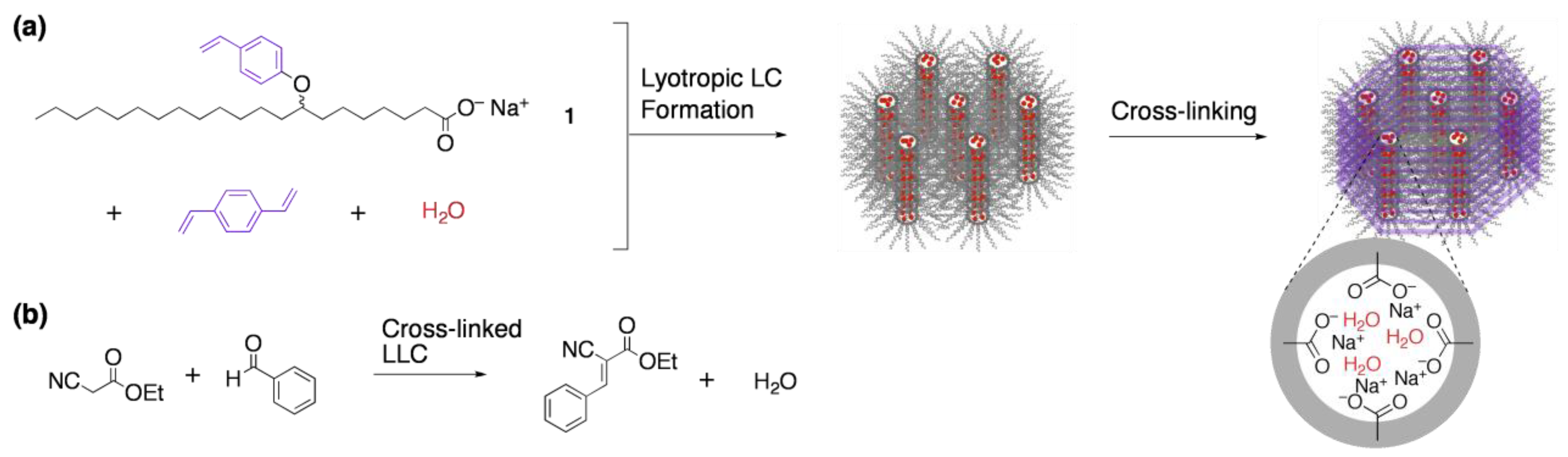
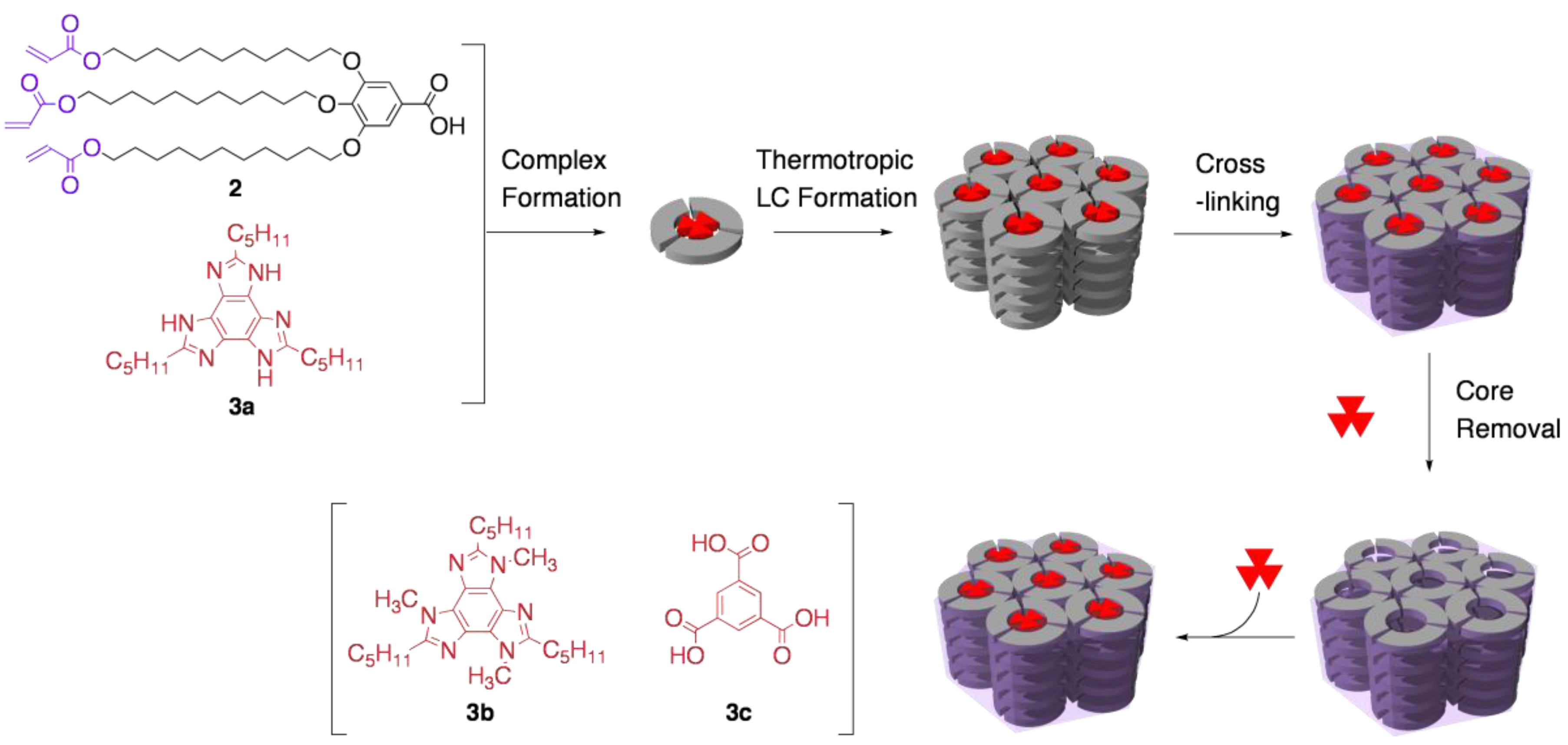
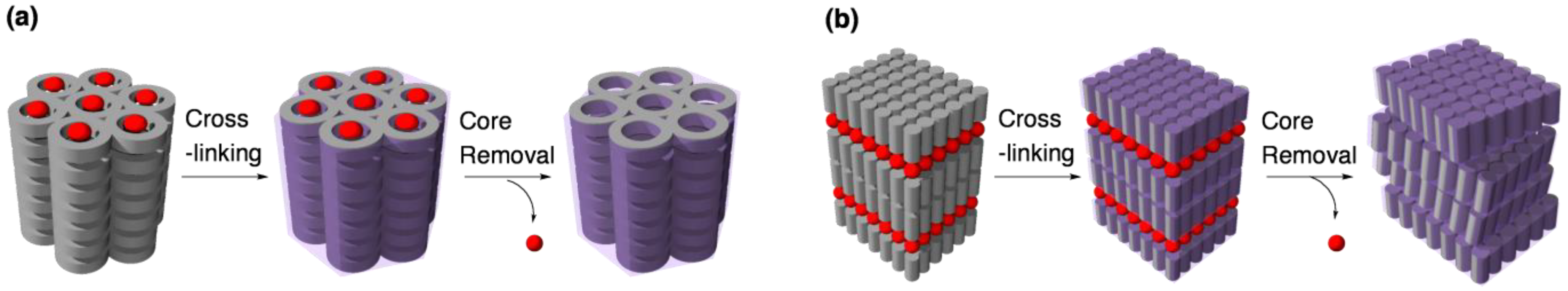
3.1. Size recognition by cross-linked LCs
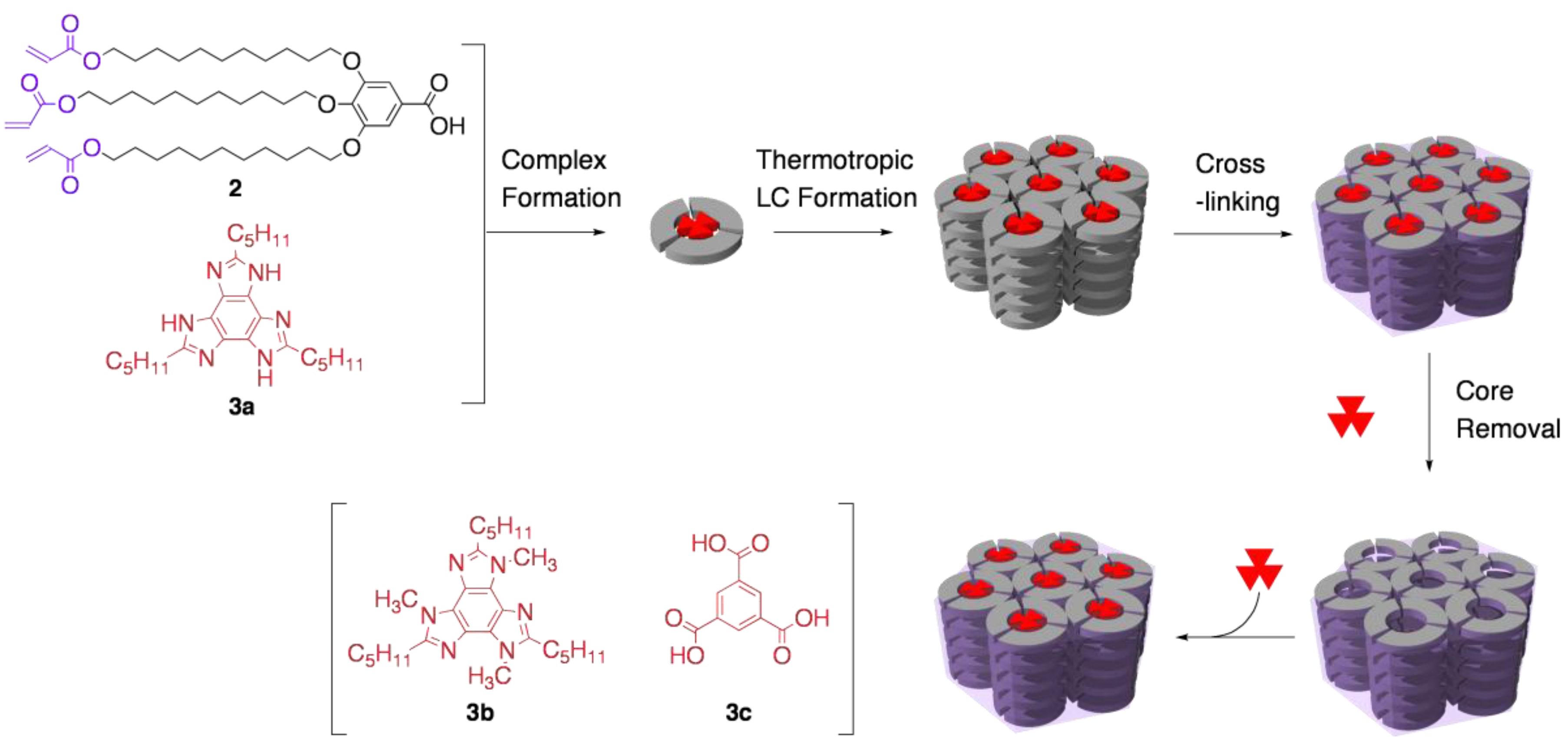
3.2. Functional group recognition by cross-linked LCs
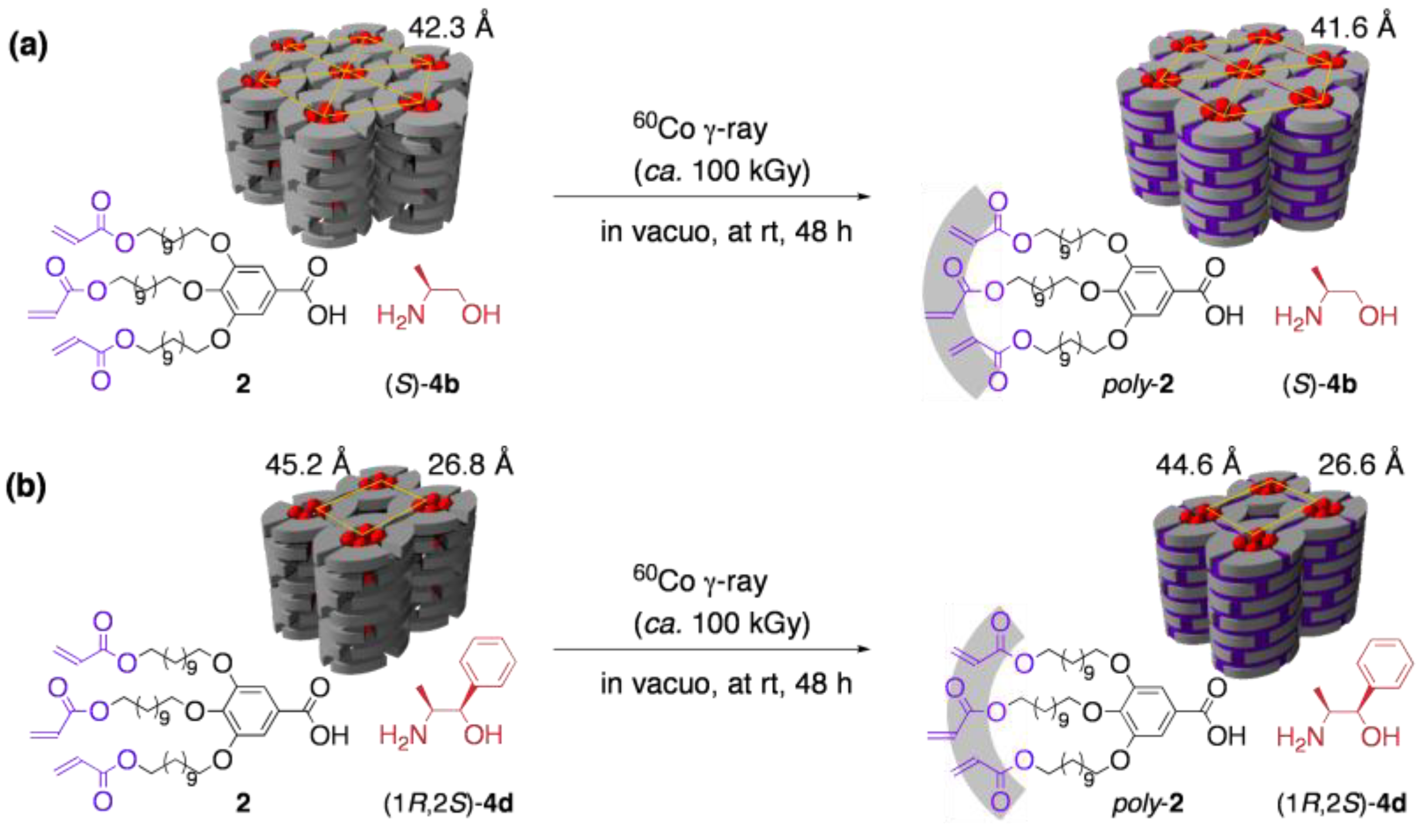
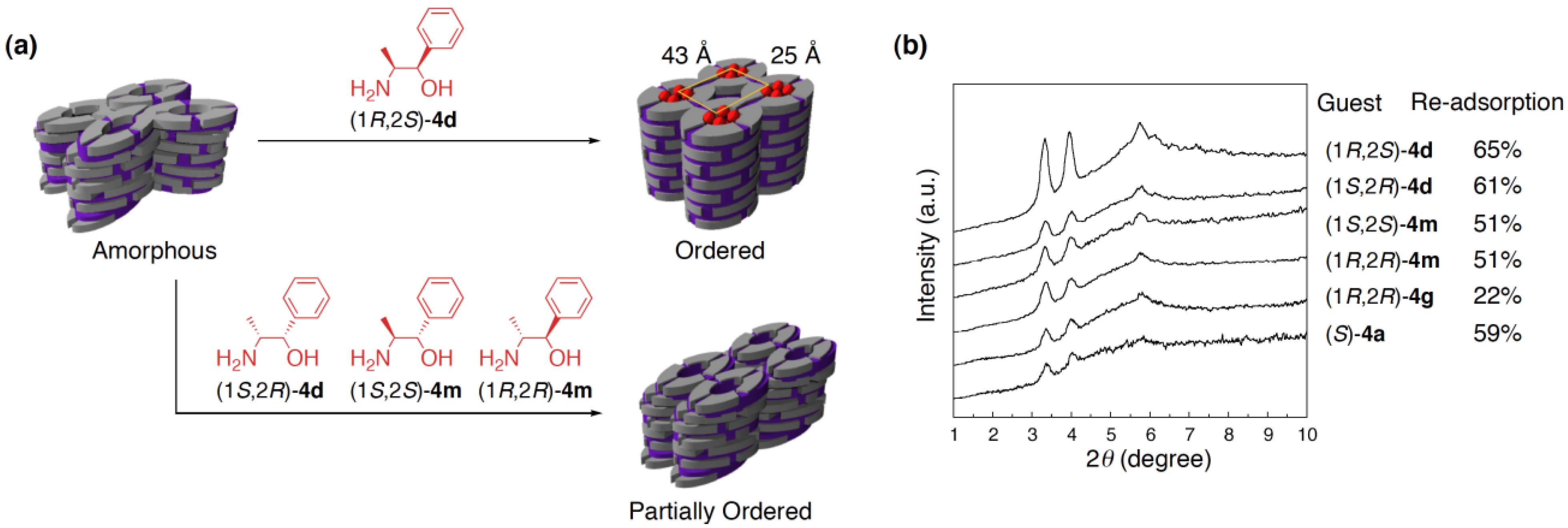
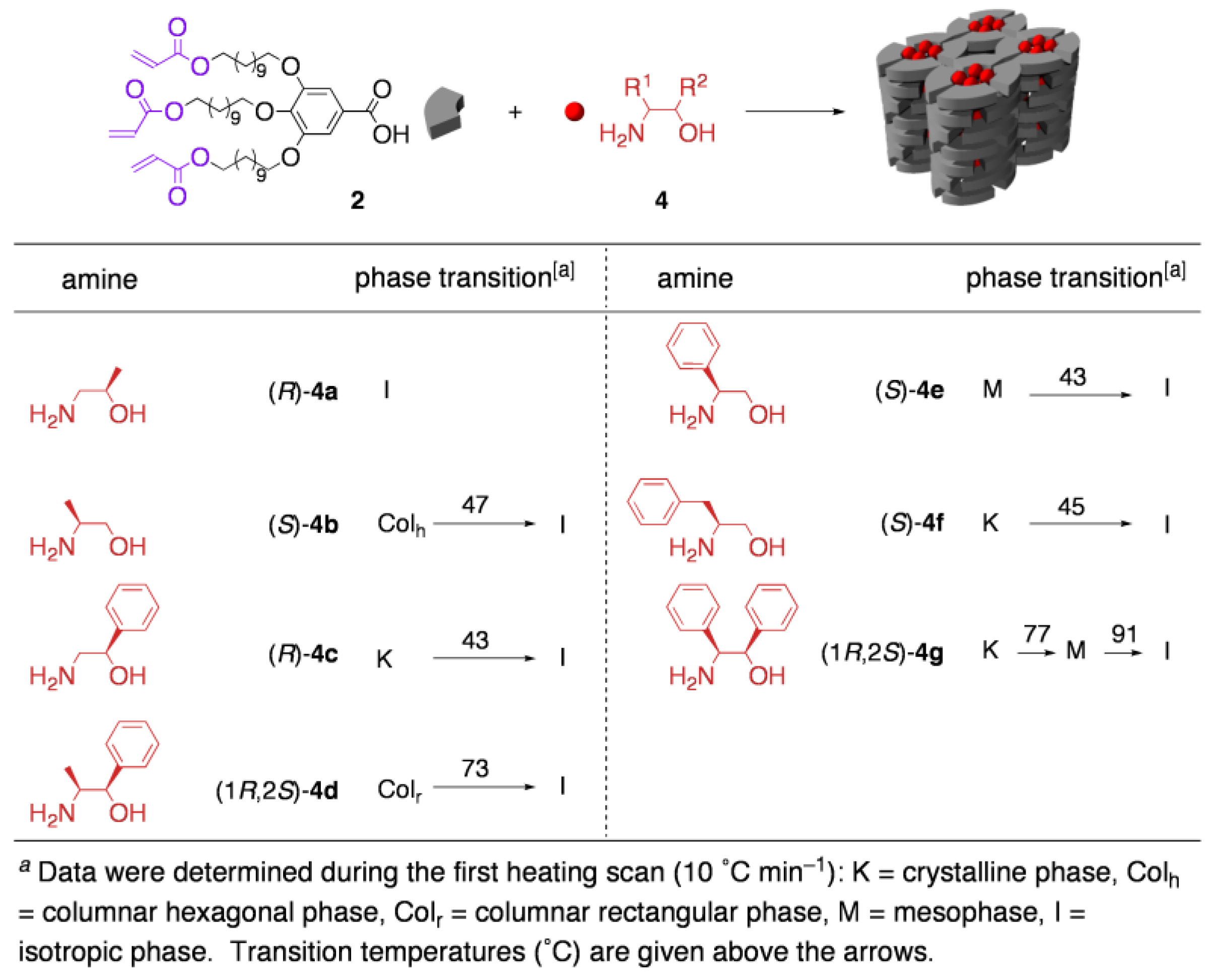
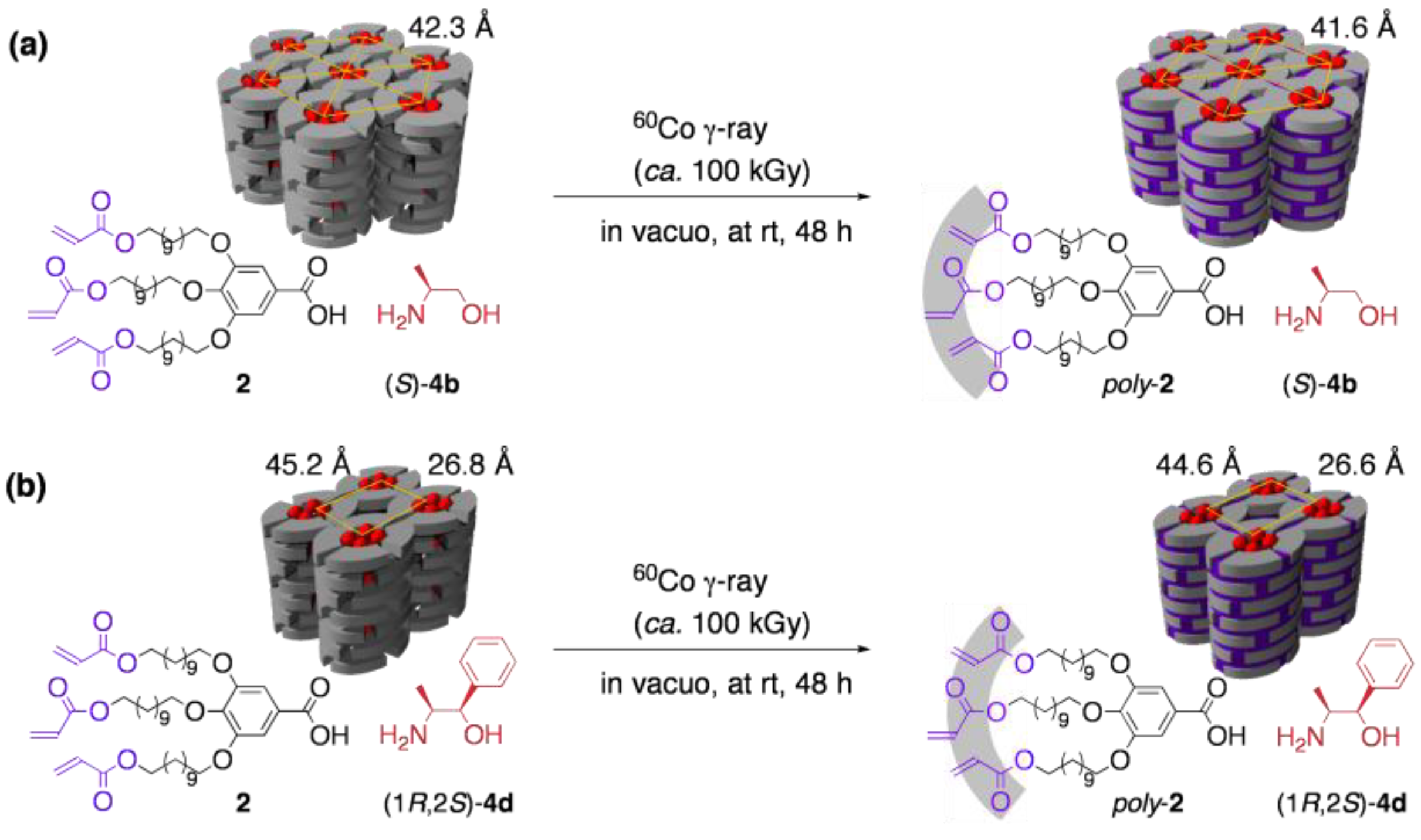
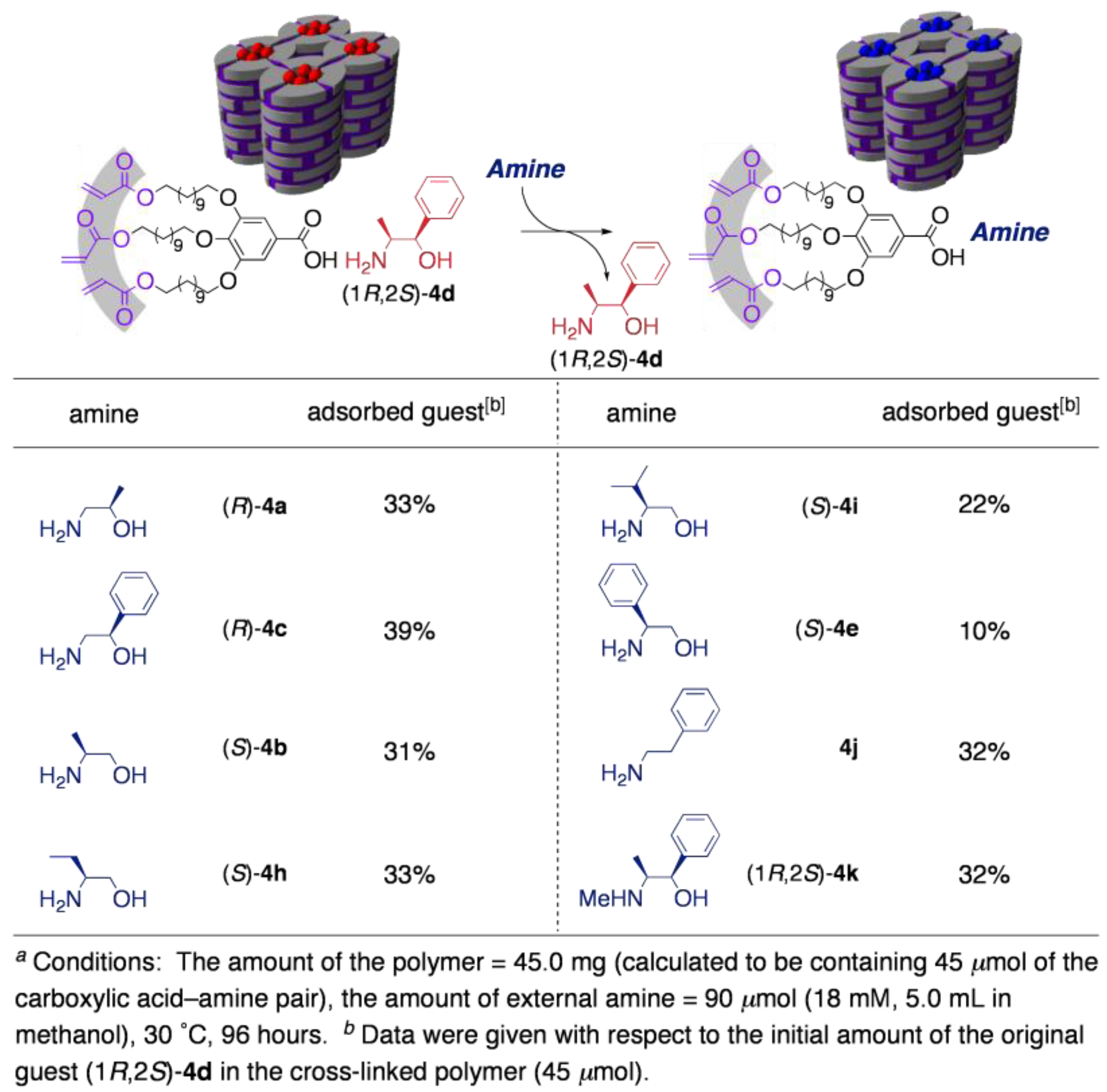
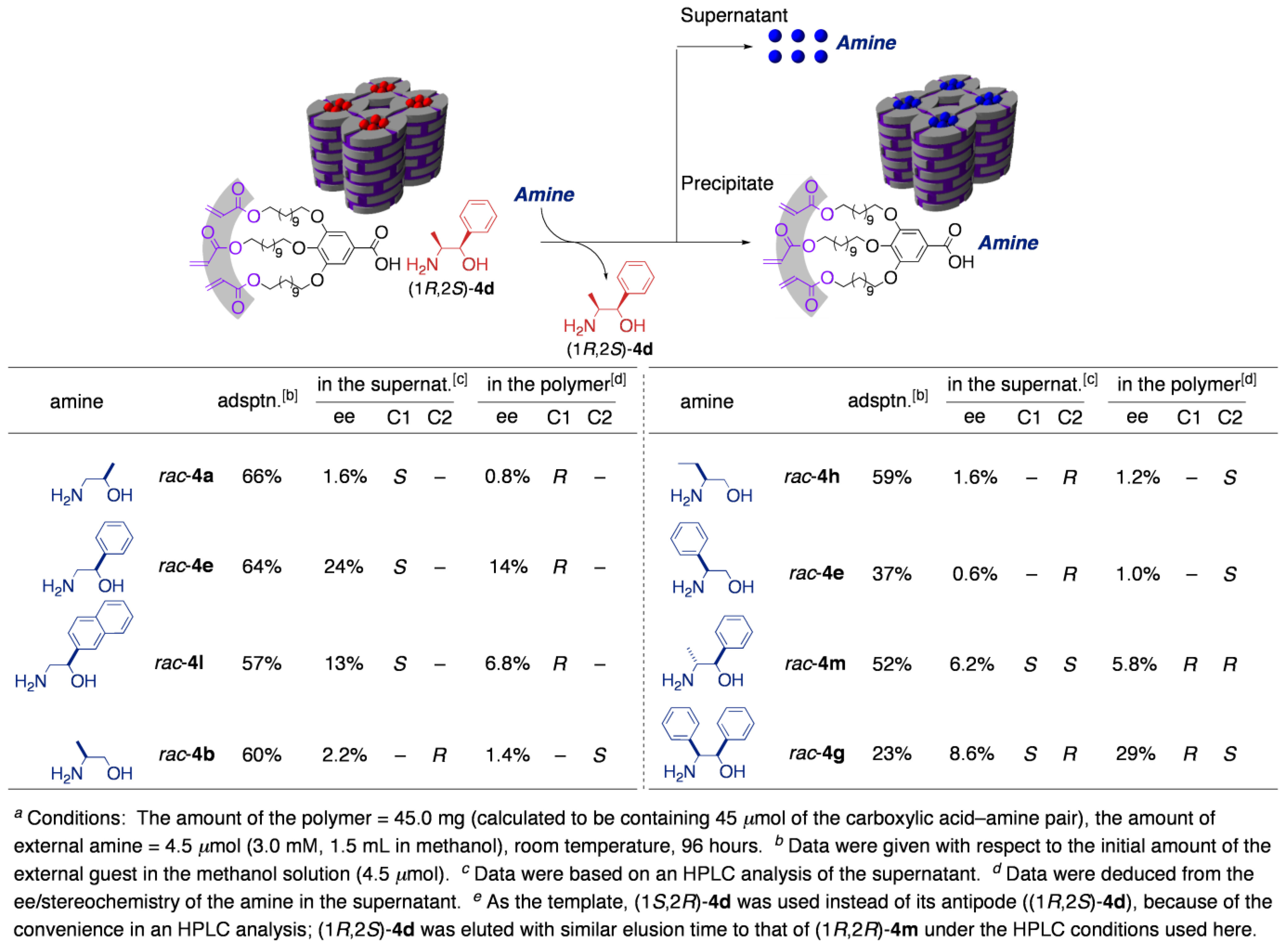
3.3. Chirality recognition by cross-linked LCs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
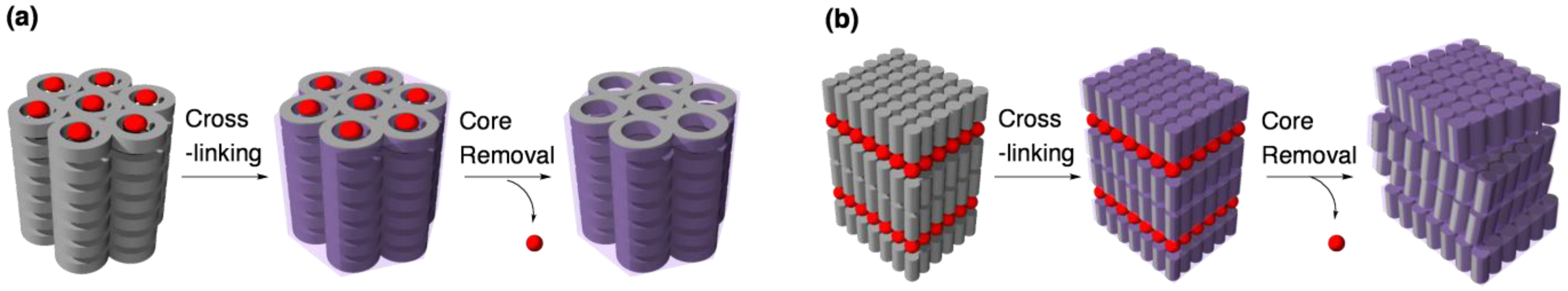
4. Guest-Responsive Frameworks
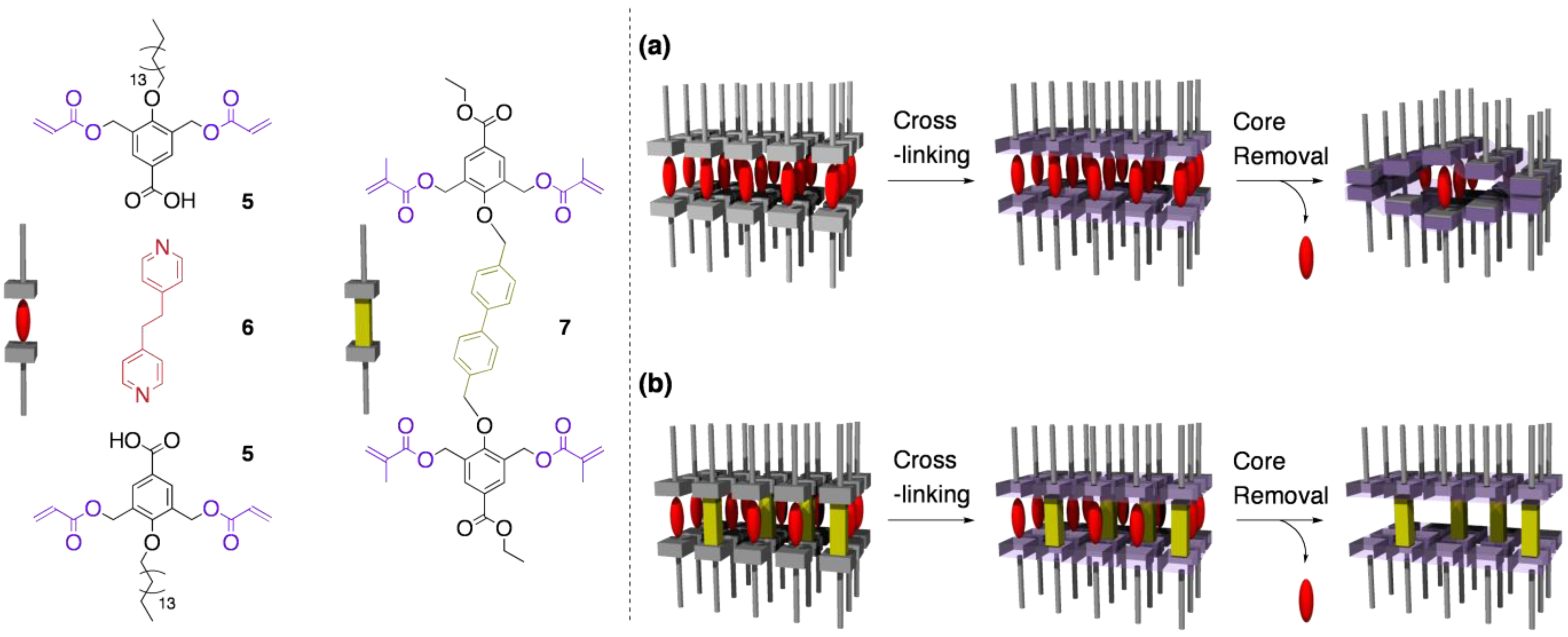
4.1. Relationship between LC packing mode and structural flexibility
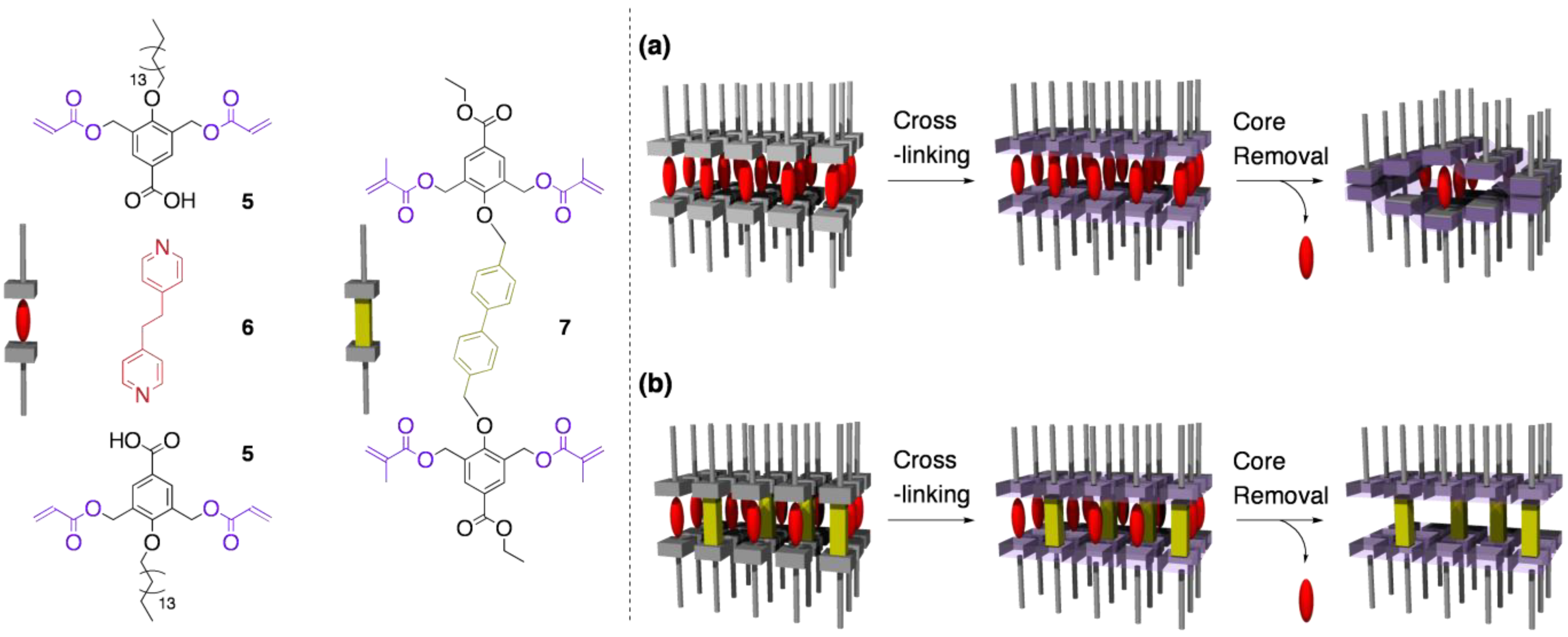
4.2. Tuning the structural rigidity of cross-linked LCs
4.3. Reversible structural switching of cross-linked LCs
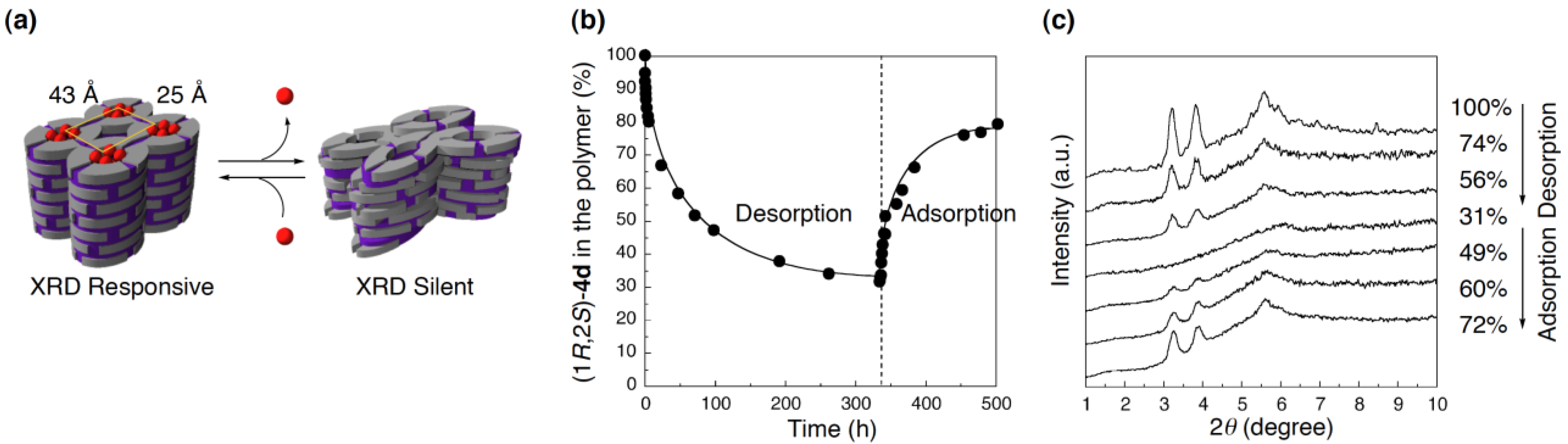
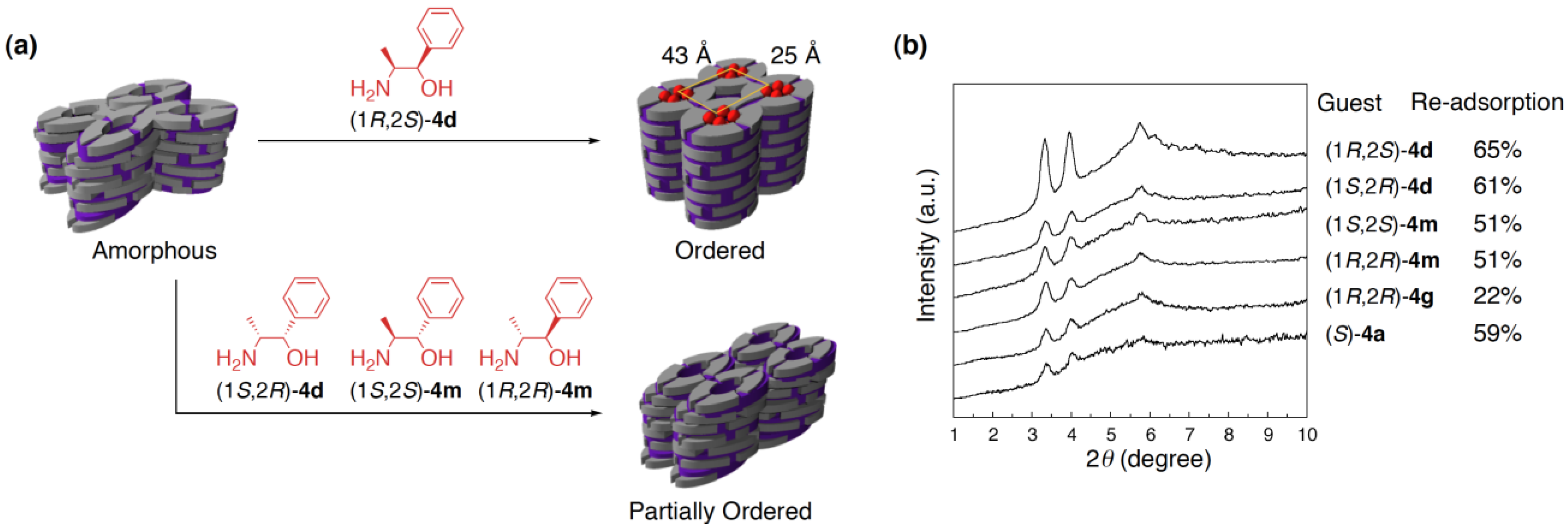
4.4. Guest-selective structural switching of cross-linked LCs
5. Tailored Reaction Media
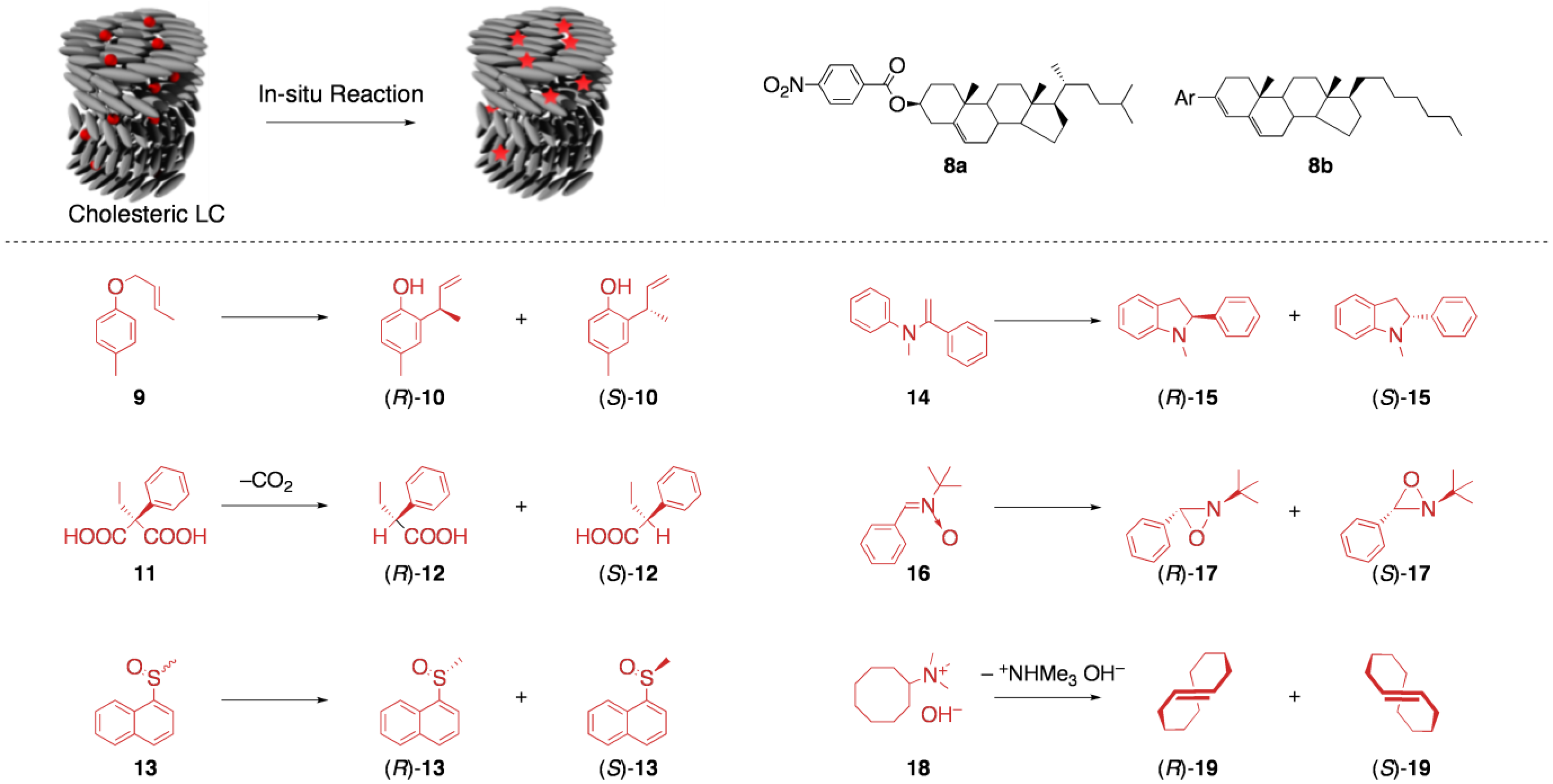
5.1. Asymmetric synthesis in cholesteric LCs
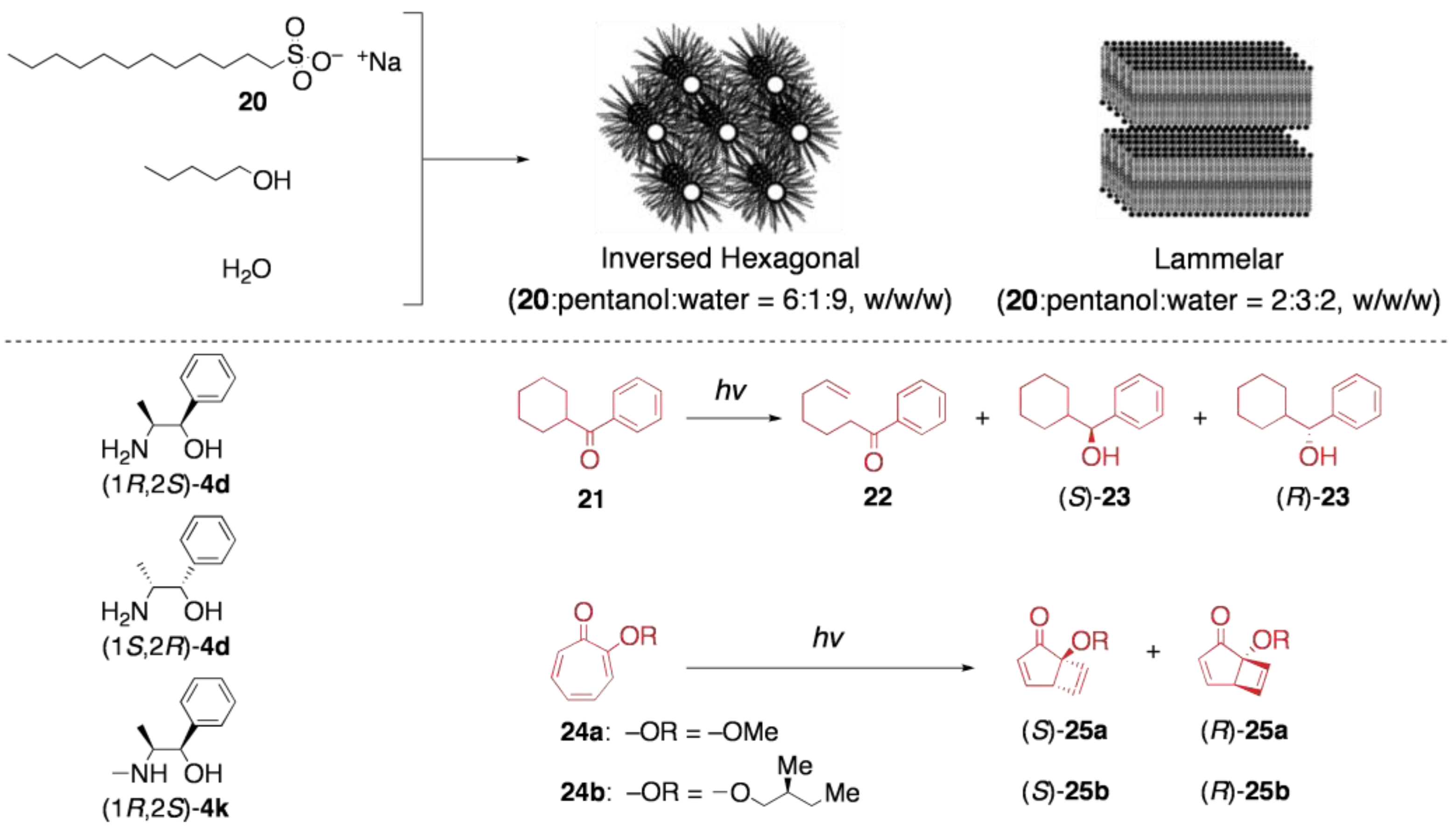
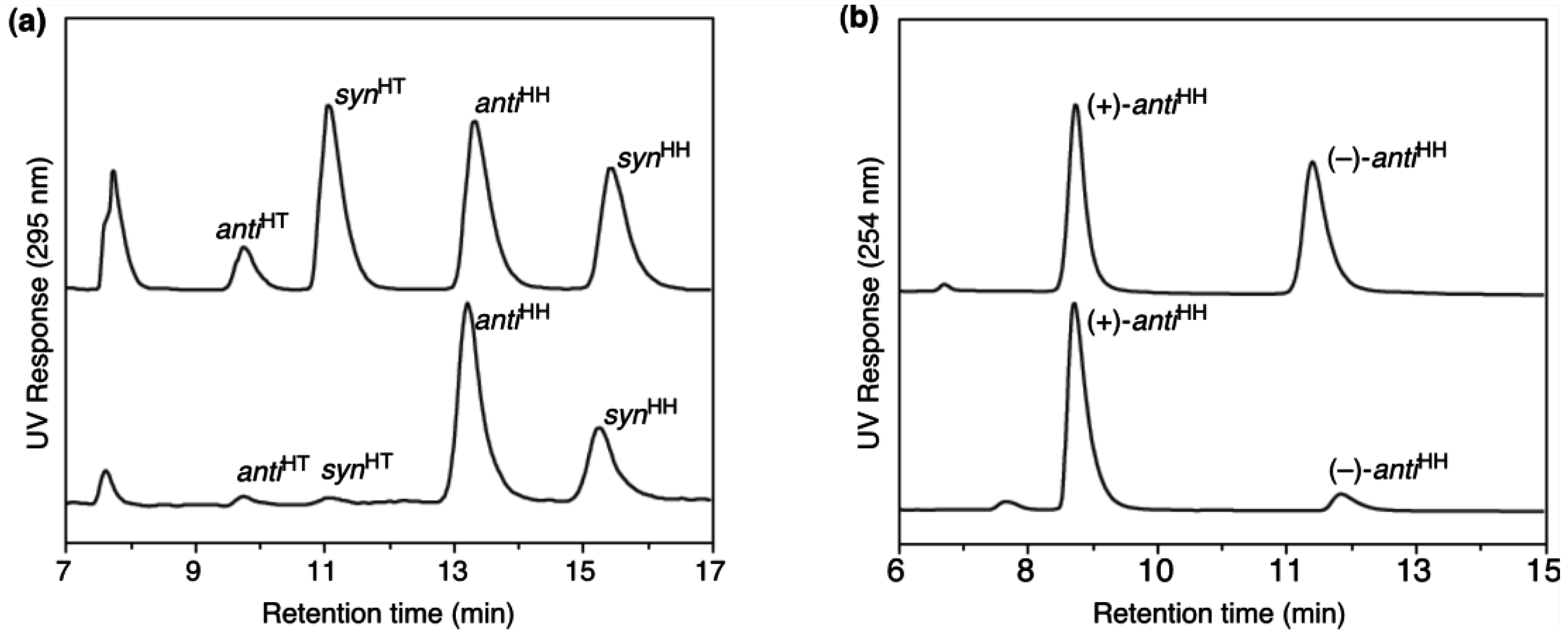
5.2. Asymmetric transformation of organic molecules in lyotropic LCs
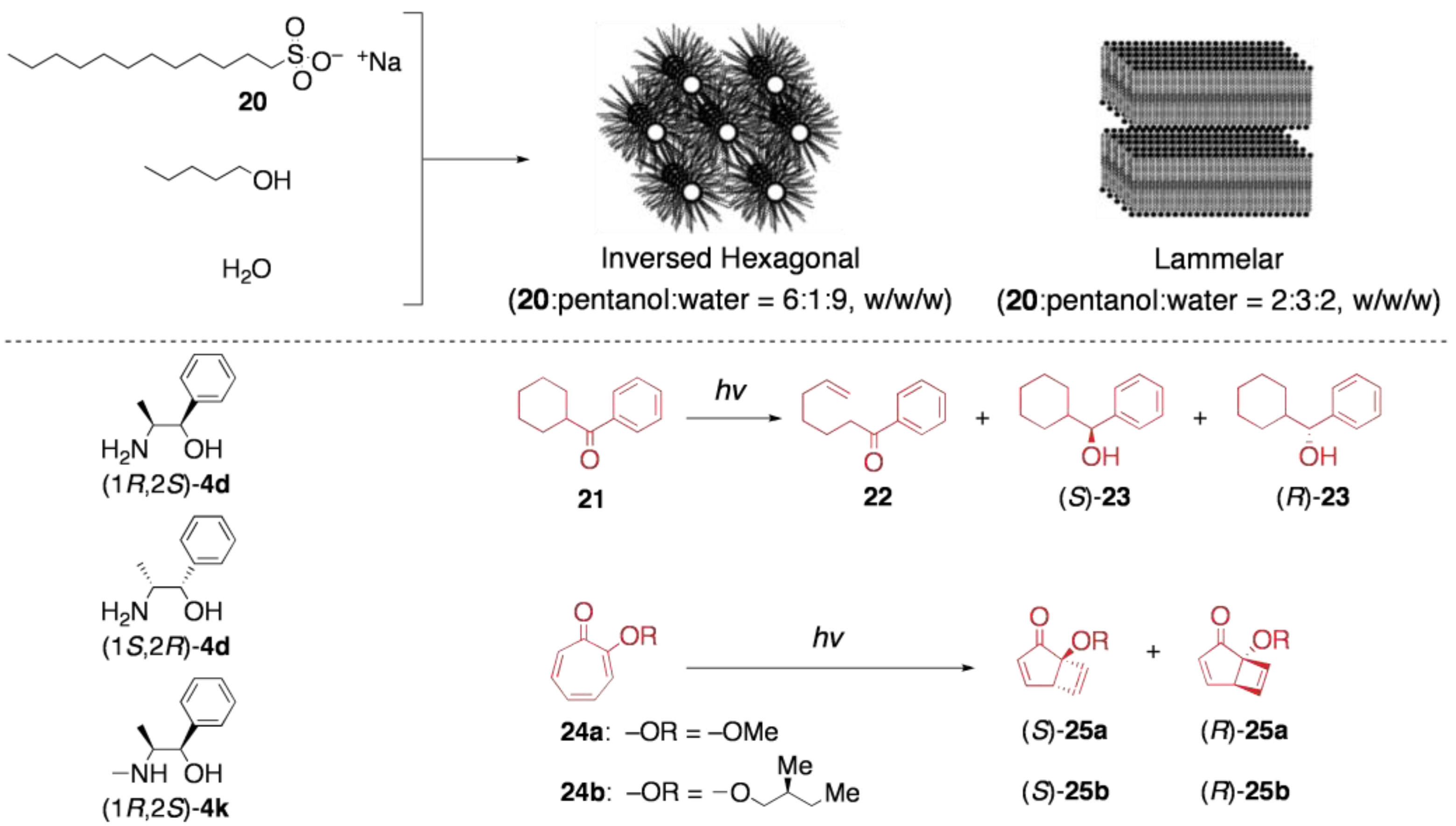
5.3. Asymmetric transformation of organic molecules in two-component thermotropic LCs
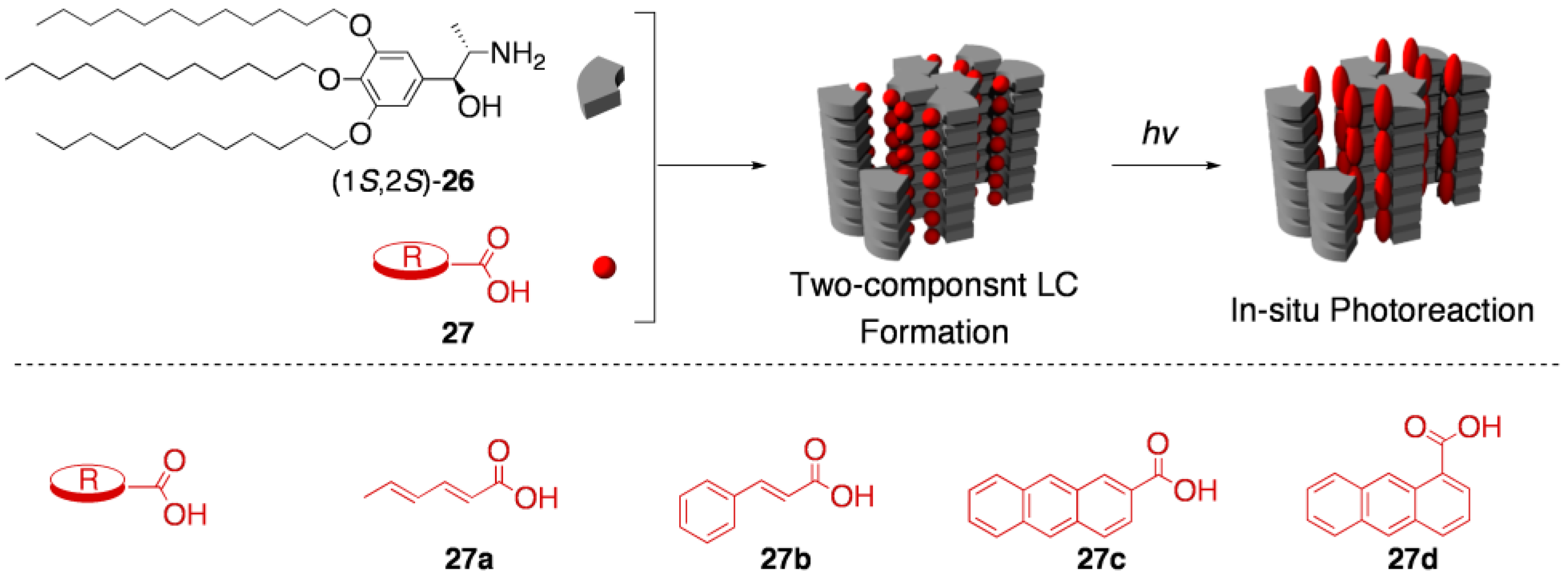
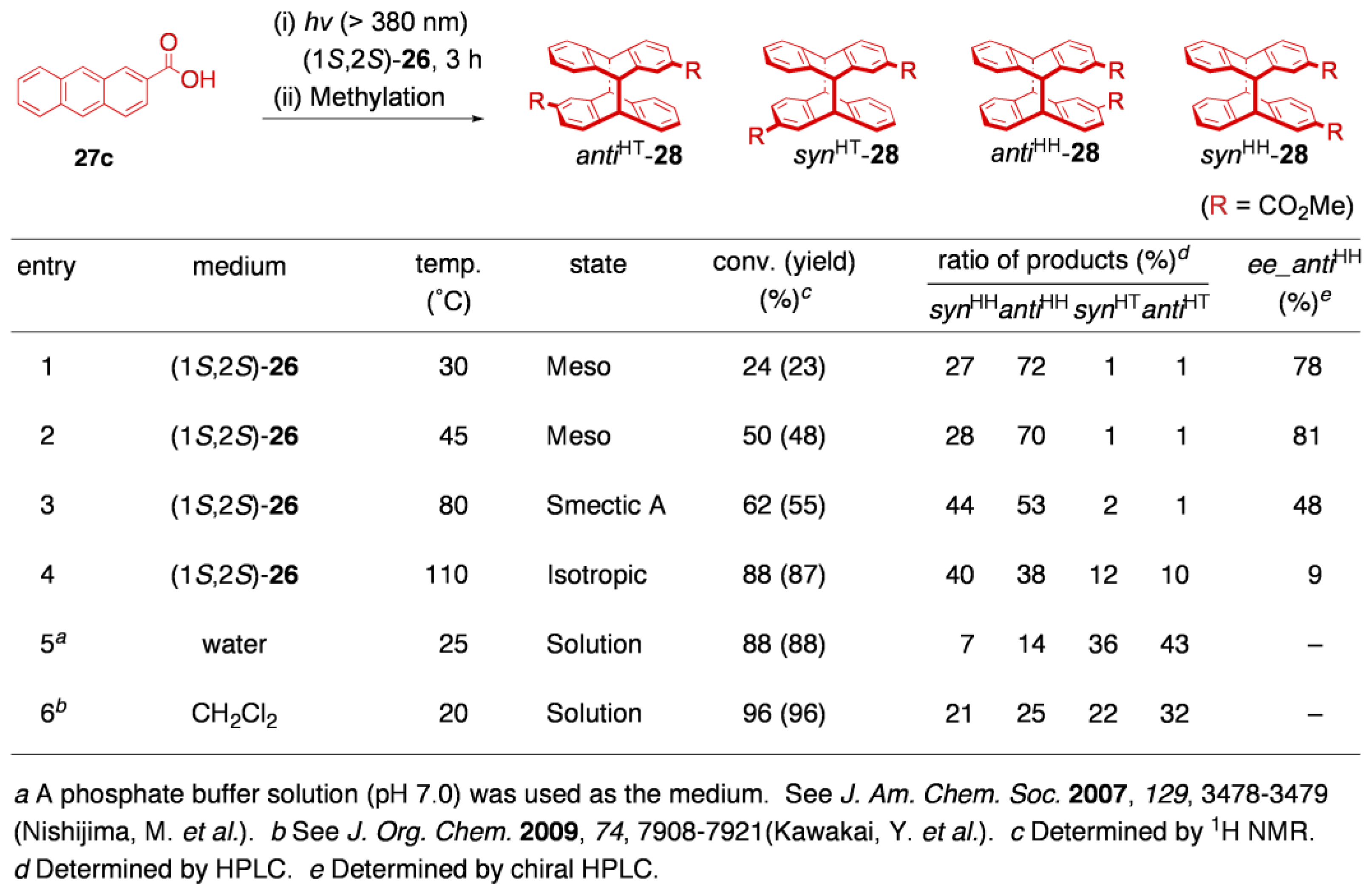
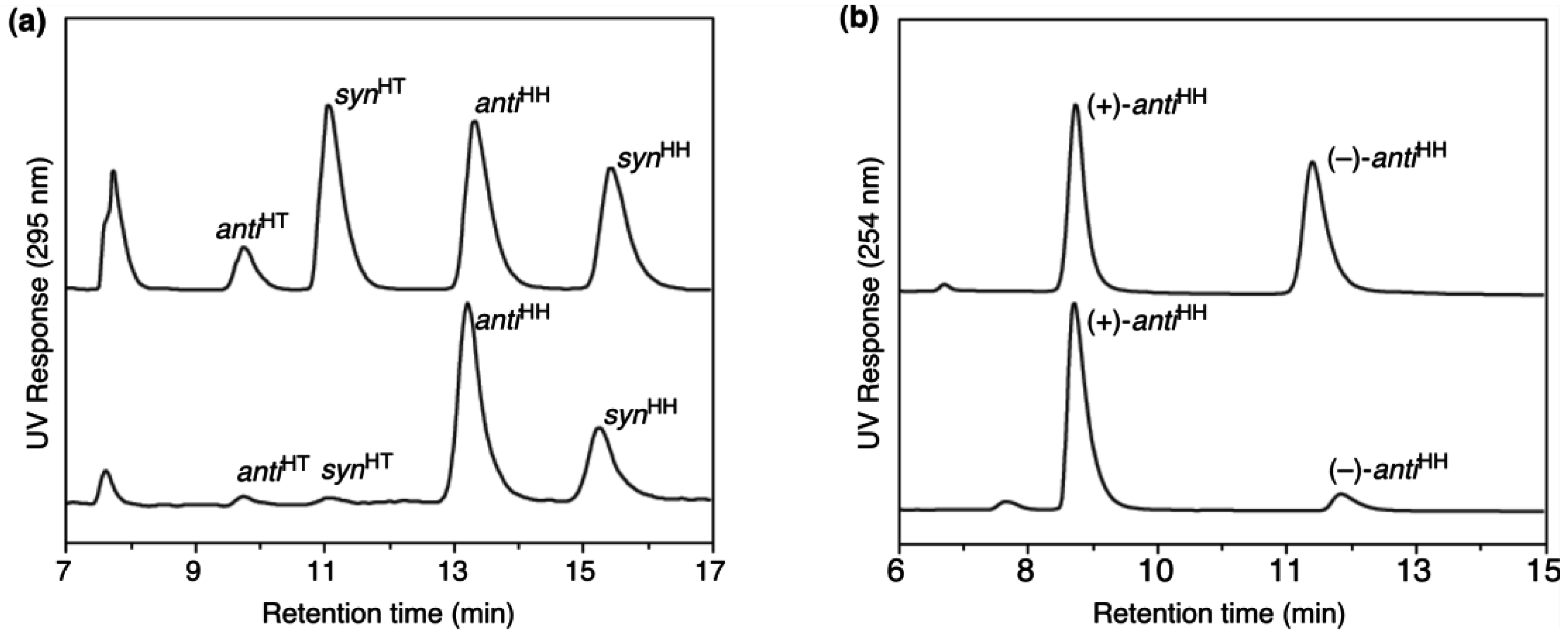
6. Conclusions
Acknowledgements
References and Notes
- Tanaka, K.; Toda, F. Solvent-free organic synthesis. Chem. Rev. 2000, 100, 1025–1074. [Google Scholar] [CrossRef] [PubMed]
- Toda, F. Isolation and optical resolution of materials utilizing inclusion crystallization. Top. Curr. Chem. 1987, 140, 43–69. [Google Scholar]
- Braga, D.; Grepioni, F. Reactions between or within molecular crystals. Angew. Chem. Int. Ed. 2004, 43, 4002–4011. [Google Scholar] [CrossRef]
- Moulton, B.; Zaworotko, M.J. From molecules to crystal engineering: Supramolecular isomerism and polymorphism in network solids. Chem. Rev. 2001, 101, 1629–1658. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, S.; Kitaura, R.; Noro, S. Functional porous coordination polymers. Angew. Chem. Int. Ed. 2004, 43, 2334–2375. [Google Scholar] [CrossRef]
- Rowsell, J.L.C.; Yaghi, O.M. Strategies for hydrogen storage in metal–organic frameworks. Angew. Chem., Int. Ed. 2005, 44, 4670. [Google Scholar] [CrossRef]
- Joy, A.; Ramamurthy, V. Chiral photochemistry within zeolites. Chem. Eur. J. 2000, 6, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.-H.; Wu, L.-Z.; Zhang, L.-P.; Chen, B. Supramolecular systems as microreactors: Control of product selectivity in organic phototransformation. Acc. Chem. Res. 2003, 36, 39–47. [Google Scholar] [CrossRef] [PubMed]
- MacGillivray, L.R.; Papaefstathiou, G.S.; Friscic, T.; Varshney, D.B.; Hamilton, T.D. Template-controlled synthesis in the solid-state. Top. Curr. Chem. 2004, 248, 297–310. [Google Scholar]
- Pinnavaia, T.J. Intercalated clay catalysts. Science 1983, 220, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.-M.; Biswick, T.T.; Choy, J.-H. Layered nanomaterials for green materials. J. Mater. Chem. 2009, 19, 2553–2563. [Google Scholar] [CrossRef]
- Solin, S.A. The nature and structural properties of graphite intercalation compounds. Adv. Chem. Phys. 1982, 49, 455–532. [Google Scholar]
- Lehn, J.-M. Supramolecular chemistry––Receptors, catalysts, and carriers. Science 1985, 227, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Conn, M.M.; Rebek, J. Self-assembling capsules. Chem. Rev. 1997, 97, 1647–1668. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, M.; Fujita, M. Self-assembled coordination cage as a molecular flask. Pure Appl. Chem. 2005, 77, 1107–1112. [Google Scholar] [CrossRef] [Green Version]
- Ishida, Y.; Amano, S.; Saigo, K. Template polymerization of columnar architectures based on the salts of a carboxylic acid and 2-amino alcohols: Application to the molecular recognition of 2-amino alcohols. Chem. Commun. 2003, 2338–2339. [Google Scholar]
- Ishida, Y.; Amano, S.; Iwahashi, N.; Saigo, K. Switching of structural order in a cross-linked polymer triggered by the desorption/adsorption of guest molecules. J. Am. Chem. Soc. 2006, 128, 13068–13069. [Google Scholar] [CrossRef] [PubMed]
- Amano, S.; Ishida, Y.; Saigo, K. Solid-state hosts by the template polymerization of columnar liquid crystals: Locked supramolecular architectures around chiral 2-amino alcohols. Chem. Eur. J. 2007, 13, 5186–5196. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Kai, Y.; Kato, S.; Misawa, A.; Amano, S.; Matsuoka, Y.; Saigo, K. Two-component liquid crystals as chiral reaction media: Highly enantioselective photo-dimerization of an anthracene derivative driven by the ordered micro-environment. Angew. Chem. Int. Ed. 2008, 47, 8241–8245. [Google Scholar] [CrossRef]
- Ishida, Y.; Achalkumar, A.S.; Kato, S.; Kai, Y.; Misawa, A.; Hayashi, Y.; Yamada, K.; Matsuoka, Y.; Shiro, M.; Saigo, K. Tunable chiral reaction media based on two-component liquid crystals: Regio-, diastereo-, and enantiocontrolled photodimerization of anthracenecarboxylic acids. J. Am. Chem. Soc. 2010, 132. in press. [Google Scholar]
- Broer, D.J.; Lub, J.; Mol, G.N. 3-D molecular architectures in thin film. In The Wiley Polymer Network Group Review Series; Nijenhuis, K.T., Mijs, W.J., Eds.; Wiley: New York, NY, USA, 1998; Volume 1, pp. 361–375. [Google Scholar]
- Burland, D.M.; Miller, R.D.; Walsh, C.A. Second-order nonlinearity in poled-polymer systems. Chem. Rev. 1994, 94, 31–75. [Google Scholar] [CrossRef]
- Ichimura, K. Photoalignment of liquid-crystal systems. Chem. Rev. 2000, 100, 1847–1874. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.G. Thermotropic liquid crystals as reaction media for mechanistic investigations. Tetrahedron 1988, 44, 3413–3475. [Google Scholar] [CrossRef]
- Brienne, M.-J.; Gabard, J.; Lehn, J.-M.; Stibor, I. Macroscopic expression of molecular recognition. Supramolecular liquid crystalline phases induced by association of complementary heterocyclic components. J. Chem. Soc. Chem. Commun. 1989, 1868–1870. [Google Scholar]
- Kato, T.; Fréchet, J.M.J. New approach to mesophase stabilization through hydrogen-bonding molecular interactions in binary mixtures. J. Am. Chem. Soc. 1989, 111, 8533–8534. [Google Scholar] [CrossRef]
- Haupt, K.; Mosbach, K. Molecularly imprinted polymers and their use in biomimetic sensors. Chem. Rev. 2000, 100, 2495–2504. [Google Scholar] [CrossRef] [PubMed]
- Wulff, G. Enzyme-like catalysis by molecularly imprinted polymers. Chem. Rev. 2002, 102, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Wulff, G. Molecular imprinting in cross-linked materials with the aid of molecular templates––A way towards artificial antibodies. Angew. Chem. Int. Ed. 1995, 34, 1812–1832. [Google Scholar] [CrossRef]
- Matsumoto, A.; Nagahama, S.; Odani, T. Molecular design and polymer structure control based on polymer crystal engineering. Topochemical polymerization of 1,3-diene mono- and dicarboxylic acid derivatives bearing a naphthylmethylammonium group as the countercation. J. Am. Chem. Soc. 2000, 122, 9109–9119. [Google Scholar] [CrossRef]
- Matsumoto, A.; Oshita, S.; Fujioka, D. A Novel organic intercalation system with layered polymer crystals as the host compounds derived from 1,3-diene carboxylic acids. J. Am. Chem. Soc. 2002, 124, 13749–13756. [Google Scholar] [PubMed]
- Tiddy, G.J.T. Surfactant––water liquid crystal phases. Phys. Rep. 1980, 57, 1–46. [Google Scholar] [CrossRef]
- Miller, S.A.; Kim, E.; Gray, D.H.; Gin, D.L. Heterogeneous catalysis with cross-linked lyotropic liquid crystal assemblies: Organic analogues to zeolites and mesoporous sieves. Angew. Chem. Int. Ed. 1999, 38, 3021–3026. [Google Scholar] [CrossRef]
- Xu, Y.; Gu, W.; Gin, D.L. Heterogeneous catalysis using a nanostructured solid acid resin based on lyotropic liquid crystals. J. Am. Chem. Soc. 2004, 126, 1616–1617. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Zhou, W.-J.; Gin, D.L. A Nanostructured, scandium-containing polymer for heterogeneous Lewis acid catalysis in water. Chem. Mater. 2001, 13, 1949–1951. [Google Scholar] [CrossRef]
- Pecinovsky, C.S.; Nicodemus, G.D.; Gin, D.L. Nanostructured, solid-state organic, chiral Diels-Alder catalysts via acid-induced liquid crystal assembly. Chem. Mater. 2005, 17, 4889–4891. [Google Scholar] [CrossRef]
- Lee, H.-K.; Lee, H.; Ko, Y.H.; Chang, Y.J.; Oh, N.-K.; Zin, W.-C.; Kim, K. Synthesis of a nanoporous polymer with hexagonal channels from supramolecular discotic liquid crystals. Angew. Chem. Int. Ed. 2001, 14, 2669–2671. [Google Scholar] [CrossRef]
- Kinbara, K.; Hashimoto, Y.; Sukegawa, M.; Nohira, H.; Saigo, K. Crystal structures of the salts of chiral primary amines with achiral carboxylic acids: Recognition of the commonly-occurring supramolecular assemblies of hydrogen-bond networks and their role in the formation of conglomerates. J. Am. Chem. Soc. 1996, 118, 3441–3449. [Google Scholar] [CrossRef]
- Yuge, T.; Sakai, T.; Kai, N.; Hisaki, I.; Miyata, M.; Tohnai, N. Topological classification and supramolecular chiralityof 21-helical ladder-type hydrogen-bond networks composed of primary ammonium carboxylates: Bundle control in 21-helical assemblies. Chem. Eur. J. 2008, 14, 2984–2993. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Yashima, E. Polysaccharide derivatives for chromatographic separation of enantiomers. Angew. Chem. Int. Ed. 1998, 37, 1020–1043. [Google Scholar] [CrossRef]
- Kishikawa, K.; Hirai, A.; Kohmto, S. Fixation of multilayered structures of liquid crystalline 2:1 complexes of benzoic acid derivatives and dipyridyl compounds and the effect of nanopillars on removal of the dipyridyl molecules from the polymers. Chem. Mater. 2008, 20, 1931–1935. [Google Scholar] [CrossRef]
- Muthuramu, K.; Ramamurthy, V. Photodimerization of coumarin in aqueous and micellar media. J. Org. Chem. 1982, 47, 3976–3979. [Google Scholar]
- Wolff, T.; Müller, N.; von Bünau, G. Regioselective photodimerization of 9-methylanthracene in homogeneous and micellar solutions. J. Photochem. 1983, 22, 61–70. [Google Scholar] [CrossRef]
- Lin, Y.C.; Weiss, R.G. Liquid-crystalline solvents as mechanistic probes. 24. A novel gelator of organic liquids and the properties of its gels. Macromolecules 1987, 20, 414–417. [Google Scholar] [CrossRef]
- Bhat, S.; Maitra, U. Hydrogels as reaction vessels: Acenaphthylene dimerization in hydrogels derived from bile acid analogues. Molecules 2007, 12, 2181–2189. [Google Scholar] [CrossRef] [PubMed]
- Dawn, A.; Fijita, N.; Haraguchi, S.; Sato, H.; Sada, K.; Shinkai, S. An organogel system can control the stereochemical course of anthracene photodimerization. Chem. Commun. 2009, 2100–2102. [Google Scholar]
- Imae, T.; Tsubota, T.; Okamura, H.; Mori, O.; Takagi, K.; Itoh, M; Sawaki, Y. Photocyclodimerization of cinnamic acid on a reaction matrix: Structural effect of molecular assemblies constructed by amphiphilic compounds. J. Phys. Chem. 1995, 99, 6046–6053. [Google Scholar] [CrossRef]
- Li, H.-R.; Wu, L.-Z.; Tung, C.-H. Reactions of singlet oxygen with olefins and sterically hindered amine in mixed surfactant vesicles. J. Am. Chem. Soc. 2000, 122, 2446–2451. [Google Scholar] [CrossRef]
- Weiss, R.G. Photochemical processes in liquid crystals. In Photochemistry in Organized and Constrained Media; Ramamurthy, V., Ed.; VCH: New York, NY, USA, 1991; Chapter 14. [Google Scholar]
- Leigh, W.J.; Workentin, M.S. Liquid crystals as solvents for spectroscopic, chemical reaction, and gas chromatographic applications. In Handbook of Liquid Crystals; Demun, D., Ed.; Wiley-VCH: Weinheim, Germany, 1998; pp. 839–895. [Google Scholar]
- Saeva, F.D.; Sharpe, P.E.; Olin, G.R. Asymmetric synthesis in a cholesteric liquid crystal solvent. J. Am. Chem. Soc. 1975, 97, 204–205. [Google Scholar]
- Verbit, L.; Halbert, T.R.; Patterson, R.B. Asymmetric decarboxylation of ethylphenylmalonic acid in cholesteric liquid crystal solvent. J. Org. Chem. 1975, 40, 1649–1650. [Google Scholar] [CrossRef]
- Pirkle, W.H.; Rinaldi, P.L. Asymmetric synthesis in liquid crystals: Independence of stereochemistry on the handedness of the cholesteric liquid crystal. J. Am. Chem. Soc. 1977, 99, 3510–3511. [Google Scholar] [CrossRef] [PubMed]
- Eskenazi, C.; Nicoud, J.F.; Kagan, H.B. Asymmetric induction in cholesteric media revisited. J. Org. Chem. 1979, 44, 995–999. [Google Scholar] [CrossRef]
- Seuron, P.; Solladie, G. Asymmetric induction in liquid crystals: Optically active trans-cyclooctene from Hofmann elimination in new cholesteric mesophases. J. Org. Chem. 1980, 45, 715–719. [Google Scholar] [CrossRef]
- Lv, F.-F.; Li, X.-W.; Wu, L.-Z.; Tung, C.-H. Photochemical reaction of cyclohexyl phenyl ketone within lyotropic liquid crystals. Tetrahedron 2008, 64, 1918–1923. [Google Scholar] [CrossRef]
- Lv, F.-F.; Chen, B.; Wu, L.-Z.; Zhang, L.-P.; Tung, C.-H. Enhanced stereoselectivity in photoelectrocyclization of tropolone ethers via confinement in chiral inductor-modified lyotropic liquid crystals. Org. Lett. 2008, 10, 3473–3476. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, T. Reversible photodimerization of water-soluble anthracenes included in γ-cyclodextrin. Chem. Lett. 1984, 13, 53–56. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ishida, Y. Organic Zeolite Analogues Based on Multi-Component Liquid Crystals: Recognition and Transformation of Molecules within Constrained Environments. Materials 2011, 4, 183-205. https://doi.org/10.3390/ma4010183
Ishida Y. Organic Zeolite Analogues Based on Multi-Component Liquid Crystals: Recognition and Transformation of Molecules within Constrained Environments. Materials. 2011; 4(1):183-205. https://doi.org/10.3390/ma4010183
Chicago/Turabian StyleIshida, Yasuhiro. 2011. "Organic Zeolite Analogues Based on Multi-Component Liquid Crystals: Recognition and Transformation of Molecules within Constrained Environments" Materials 4, no. 1: 183-205. https://doi.org/10.3390/ma4010183
APA StyleIshida, Y. (2011). Organic Zeolite Analogues Based on Multi-Component Liquid Crystals: Recognition and Transformation of Molecules within Constrained Environments. Materials, 4(1), 183-205. https://doi.org/10.3390/ma4010183