
Theoretical Investigation of Single-Atom Catalysts for Hydrogen Evolution Reaction Based on Two-Dimensional Tetragonal V2C2 and V3C3


Abstract
1. Introduction
2. Computational Details
3. Results and Discussion
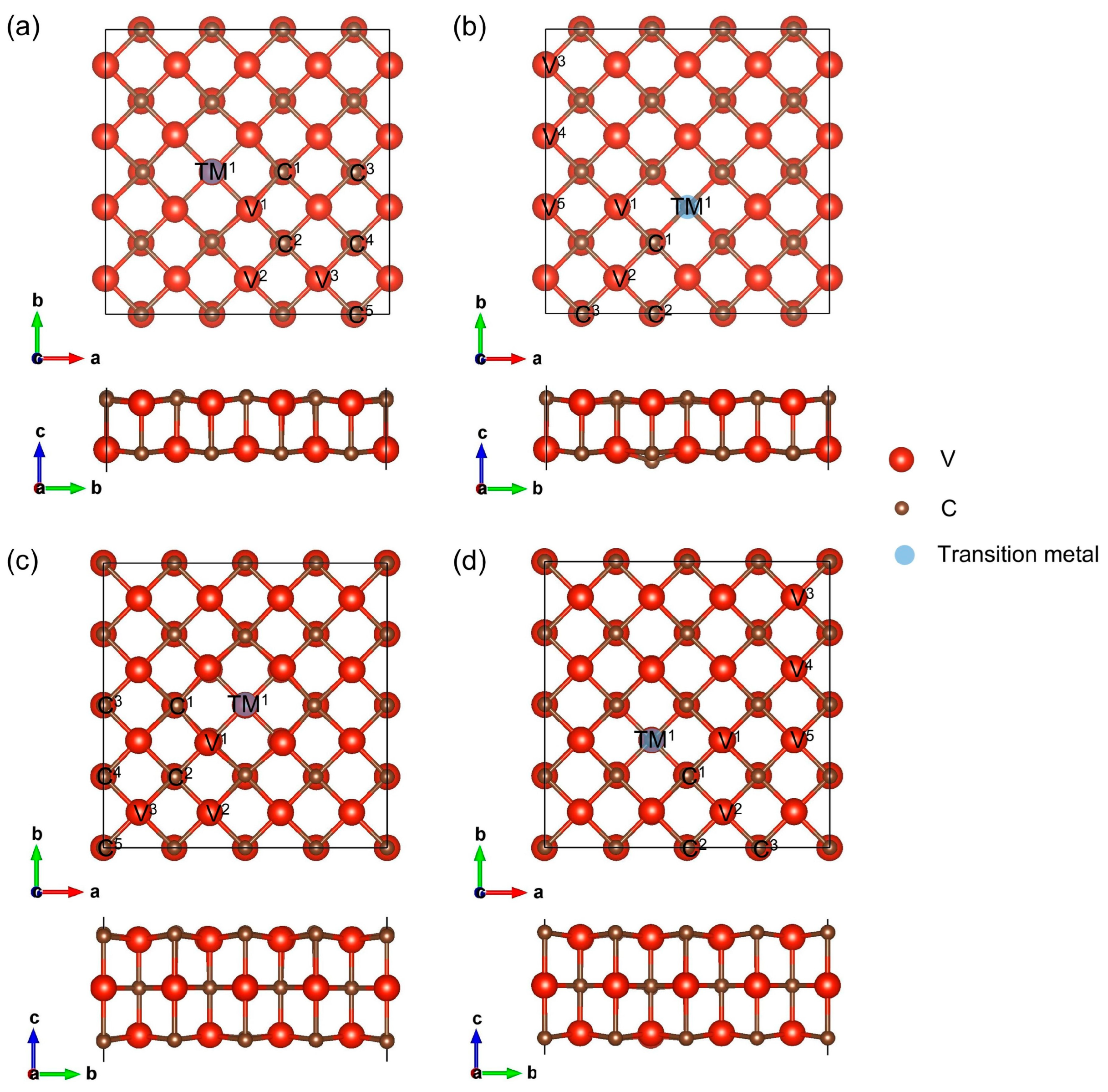
3.1. Structures, Stability, and Active Sites
3.2. Electronic Conductivity
3.3. Hydrogen Evolution Reaction Activity of V2C2-Based Catalysts
3.4. Hydrogen Evolution Reaction Activity of V3C3-Based Catalysts
4. Summary
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, Y.; Wang, R.; Shi, Y.; Wu, X. Can new energy become a breakthrough for economic development-based on clean development mechanism projects in less developed coastal cities. Sustainability 2024, 16, 8895. [Google Scholar] [CrossRef]
- Omer, A.M. Energy, environment and sustainable development. Renew. Sust. Energ. Rev. 2008, 12, 2265–2300. [Google Scholar] [CrossRef]
- Wang, S.; Lu, A.; Zhong, C.J. Hydrogen production from water electrolysis: Role of catalysts. Nano Converg. 2021, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Hu, L.; Zhao, P.; Lee, L.Y.S.; Wong, K.Y. Recent advances in electrocatalytic hydrogen evolution using nanoparticles. Chem. Rev. 2020, 120, 851–918. [Google Scholar] [CrossRef]
- Gielen, D.; Boshell, F.; Saygin, D.; Bazilian, M.D.; Wagner, N.; Gorini, R. The role of renewable energy in the global energy transformation. Energy Strateg. Rev. 2019, 24, 38–50. [Google Scholar] [CrossRef]
- Hassan, Q.; Algburi, S.; Sameen, A.Z.; Jaszczur, M.; Salman, H.M. Hydrogen as an energy carrier: Properties, storage methods, challenges, and future implications. Environ. Syst. Decis. 2024, 44, 327–350. [Google Scholar] [CrossRef]
- Yao, R.; Sun, K.; Zhang, K.; Wu, Y.; Du, Y.; Zhao, Q.; Liu, G.; Chen, C.; Sun, Y.; Li, J. Stable hydrogen evolution reaction at high current densities via designing the Ni single atoms and Ru nanoparticles linked by carbon bridges. Nat. Commun. 2024, 15, 2218. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Sherpa, K.; Chen, C.W.; Chen, L.; Dong, C.D. Breakthroughs and prospects in ruthenium-based electrocatalyst for hydrogen evolution reaction. J. Alloys Compd. 2023, 968, 172020. [Google Scholar] [CrossRef]
- Cheng, N.; Stambula, S.; Wang, D.; Banis, M.N.; Liu, J.; Riese, A.; Xiao, B.; Li, R.; Sham, T.K.; Liu, L.M.; et al. Platinum single-atom and cluster catalysis of the hydrogen evolution reaction. Nat. Commun. 2016, 7, 13638. [Google Scholar] [CrossRef]
- Wang, R.; Wang, H.; Luo, F.; Liao, S. Core-shell-structured low-platinum electrocatalysts for fuel cell applications. Electrochem. Energ. Rev. 2018, 1, 324–387. [Google Scholar] [CrossRef]
- Liang, S.; Hu, H.; Liu, J.; Shen, H.; Li, Q.; Qiu, N.; Guo, H.; Guo, X.; Du, S.; Zhu, Y.; et al. Nickel-nitride composite: An eco-friendly and efficient alternative to platinum for electrocatalytic hydrogen production. Appl. Catal. B Environ. 2023, 337, 123008. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, J.; Adimi, S.; Shen, H.; Thomas, T.; Ma, R.; Attfield, J.P.; Yang, M. Zirconium nitride catalysts surpass platinum for oxygen reduction. Nat. Mater. 2020, 19, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, S.; Feng, C.; Wu, H.; Zhang, L.; Zhang, J. Novel cobalt-doped Ni0.85Se chalcogenides (CoxNi0.85-xSe) as high active and stable electrocatalysts for hydrogen evolution reaction in electrolysis water splitting. ACS Appl. Mater. Interfaces 2018, 10, 40491–40499. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Cong, W.; Fujita, T.; Tang, Z.; Chen, M. High catalytic activity of nitrogen and sulfur Co-doped nanoporous graphene in the hydrogen evolution reaction. Angew. Chem. Int. Edit. 2015, 54, 2131–2136. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wan, J.; Zhang, H.; Fang, L.; Liu, L.; Huang, Z.; Li, J.; Gu, X.; Wang, Y. A new platinum-like efficient electrocatalyst for hydrogen evolution reaction at all pH: Single-crystal metallic interweaved V8C7 networks. Adv. Energy Mater. 2018, 8, 1800575. [Google Scholar] [CrossRef]
- Siahrostami, S.; Tsai, C.; Karamad, M.; Koitz, R.; García-Melchor, M.; Bajdich, M.; Vojvodic, A.; Abild-Pedersen, F.; Nørskov, J.K.; Studt, F. Two-Dimensional materials as catalysts for energy conversion. Catal. Lett. 2016, 146, 1917–1921. [Google Scholar] [CrossRef]
- Guo, Q.; Zhang, Q.; Zhang, T.; Zhou, J.; Xiao, S.; Wang, S.; Feng, Y.P.; Qiu, C.W. Colossal in-plane optical anisotropy in a two-dimensional van der Waals crystal. Nat. Photon. 2024, 18, 1170–1175. [Google Scholar] [CrossRef]
- Jiang, H.; Zheng, L.; Liu, Z.; Wang, X. Two-dimensional materials: From mechanical properties to flexible mechanical sensors. InfoMat 2020, 2, 1077–1094. [Google Scholar] [CrossRef]
- Cho, Y.S.; Kang, J. Two-dimensional materials as catalysts, interfaces, and electrodes for an efficient hydrogen evolution reaction. Nanoscale 2024, 16, 3936–3950. [Google Scholar] [CrossRef]
- Bai, S.; Yang, M.; Jiang, J.; He, X.; Zou, J.; Xiong, Z.; Liao, G.; Liu, S. Recent advances of MXenes as electrocatalysts for hydrogen evolution reaction. npj 2D Mater. Appl. 2021, 5, 78. [Google Scholar] [CrossRef]
- Yu, Y.; Zhou, J.; Sun, Z. Novel 2D transition-metal carbides: Ultrahigh performance electrocatalysts for overall water splitting and oxygen reduction. Adv. Funct. Mater. 2020, 30, 2000570. [Google Scholar] [CrossRef]
- Wang, L.; Huang, L.; Liang, F.; Liu, S.; Wang, Y.; Zhang, H. Preparation, characterization and catalytic performance of single-atom catalysts. Chin. J. Catal. 2017, 38, 1528–1539. [Google Scholar] [CrossRef]
- Pu, Z.; Amiinu, I.S.; Cheng, R.; Wang, P.; Zhang, C.; Mu, S.; Zhao, W.; Su, F.; Zhang, G.; Liao, S.; et al. Single-atom catalysts for electrochemical hydrogen evolution reaction: Recent advances and future perspectives. Nano-Micro Lett. 2020, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Liu, S.; Sun, G.; Zhang, C.; Pan, Y. Single-atom catalysts for hydrogen activation. Small 2023, 19, 2300956. [Google Scholar] [CrossRef]
- Wang, Z.; Yuan, X.; Guo, H.; Zhang, X.; Peng, J.; Pan, Y. Rational design principles of single-atom catalysts for hydrogen production and hydrogenation. Energy Environ. Sci. 2024, 17, 8019–8056. [Google Scholar] [CrossRef]
- Liu, J. Catalysis by supported single metal atoms. ACS Catal. 2017, 7, 34–59. [Google Scholar] [CrossRef]
- Qiu, H.J.; Ito, Y.; Cong, W.; Tan, Y.; Liu, P.; Hirata, A.; Fujita, T.; Tang, Z.; Chen, M. Nanoporous graphene with single-atom nickel dopants: An efficient and stable catalyst for electrochemical hydrogen production. Angew. Chem. Int. Edit. 2015, 54, 14031–14035. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, Y.; Guo, X.; Chen, C.; Dong, C.L.; Liu, R.S.; Han, C.P.; Li, Y.; Gogotsi, Y.; Wang, G. Single platinum atoms immobilized on an MXene as an efficient catalyst for the hydrogen evolution reaction. Nat. Catal. 2018, 1, 985–992. [Google Scholar] [CrossRef]
- Deng, J.; Li, H.; Xiao, J.; Tu, Y.; Deng, D.; Yang, H.; Tian, H.; Li, J.; Ren, P.; Bao, X. Triggering the electrocatalytic hydrogen evolution activity of the inert two-dimensional MoS2 surface via single-atom metal doping. Energy Environ. Sci. 2015, 8, 1594–1601. [Google Scholar] [CrossRef]
- Fei, H.; Dong, J.; Arellano-Jiménez, M.J.; Ye, G.; Kim, N.D.; Samuel, E.L.G.; Peng, Z.; Zhu, Z.; Qin, F.; Bao, J.; et al. Atomic cobalt on nitrogen-doped graphene for hydrogen generation. Nat. Commun. 2015, 6, 8668. [Google Scholar] [CrossRef]
- Zhang, L.; Jia, Y.; Gao, G.; Yan, X.; Chen, N.; Chen, J.; Soo, M.T.; Wood, B.; Yang, D.; Du, A.; et al. Graphene defects trap atomic Ni species for hydrogen and oxygen evolution reactions. Chem 2018, 4, 285–297. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, B.; Peng, Q.; Zhou, J.; Sun, Z. Mo2B2 MBene-supported single-atom catalysts as bifunctional HER/OER and OER/ORR electrocatalysts. J. Mater. Chem. A 2021, 9, 433. [Google Scholar] [CrossRef]
- Ullah, F.; Ayub, K.; Mahmood, T. High performance SACs for HER process using late first-row transition metals anchored on graphyne support: A DFT insight. Int. J. Hydrogen Energy 2021, 46, 37814–37823. [Google Scholar] [CrossRef]
- Fung, V.; Hu, G.; Wu, Z.; Jiang, D.E. Descriptors for hydrogen evolution on single atom catalysts in nitrogen-doped graphene. J. Phys. Chem. C 2020, 124, 19571–19578. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Bo, T.; Zhang, J.; Lu, Z.; Wang, Z.; Li, Y.; Wang, B.T. Novel two-dimensional tetragonal vanadium carbides and nitrides as promising materials for Li-ion batteries. Phys. Chem. Chem. Phys. 2019, 21, 19513–19520. [Google Scholar] [CrossRef]
- Xue, B.; Xing, H.; Zhao, W.; Xie, J.; Zeng, Q.; Yu, S.; Su, K. Structural properties and hydrogen evolution reactions of predicted two-dimensional VnCn(n = 2, 3, 6) with first-principles calculations. Phys. Scr. 2023, 98, 115906. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dis-persion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Andersen, H.C. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 1980, 72, 2384–2393. [Google Scholar] [CrossRef]
- Lasia, A. Mechanism and kinetics of the hydrogen evolution reaction. Int. J. Hydrogen Energy 2019, 44, 19484–19518. [Google Scholar] [CrossRef]
- Liao, X.; Lu, R.; Xia, L.; Liu, Q.; Wang, H.; Zhao, K.; Wang, Z.; Zhao, Y. Density functional theory for electrocatalysis. Energy Environ. Mater. 2022, 5, 157–185. [Google Scholar] [CrossRef]
- Li, Q.; Lu, Y.; Luo, Q.; Yang, X.; Yang, Y.; Tan, J.; Dong, Z.; Dang, J.; Li, J.; Chen, Y.; et al. Thermodynamics and kinetics of hydriding and dehydriding reactions in Mg-based hydrogen storage materials. J. Magnes. Alloys 2021, 9, 1922–1941. [Google Scholar] [CrossRef]
- Cheng, R.; Min, Y.; Li, H.; Fu, C. Electronic structure regulation in the design of low-cost efficient electrocatalysts: From theory to applications. Nano Energy 2023, 115, 108718. [Google Scholar] [CrossRef]
- Liu, R.; Fei, H.L.; Ye, G.L. Recent advances in single metal atom-doped MoS2 as catalysts for hydrogen evolution reaction. Tungsten 2020, 2, 147–161. [Google Scholar] [CrossRef]
- Gao, G.; O’Mullane, A.P.; Du, A. 2D MXenes: A new family of promising catalysts for the hydrogen evolution reaction. ACS Catal. 2017, 7, 494–500. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 2005, 152, J23. [Google Scholar] [CrossRef]
- Chen, X.; Sun, J.; Guan, J.; Ji, J.; Zhou, M.; Meng, L.; Chen, M.; Zhou, W.; Liu, Y.; Zhang, X. Enhanced hydrogen evolution reaction performance of MoS2 by dual metal atoms doping. Int. J. Hydrogen Energy 2022, 47, 23191–23200. [Google Scholar] [CrossRef]
- Jing, T.; Liang, D.; Hao, J.; Deng, M.; Cai, S. Single Pt atoms stabilized on Mo2TiC2O2 for hydrogen evolution: A first-principles investigation. J. Chem. Phys. 2019, 151, 024702. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, X.; Li, Y.; Wang, Y.; Hu, R.; Liu, B.; Zhang, P.; Xu, B.; Li, Y. Ru single atoms and nanoparticles immobilized on hierarchically porous carbon for robust dual-pH hydrogen evolution. J. Colloid Interf. Sci. 2025, 683, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.W.; Dai, J.H.; Zhang, Y.M.; Song, Y. Two-dimensional, ordered, double transition metal carbides (MXenes): A new family of promising catalysts for the hydrogen evolution reaction. J. Phys. Chem. C 2018, 122, 28113–28122. [Google Scholar] [CrossRef]

{kind=link}
| Ti | V | Cr | Mn | Fe | Co | Ni | Cu | |
|---|---|---|---|---|---|---|---|---|
| C monovacancy | 1.62 | 2.22 | 2.27 | 2.12 | 1.51 | 1.58 | 1.11 | 0.92 |
| V monovacancy | −5.02 | −3.81 | −3.34 | −2.84 | −2.49 | −2.14 | −1.14 |
| Ti | V | Cr | Mn | Fe | Co | Ni | Cu | |
|---|---|---|---|---|---|---|---|---|
| Csurf monovacancy | 1.72 | 2.62 | 2.90 | 2.78 | 2.41 | 1.86 | 1.04 | 0.77 |
| Vsurf monovacancy | −5.61 | −3.90 | −3.38 | −2.80 | −2.31 | −2.03 | −1.17 |
| C1 | C2 | C3 | C4 | C5 | V1 | V2 | V3 | TM1 | |
|---|---|---|---|---|---|---|---|---|---|
| Ti@(4 × 4)-V2C2-VC | 0.38 | 0.47 | 0.53 | 0.45 | 0.42 | −0.33 | 0.52 | 0.53 | 0.30 |
| V@(4 × 4)-V2C2-VC | 0.43 | 0.45 | 0.51 | 0.47 | 0.43 | −0.23 | 0.52 | 0.53 | 0.13 |
| Cr@(4 × 4)-V2C2-VC | 0.45 | 0.48 | 0.45 | 0.48 | 0.47 | −0.22 | 0.53 | 0.53 | −0.06 |
| Mn@(4 × 4)-V2C2-VC | −0.13 | 0.00 | 0.02 | −0.04 | −0.05 | −0.78 | 0.07 | 0.06 | −0.04 |
| Fe@(4 × 4)-V2C2-VC | 0.40 | 0.49 | 0.49 | 0.45 | 0.43 | −0.14 | 0.54 | 0.53 | 0.04 |
| Co@(4 × 4)-V2C2-VC | 0.35 | 0.43 | 0.43 | 0.41 | 0.40 | −0.23 | 0.50 | 0.48 | −0.23 |
| Ni@(4 × 4)-V2C2-VC | 0.39 | 0.47 | 0.48 | 0.46 | 0.41 | −0.18 | 0.56 | 0.52 | −0.19 |
| Cu@(4 × 4)-V2C2-VC | 0.35 | 0.51 | 0.49 | 0.42 | 0.42 | −0.04 | 0.53 | 0.51 | 0.19 |
| C1 | C2 | C3 | V1 | V2 | V3 | V4 | V5 | TM1 | |
|---|---|---|---|---|---|---|---|---|---|
| Ti@(4 × 4)-V2C2-VV | 0.43 | 0.46 | 0.47 | 0.52 | 0.54 | 0.52 | 0.52 | 0.51 | 0.78 |
| Cr@(4 × 4)-V2C2-VV | 0.46 | 0.46 | 0.47 | 0.51 | 0.50 | 0.52 | 0.50 | 0.51 | 0.31 |
| Mn@(4 × 4)-V2C2-VV | 0.42 | 0.45 | 0.47 | 0.57 | 0.50 | 0.51 | 0.51 | 0.53 | 0.31 |
| Fe@(4 × 4)-V2C2-VV | 0.38 | 0.46 | 0.51 | 0.59 | 0.50 | 0.53 | 0.50 | 0.54 | 0.35 |
| Co@(4 × 4)-V2C2-VV | 0.33 | 0.51 | 0.55 | 0.67 | 0.54 | 0.58 | 0.56 | 0.60 | 0.47 |
| Ni@(4 × 4)-V2C2-VV | 0.34 | 0.46 | 0.53 | 0.67 | 0.51 | 0.57 | 0.52 | 0.57 | 0.91 |
| Cu@(4 × 4)-V2C2-VV | 0.44 | 0.47 | 0.51 | 0.63 | 0.50 | 0.62 | 0.52 | 0.56 | 1.48 |
| C1 | C2 | C3 | C4 | C5 | V1 | V2 | V3 | TM1 | |
|---|---|---|---|---|---|---|---|---|---|
| Ti@(4 × 4)-V3C3-Vsurf-C | −0.16 | 0.19 | 0.17 | 0.09 | 0.02 | −0.21 | 0.66 | 0.66 | 0.18 |
| V@(4 × 4)-V3C3-Vsurf-C | −0.36 | 0.13 | 0.17 | 0.09 | 0.03 | −0.21 | 0.66 | 0.66 | 0.05 |
| Cr@(4 × 4)-V3C3-Vsurf-C | −0.91 | −0.70 | −0.77 | −0.87 | −0.88 | −1.14 | −0.30 | −0.32 | −0.09 |
| Mn@(4 × 4)-V3C3-Vsurf-C | −1.23 | −1.04 | −1.10 | −1.18 | −1.20 | −1.46 | −0.62 | −0.63 | −1.07 |
| Fe@(4 × 4)-V3C3-Vsurf-C | −0.71 | −0.63 | −0.64 | −0.73 | −0.75 | −0.89 | −0.14 | −0.16 | −0.01 |
| Co@(4 × 4)-V3C3-Vsurf-C | −0.14 | 0.02 | 0.02 | −0.06 | 0.06 | −0.23 | 0.51 | 0.49 | −0.08 |
| Ni@(4 × 4)-V3C3-Vsurf-C | 0.15 | 0.21 | 0.19 | 0.10 | 0.09 | −0.03 | 0.68 | 0.66 | 0.10 |
| Cu@(4 × 4)-V3C3-Vsurf-C | 0.23 | 0.28 | 0.21 | 0.12 | 0.11 | 0.04 | 0.68 | 0.66 | 0.41 |
| C1 | C2 | C3 | V1 | V2 | V3 | V4 | V5 | TM1 | |
|---|---|---|---|---|---|---|---|---|---|
| Ti@(4 × 4)-V3C3-Vsurf-V | 0.15 | 0.12 | 0.07 | 0.63 | 0.68 | 0.63 | 0.63 | 0.63 | 0.94 |
| Cr@(4 × 4)-V3C3-Vsurf-V | −0.04 | 0.04 | 0.08 | 0.65 | 0.61 | 0.64 | 0.63 | 0.63 | 0.45 |
| Mn@(4 × 4)-V3C3-Vsurf-V | −0.05 | 0.08 | 0.10 | 0.65 | 0.65 | 0.65 | 0.64 | 0.63 | 0.57 |
| Fe@(4 × 4)-V3C3-Vsurf-V | −0.12 | 0.08 | 0.12 | 0.66 | 0.65 | 0.65 | 0.64 | 0.63 | 0.59 |
| Co@(4 × 4)-V3C3-Vsurf-V | −0.25 | 0.03 | 0.08 | 0.64 | 0.61 | 0.66 | 0.61 | 0.63 | 0.60 |
| Ni@(4 × 4)-V3C3-Vsurf-V | −0.18 | 0.08 | 0.13 | 0.66 | 0.65 | 0.67 | 0.64 | 0.62 | 0.98 |
| Cu@(4 × 4)-V3C3-Vsurf-V | −0.04 | 0.10 | 0.10 | 0.64 | 0.67 | 0.67 | 0.65 | 0.63 | 1.37 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, B.; Zeng, Q.; Yu, S.; Su, K. Theoretical Investigation of Single-Atom Catalysts for Hydrogen Evolution Reaction Based on Two-Dimensional Tetragonal V2C2 and V3C3. Materials 2025, 18, 931. https://doi.org/10.3390/ma18050931
Xue B, Zeng Q, Yu S, Su K. Theoretical Investigation of Single-Atom Catalysts for Hydrogen Evolution Reaction Based on Two-Dimensional Tetragonal V2C2 and V3C3. Materials. 2025; 18(5):931. https://doi.org/10.3390/ma18050931
Chicago/Turabian StyleXue, Bo, Qingfeng Zeng, Shuyin Yu, and Kehe Su. 2025. "Theoretical Investigation of Single-Atom Catalysts for Hydrogen Evolution Reaction Based on Two-Dimensional Tetragonal V2C2 and V3C3" Materials 18, no. 5: 931. https://doi.org/10.3390/ma18050931
APA StyleXue, B., Zeng, Q., Yu, S., & Su, K. (2025). Theoretical Investigation of Single-Atom Catalysts for Hydrogen Evolution Reaction Based on Two-Dimensional Tetragonal V2C2 and V3C3. Materials, 18(5), 931. https://doi.org/10.3390/ma18050931