
Robust Self-Trapped Exciton Emission in Sb3+-Engineered Lead-Free Cs4SnBr6 Zero-Dimensional Perovskites


Abstract
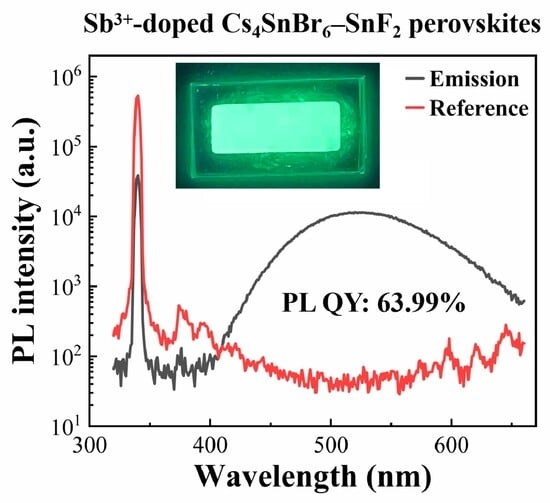
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Synthesis of Sb3+-Doped Cs4SnBr6–SnF2 Powders
2.3. Characterization
3. Results and Discussion
4. Conclusion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dong, H.; Ran, C.; Gao, W.; Zhang, Z.; Li, N.; Xia, Y.; Huang, B.; Liu, Z.; Chen, Y.; Li, X. Metal Halide Perovskite for Next-Generation Optoelectronics: Progresses and Prospects. eLight 2023, 3, 19. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, S.; Satapathi, S. Impact of Indium Doping in Lead-Free (CH3NH3)3Bi2−xInxI9 Perovskite Photovoltaics for Indoor and Outdoor Light Harvesting. ACS Appl. Electron. Mater. 2024, 6, 8360–8368. [Google Scholar] [CrossRef]
- Li, C.; Ma, J.; Li, P.; Song, Y.; Huang, W. Sensing and Ionovoltaic Power Generation of Two-Dimensional Cs3Sb2X9 (X = Cl/Br/I) Perovskite Microcrystals. ACS Appl. Electron. Mater. 2024, 6, 5255–5268. [Google Scholar]
- Dávid, A.; Morát, J.; Chen, M.; Gao, F.; Fahlman, M.; Liu, X. Mapping Uncharted Lead-Free Halide Perovskites and Related Low-Dimensional Structures. Materials 2024, 17, 491. [Google Scholar] [CrossRef]
- Li, M.; Xia, Z. Recent Progress of Zero-Dimensional Luminescent Metal Halides. Chem. Soc. Rev. 2021, 50, 2626–2662. [Google Scholar] [CrossRef]
- Sun, S.; Lu, M.; Gao, X.; Shi, Z.; Bai, X.; Yu, W.W.; Zhang, Y. 0D Perovskites: Unique Properties, Synthesis, and Their Applications. Adv. Sci. 2021, 8, 2102689. [Google Scholar] [CrossRef]
- Cheng, S.; Nikl, M.; Beitlerova, A.; Kucerkova, R.; Du, X.; Niu, G.; Jia, Y.; Tang, J.; Ren, G.; Wu, Y. Ultrabright and Highly Efficient All-Inorganic Zero-Dimensional Perovskite Scintillators. Adv. Opt. Mater. 2021, 9, 2100460. [Google Scholar] [CrossRef]
- Yakunin, S.; Benin, B.M.; Shynkarenko, Y.; Nazarenko, O.; Bodnarchuk, M.I.; Dirin, D.N.; Hofer, C.; Cattaneo, S.; Kovalenko, M.V. High-Resolution Remote Thermometry and Thermography Using Luminescent Low-Dimensional Tin-Halide Perovskites. Nat. Mater. 2019, 18, 846–852. [Google Scholar] [CrossRef]
- Benin, B.M.; Dirin, D.N.; Morad, V.; Wörle, M.; Yakunin, S.; Rainò, G.; Nazarenko, O.; Fischer, M.; Infante, I.; Kovalenko, M.V. Highly Emissive Self-Trapped Excitons in Fully Inorganic Zero-Dimensional Tin Halides. Angew. Chem. Int. Ed. 2018, 57, 11329–11333. [Google Scholar] [CrossRef]
- Tan, L.; Wang, W.; Li, Q.; Huang, Y.; Liu, J.; Sun, Z. Colloidal Syntheses of Zero-Dimensional Cs4SnX6 (X = Br, I) Nanocrystals with High Emission Efficiencies. Chem. Commun. 2020, 56, 387–390. [Google Scholar] [CrossRef]
- Lin, H.; Zhou, C.; Tian, Y.; Siegrist, T.; Ma, B. Low-Dimensional Organometal Halide Perovskites. ACS Energy Lett. 2017, 3, 54–62. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, H.; Wang, S.; Wang, Y.; Yang, Q.; Li, X.; Wang, H.; Gu, L.; Yu, Y.; Huang, K.; et al. Room Temperature Synthesis of All Inorganic Lead-Free Zero-Dimensional Cs4SnBr6 and Cs3KSnBr6 Perovskites. Inorg. Chem. 2019, 59, 533–538. [Google Scholar] [CrossRef]
- Chen, D.; Wan, Z.; Chen, X.; Yuan, Y.; Zhong, J. Large-Scale Room-Temperature Synthesis and Optical Properties of Perovskite-Related Cs4PbBr6 Fluorophores. J. Mater. Chem. C 2016, 4, 10646–10653. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, S.; He, M.; Zheng, W.; Wan, Q.; Liu, M.; Liao, X.; Zhan, W.; Yuan, C.; Liu, J.; et al. Stable Lead-Free Tin Halide Perovskite with Operational Stability >1200 h by Suppressing Tin (II) Oxidation. Angew. Chem. Int. Ed. 2022, 61, e202205463. [Google Scholar] [CrossRef] [PubMed]
- Jellicoe, T.C.; Richter, J.M.; Glass, H.F.J.; Tabachnyk, M.; Brady, R.; Dutton, S.E.; Rao, A.; Friend, R.H.; Credgington, D.; Greenham, N.C.; et al. Synthesis and Optical Properties of Lead-Free Cesium Tin Halide Perovskite Nanocrystals. J. Am. Chem. Soc. 2016, 138, 2941–2944. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Rao, H.; Fang, Y.; Zeng, J.; Pan, Z.; Zhong, X. Antioxidative Stannous Oxalate Derived Lead-Free Stable CsSnX3 (X = Cl, Br, and I) Perovskite Nanocrystals. Angew. Chem. Int. Ed. 2021, 60, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lin, Z.; Song, J.; Huang, Y.; Lu, X.; Xu, J.; Zhong, Y.; Li, Y.; Chen, J. Boosting the Self-Trapped Exciton Emission in Cs4SnBr6 Zero-Dimensional Perovskite via Rapid Heat Treatment. Nanomaterials 2023, 13, 2259. [Google Scholar] [CrossRef]
- He, C.; Qiu, J.; Mu, Z.; Zhang, Z.; Xia, Z.; Guo, X. Unlocking the Potential of Halide Perovskites Through Doping. CCS Chem. 2023, 5, 1961–1972. [Google Scholar] [CrossRef]
- Stroyuk, O.; Raievska, O.; Hauch, J.; Brabec, C.J. Doping/Alloying Pathways to Lead-Free Halide Perovskites with Ultimate Photoluminescence Quantum Yields. Angew. Chem. Int. Ed. 2023, 62, e202212668. [Google Scholar] [CrossRef]
- Chang, T.; Wei, Q.; Zeng, R.; Wang, S.; Chui, Y.; Zhao, Z.; Tang, Z.; Lu, C.; Huang, X. Efficient Energy Transfer in Te4+-Doped Cs2ZrCl6 Vacancy-Ordered Perovskites and Ultrahigh Moisture Stability via A-Site Rb-Alloying Strategy. J. Phys. Chem. Lett. 2021, 12, 1829–1837. [Google Scholar] [CrossRef]
- Luo, J.; Wang, X.; Li, S.; Liu, J.; Guo, Y.; Niu, G.; Yao, L.; Fu, Y.; Gao, L.; Dong, Q.; et al. Efficient and Stable Emission of Warm-White Light from Lead-Free Halide Double Perovskites. Nature 2018, 563, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shen, Q.; Chen, Y.; Zhang, Y.; Ding, J.; Bai, Y.; Gélvez-Rueda, M.C.; Grozema, F.C.; Gao, X. Bright Luminescence of Sb Doping in All-Inorganic Zinc Halide Perovskite Variant. J. Alloys Compd. 2022, 895, 162610. [Google Scholar] [CrossRef]
- Lin, Z.; Wang, A.; Huang, R.; Wu, H.; Huang, Y.; Lu, X.; Xu, J.; Zhong, Y.; Li, Y.; Chen, J. Manipulating the Sublattice Distortion Induced by Mn2+ Doping for Boosting the Emission Characteristics of Self-Trapped Excitons in Cs4SnBr6. J. Mater. Chem. C 2023, 11, 5680–5687. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, W.; Xu, R.; Yang, Y.; Yang, Z.; Ma, Z.; Zhao, J.; Zhao, C. Bright Tunable Luminescence of Sb3+ Doping in Zero-Dimensional Lead-Free Halide Cs3ZnCl5 Perovskite Crystals. Dalton Trans. 2022, 51, 10029–10035. [Google Scholar] [CrossRef]
- Jing, Y.; Liu, Y.; Jiang, X.; Li, M.; Mao, X.; Li, M.; Li, Y.; Wang, S.; Zhao, J.; Li, X. Sb3+ Dopant and Halogen Substitution Triggered Highly Efficient and Tunable Emission in Lead-Free Metal Halide Single Crystals. Chem. Mater. 2020, 32, 5327–5334. [Google Scholar] [CrossRef]
- Han, P.; Luo, C.; Yang, S.; Yang, Y.; Deng, W.; Han, K. All-Inorganic Lead-Free 0D Perovskites by a Doping Strategy to Achieve a PLQY Boost from <2% to 90%. Angew. Chem. Int. Ed. 2020, 59, 12709–12713. [Google Scholar]
- Su, B.; Li, M.; Song, E.; Wang, Z.; Zhang, Q.; Moon, C.; Liu, C.; Zhou, G.; Wang, G.; Xia, Z. Sb3+-Doping in Cesium Zinc Halides Single Crystals Enabling High-Efficiency Near-Infrared Emission. Adv. Funct. Mater. 2021, 31, 2105316. [Google Scholar] [CrossRef]
- Jin, J.; Peng, Y.; Xu, Y.; Li, T.; Li, L.; Wang, S.; Li, S.; Lu, X.; Zhang, J.; Wang, Y.; et al. Bright Green Emission from Self-Trapped Excitons Triggered by Sb3+ Doping in Rb4CdCl6. Chem. Mater. 2022, 34, 5717–5725. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, L.; Li, H.; Chen, R.; Wang, J.; Hu, J.; Li, X.J.; Jiao, H.; Zhang, Z.; Zhai, T.; et al. Defect Passivation in Air-Stable Tin(IV)-Halide Single Crystal for Emissive Self-Trapped Excitons. Adv. Funct. Mater. 2021, 31, 2108561. [Google Scholar] [CrossRef]
- Lin, K.H.; Liou, S.C.; Chen, W.L.; Cheng, H.C.; Chen, C.C.; Dong, C.L.; Chen, J.F.; Chen, K.H.; Chen, L.C. Tunable and Stable UV-NIR Photoluminescence from Annealed SiOx with Si Nanoparticles. Opt. Express 2013, 21, 23416–23424. [Google Scholar] [CrossRef]
- Chiara, R.; Ciftci, Y.O.; Queloz, V.I.E.; Törmä, P.; Scopelliti, R.; El Kazzi, M.; Ongaro, N.; Sessolo, M.; Bolink, H.J.; Palazon, F.; et al. Green-Emitting Lead-Free Cs4SnBr6 Zero-Dimensional Perovskite Nanocrystals with Improved Air Stability. J. Phys. Chem. Lett. 2020, 11, 618–623. [Google Scholar] [CrossRef]
- Li, S.; Luo, J.; Liu, J.; Tang, J. Self-Trapped Excitons in All-Inorganic Halide Perovskites: Fundamentals, Status, and Potential Applications. J. Phys. Chem. Lett. 2019, 10, 1999–2007. [Google Scholar] [CrossRef]
- Ulloa, J.M.; Llorens, J.M.; Alén, B.; Reyes, D.F.; Sales, D.L.; González, D.; Hierro, A. High Efficient Luminescence in Type-II GaAsSb-Capped InAs Quantum Dots upon Annealing. Appl. Phys. Lett. 2012, 101, 253112. [Google Scholar] [CrossRef]
- Bergman, L.; Chen, X.-B.; Morrison, J.L.; Huso, J.; Purdy, A.P. Photoluminescence Dynamics in Ensembles of Wide-Band-Gap Nanocrystallites and Powders. J. Appl. Phys. 2004, 96, 675–682. [Google Scholar] [CrossRef]
- Kuok, M.H.; Tan, L.S.; Shen, Z.X.; Huan, C.H.; Mok, K.F. A Raman Study of RbSnBr3. Solid State Commun. 1996, 97, 497–501. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, H.; Zhou, W.; Huang, R.; Song, J.; Lin, Z.; Zhang, Y.; Qiu, T.; Li, H. Robust Self-Trapped Exciton Emission in Sb3+-Engineered Lead-Free Cs4SnBr6 Zero-Dimensional Perovskites. Materials 2025, 18, 5324. https://doi.org/10.3390/ma18235324
Wu H, Zhou W, Huang R, Song J, Lin Z, Zhang Y, Qiu T, Li H. Robust Self-Trapped Exciton Emission in Sb3+-Engineered Lead-Free Cs4SnBr6 Zero-Dimensional Perovskites. Materials. 2025; 18(23):5324. https://doi.org/10.3390/ma18235324
Chicago/Turabian StyleWu, Haixia, Wendi Zhou, Rui Huang, Jie Song, Zhenxu Lin, Yi Zhang, Tianpei Qiu, and Hongliang Li. 2025. "Robust Self-Trapped Exciton Emission in Sb3+-Engineered Lead-Free Cs4SnBr6 Zero-Dimensional Perovskites" Materials 18, no. 23: 5324. https://doi.org/10.3390/ma18235324
APA StyleWu, H., Zhou, W., Huang, R., Song, J., Lin, Z., Zhang, Y., Qiu, T., & Li, H. (2025). Robust Self-Trapped Exciton Emission in Sb3+-Engineered Lead-Free Cs4SnBr6 Zero-Dimensional Perovskites. Materials, 18(23), 5324. https://doi.org/10.3390/ma18235324