
Substituent Effects on Crystal Engineering of DNBT-Based Energetic Cocrystals: Insights from Multiscale Computational Analysis

 ,
,
Abstract

1. Introduction
2. Methods
3. Results and Discussion
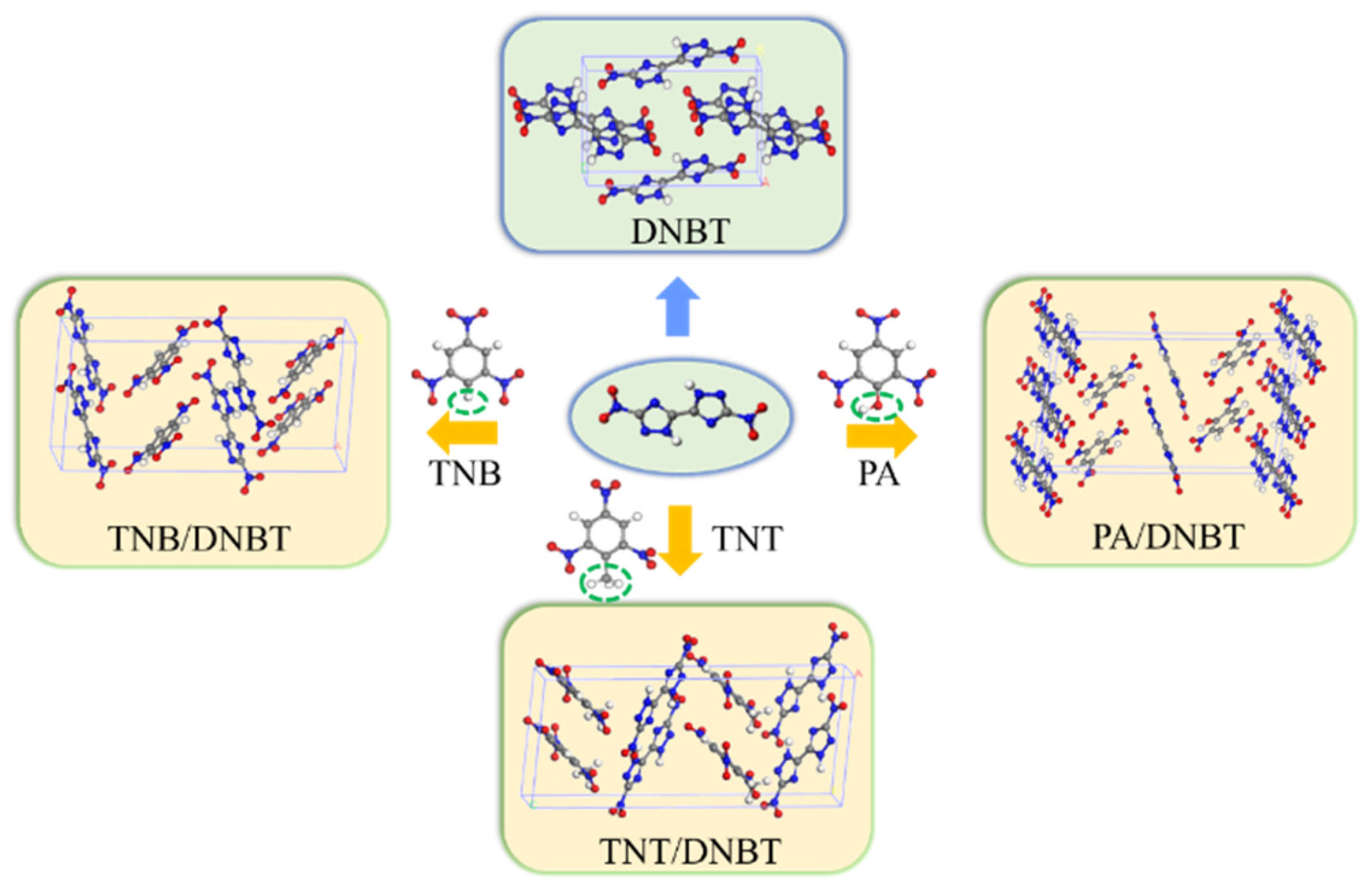
3.1. Crystal Structure Characteristics
3.2. Electrostatic Potential and π-Electron Analysis
3.3. Intermolecular Interactions
3.4. Binding Energy
3.5. Detonation Performance
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Y.; Feng, X.; Liu, H.; Hao, J.; Redfern, S.A.T.; Lei, W.; Liu, D.; Ma, Y. Route to high-energy density polymeric nitrogen t-N via HeN compounds. Nat. Commun. 2018, 9, 722. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Du, H.C.; Wang, F.; Gong, X.D.; Huang, Y.S. DFT Studies on a High Energy Density Cage Compound 4-Trinitroethyl-2,6,8,10,12-pentanitrohezaazaisowurtzitane. J. Phys. Chem. A 2011, 115, 6617–6621. [Google Scholar] [CrossRef] [PubMed]
- Huynh, M.H.V.; Hiskey, M.A.; Hartline, E.L.; Montoya, D.P.; Gilardi, R. Polyazido high-nitrogen compounds: Hydrazo- and azo-1,3,5-triazine. Angew. Chem. Int. Ed. 2010, 43, 4924–4928. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Lin, Q.; Wang, P.; Lu, M. Stabilization of the Pentazolate Anion in Three Anhydrous and Metal-Free Energetic Salts. Chem. Asian J. 2018, 13, 924–928. [Google Scholar] [CrossRef]
- Tian, L.; Xu, Y.; Lin, Q.; Wang, P.; Lu, M. Syntheses of Energetic cyclo-Pentazolate Salts. Chem. Asian J. 2019, 14, 2877–2882. [Google Scholar] [CrossRef]
- Chen, M.; Wang, W.; Chen, H.; Bai, L.; Xue, Z.; Wei, D.; Yang, H.; Niu, Y. Synthesis and Properties of Self-healing Metallopolymers with 5-Vinyltetrazole Units and Zn(II). Macromol. Res. 2019, 27, 96–104. [Google Scholar] [CrossRef]
- Bai, L.; Ma, A.; Wang, W.; Chen, H.; Xue, Z.; Cao, Y.; Niu, Y. A novel side-chain ferrocene-containing polymer by combination of Cu(0)-mediated SET-LRP of acrylonitrile and post-modification. Polym. Bull. 2019, 76, 2991–3002. [Google Scholar] [CrossRef]
- Sun, X.; Yu, M.; Mu, X.; Zhou, Z.; Wang, L.; Liu, J.; Liu, X. A facile approach to [1,2,4]triazolo[3,4-i]purine via PIDA oxidation ring-closing reaction. J. Heterocycl. Chem. 2021, 58, 2270–2279. [Google Scholar] [CrossRef]
- Wang, W.; Chen, M.; Niu, Y.; Tao, Q.; Bai, L.; Chen, H.; Cheng, Z. Facile one-pot synthesis and self-healing properties of tetrazole-based metallopolymers in the presence of iron salts. RSC Adv. 2017, 7, 47316–47323. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Göbel, M.; Klapötke, T.M.; Politzer, P. Reaction force analyses of nitro-aci tautomerizations of trinitromethane, the elusive trinitromethanol, picric acid and 2,4-dinitro-1H-imidazole. Theor. Chem. Acc. 2009, 124, 355–363. [Google Scholar] [CrossRef]
- Swain, P.K.; Singh, H.; Tewari, S.P. Energetic ionic salts based on nitrogen-rich heterocycles: A prospective study. J. Mol. Liq. 2010, 151, 87–96. [Google Scholar] [CrossRef]
- Talawar, M.B.; Sivabalan, R.; Mukundan, T.; Muthurajan, H.; Sikder, A.K.; Gandhe, B.R.; Rao, A.S. Environmentally compatible next generation green energetic materials (GEMs). J. Hazard. Mater. 2009, 161, 589–607. [Google Scholar] [CrossRef] [PubMed]
- Klapötke, T.M.; Sabaté, C.M. Nitrogen-Rich Tetrazolium Azotetrazolate Salts: A New Family of Insensitive Energetic Materials. Chem. Mater. 2008, 20, 1750–1763. [Google Scholar] [CrossRef]
- Dippold, A.A.; Klapötke, T.M.; Winter, N. Insensitive Nitrogen-Rich Energetic Compounds Based on the 5,5′-Dinitro-3,3′-bi-1,2,4-triazol-2-ide Anion. Eur. J. Inorg. Chem. 2012, 21, 3474–3484. [Google Scholar] [CrossRef]
- Lyu, R.; Huang, Z.; Deng, H.; Wei, Y.; Chen, J.; Zhong, K.; Wang, R.; Mou, C.; Wang, L. Exploration for the Optical Properties and Fluorescent Prediction of Nitrotriazole and Nitrofurazan: First-Principles and TD-DFT Calculations. ACS Omega 2022, 7, 19694–19705. [Google Scholar] [CrossRef]
- Tsyshevsky, R.V.; Pagoria, P.; Kuklja, M.M. Computational Design of Novel Energetic Materials: Dinitro-Bis-Triazolo-Tetrazine. J. Phys. Chem. C 2015, 119, 8512–8521. [Google Scholar] [CrossRef]
- Liu, G.; Gou, R.; Li, H.; Zhang, C. Polymorphism of Energetic Materials: A Comprehensive Study of Molecular Conformers, Crystal Packing, and the Dominance of Their Energetics in Governing the Most Stable Polymorph. Cryst. Growth Des. 2018, 18, 4174–4186. [Google Scholar] [CrossRef]
- Bu, R.; Li, H.; Zhang, C. Polymorphic Transition in Traditional Energetic Materials: Influencing Factors and Effects on Structure, Property, and Performance. Cryst. Growth Des. 2020, 20, 3561–3576. [Google Scholar] [CrossRef]
- Bennion, J.C.; Chowdhury, N.; Kampf, J.W.; Matzger, A.J. Hydrogen Peroxide Solvates of 2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane. Angew. Chem. 2016, 128, 13312–13315. [Google Scholar] [CrossRef]
- Jin, D.; Xu, J.; Zhang, H.; Lei, M.; Sun, J. Comparative Study of Experiments and Calculations on the Guest Molecules’ Escaping Mechanism of CL-20-Based Host–Guest Energetic Materials. J. Phys. Chem. C 2023, 127, 11641–11651. [Google Scholar] [CrossRef]
- Liu, G.; Tian, B.; Wei, S.-H.; Zhang, C. Polymorph-dependent initial thermal decay mechanism of energetic materials: A case of 1, 3, 5, 7-Tetranitro-1, 3, 5, 7-Tetrazocane. J. Phys. Chem. C 2021, 125, 10057–10067. [Google Scholar] [CrossRef]
- Bennion, J.C.; McBain, A.; Son, S.F.; Matzger, A.J. Design and Synthesis of a Series of Nitrogen-Rich Energetic Cocrystals of 5,5′-Dinitro-2H,2H′-3,3′-bi-1,2,4-triazole (DNBT). Cryst. Growth Des. 2015, 15, 2545–2549. [Google Scholar] [CrossRef]
- Bennett, A.J.; Foroughi, L.M.A.J. Matzger. Perchlorate-Free Energetic Oxidizers Enabled by Ionic Cocrystallization. J. Am. Chem. Soc. 2024, 146, 1771–1775. [Google Scholar] [CrossRef] [PubMed]
- Bennion, J.C.; Matzger, A.J. Development and Evolution of Energetic Cocrystals. Acc. Chem. Res. 2021, 54, 1699–1710. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, J.Z.; Jiang, S.L.; Yu, Y.; Chen, J. From intermolecular interactions to structures and properties of a novel cocrystal explosive: A first-principles study. Phys. Chem. Chem. Phys. 2016, 18, 26960–26969. [Google Scholar] [CrossRef]
- Ding, R.; Xu, J.; Tao, Y.; Sun, J.; Lei, M. Experimental and Theoretical Study on the Stability of CL-20-Based Host-guest Energetic Materials. J. Phys. Chem. A 2020, 124, 6389–6398. [Google Scholar] [CrossRef]
- Zhong, L.; Liu, D.; Hu, M.; Yang, X.; Liu, R.; Yao, Y. First-principles calculations of solid-phase enthalpy of formation of energetic materials. Commun. Chem. 2025, 8, 142. [Google Scholar] [CrossRef]
- Peng, Y.J.; Jiang, Y.X. Analyses of the influences of molecular vacancy defect on the geometrical structure, electronic structure, and vibration characteristics of Hexogeon energetic material. Acta Phys. Sin. 2015, 64, 243102. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, A.; Xue, X.; Jiang, D.; Zhu, Y.; Zhang, C. Crystal Packing of Impact-Sensitive High-Energy Explosives. Cryst. Growth Des. 2014, 14, 6101–6114. [Google Scholar] [CrossRef]
- Spackman, M.A.; Byrom, P.G. A novel definition of a molecule in a crystal. Chem. Phys. Lett. 1997, 267, 215–220. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Lu, T. A comprehensive electron wavefunction analysis toolbox for chemists, Multiwfn. J. Chem. Phys. 2024, 161, 082503. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q.X. Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems. J. Comput. Chem. 2022, 43, 539–555. [Google Scholar] [CrossRef]
- Li, H.; Bai, L.; Chen, X.; Xie, L.; Chen, J.; Yang, Z.; Fang, L.; Xia, Y.; Sun, G.; Gong, J. Strain-induced structural change and mechanical properties of 1,3,5-triamino-2,4,6-trinitrobenzene probed by neutron diffraction. Bull. Mater. Sci. 2021, 44, 53. [Google Scholar] [CrossRef]
- Liu, G.; Wei, S.-H.; Zhang, C. Review of the Intermolecular Interactions in Energetic Molecular Cocrystals. Cryst. Growth Des. 2020, 20, 7065–7079. [Google Scholar] [CrossRef]
- Cao, Y.; Yu, T.; Lai, W. Analysis of Intermolecular Interactions in Homologous Molecular Crystals of Energetic Materials. Energ. Mater. Front. 2020, 1, 95–102. [Google Scholar] [CrossRef]
- Liu, Y.; Fan, J.K.; Xue, Z.Q.; Lu, Y.J.; Zhao, J.N.; Hui, W.Y. Crystal Structure and Noncovalent Interactions of Heterocyclic Energetic Molecules. Molecules 2022, 27, 4969. [Google Scholar] [CrossRef]
- Li, S.J.; Bu, R.P.; Gou, R.J. Hirshfeld Surface Method and Its Application in Energetic Crystals. Cryst. Growth Des. 2021, 21, 6619–6634. [Google Scholar] [CrossRef]
- Abraham, B.M.; Vaitheeswaran, G. From van der Waals interactions to structures and properties of 3,3′-dinitro-5,5′-bis-1,2,4-triazole-1,1′-diolate based energetic materials. Mater. Chem. Phys. 2020, 240, 122175. [Google Scholar] [CrossRef]
- Eckhardt, C.J.; Gavezzotti, A. Computer simulations and analysis of structural and energetic features of some crystalline energetic materials. J. Phys. Chem. B 2007, 111, 3430–3437. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Duan, X.; Li, H. The structural evolution of CL-20-based energetic host–guest solvates at decomposition temperature according to the perceptions of THz spectroscopy. CrystEngComm 2024, 26, 2322–2332. [Google Scholar] [CrossRef]
- Sućeska, M. Calculation of the Detonation Properties of CHNO explosives. Propell. Explos. Pypot. 2010, 16, 197–202. [Google Scholar] [CrossRef]
- Politzer, P.; Martinez, J.; Murray, J.S. An electrostatic interaction correction for improved crystal density prediction. Mol. Phys. 2009, 107, 2095–2101. [Google Scholar] [CrossRef]
- Keshavarz, M.H. Simple Relationship for Predicting Impact Sensitivity of Nitroaromatics, Nitramines, and Nitroaliphatics. Propell. Explo. Pyrot. 2010, 35, 175–181. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cocrystals | EA/Ha | EB/Ha | ETotal/Ha | EBinding/kJ mol−1 |
|---|---|---|---|---|
| TNB/DNBT | −3567.13 | −3380.98 | −6948.15 | 113.38 |
| TNT/DNBT | −3567.10 | −3538.05 | −7105.19 | 118.65 |
| PA/DNBT | −3567.13 | −3681.75 | −7248.93 | 120.31 |
| Crystals | ρ (g/cm3) | Packing Coefficient | D (km/s) | P (GPa) | h50 (cm) | |||
|---|---|---|---|---|---|---|---|---|
| DFT | Ref. [23] | DFT | Ref. [38] | DFT | Ref. [12] | |||
| DNBT | 1.83 | 1.89 | 0.78 | 8.40 | 8.41 | 32.86 | 26.38 | |
| TNB/DNBT | 1.82 | 1.829 | 0.68 | 8.17 | 29.52 | 44.30 | ||
| TNT/DNBT | 1.78 | 1.768 | 0.68 | 7.89 | 27.30 | 45.30 | ||
| PA/DNBT | 1.84 | 1.870 | 0.74 | 8.33 | 30.98 | 42.05 | ||
| TNT a | 1.78 | 6.80 | 19.00 | >170 | ||||
| RDX a | 1.82 | 8.75 | 34.00 | 46 | ||||
| HMX a | 1.91 | 9.10 | 39.00 | 36 | ||||
| CL-20 a | 2.04 | 9.40 | 42.00 | 24 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, L.; Liu, M.; Xie, S.; Li, S.; Liu, S.; Yuan, S.; Duan, X.; Li, H. Substituent Effects on Crystal Engineering of DNBT-Based Energetic Cocrystals: Insights from Multiscale Computational Analysis. Materials 2025, 18, 2995. https://doi.org/10.3390/ma18132995
Shi L, Liu M, Xie S, Li S, Liu S, Yuan S, Duan X, Li H. Substituent Effects on Crystal Engineering of DNBT-Based Energetic Cocrystals: Insights from Multiscale Computational Analysis. Materials. 2025; 18(13):2995. https://doi.org/10.3390/ma18132995
Chicago/Turabian StyleShi, Lu, Min Liu, Shangrui Xie, Song Li, Shuxin Liu, Shen Yuan, Xiaohui Duan, and Hongzhen Li. 2025. "Substituent Effects on Crystal Engineering of DNBT-Based Energetic Cocrystals: Insights from Multiscale Computational Analysis" Materials 18, no. 13: 2995. https://doi.org/10.3390/ma18132995
APA StyleShi, L., Liu, M., Xie, S., Li, S., Liu, S., Yuan, S., Duan, X., & Li, H. (2025). Substituent Effects on Crystal Engineering of DNBT-Based Energetic Cocrystals: Insights from Multiscale Computational Analysis. Materials, 18(13), 2995. https://doi.org/10.3390/ma18132995