
Organic and Ionic Liquids Electrolyte Solutions as Versatile Media for Metallic Lithium Recovery
 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussions
3.1. Study of Lithium Salts Dissolution in the Chosen Solvents
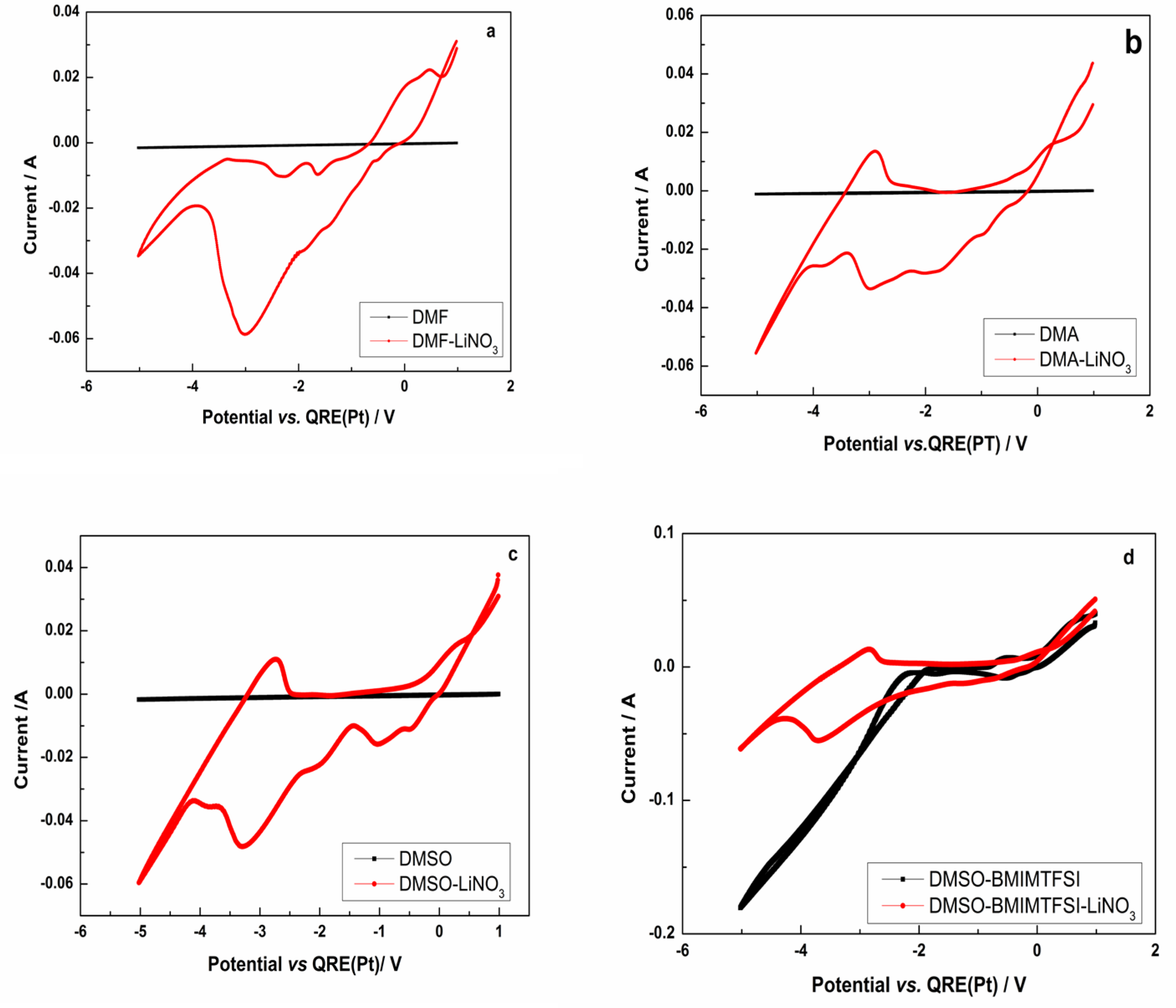
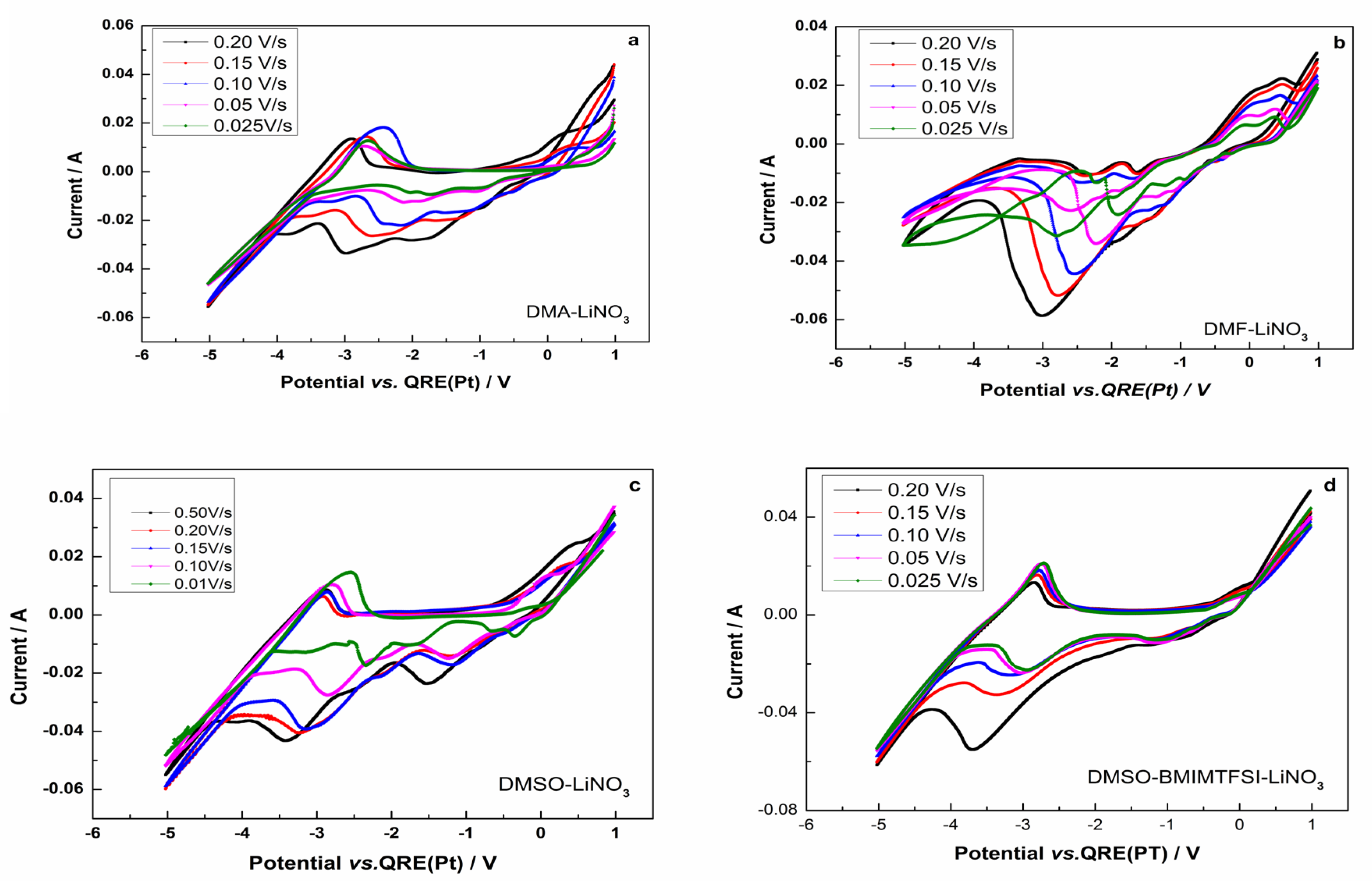
3.2. Cyclic Voltammetry of the Lithium Electrodeposition
3.3. Electrodeposition of Metallic Lithium
3.3.1. System DMA-LiNO3
3.3.2. System DMF-LiNO3
3.3.3. System DMSO-LiNO3
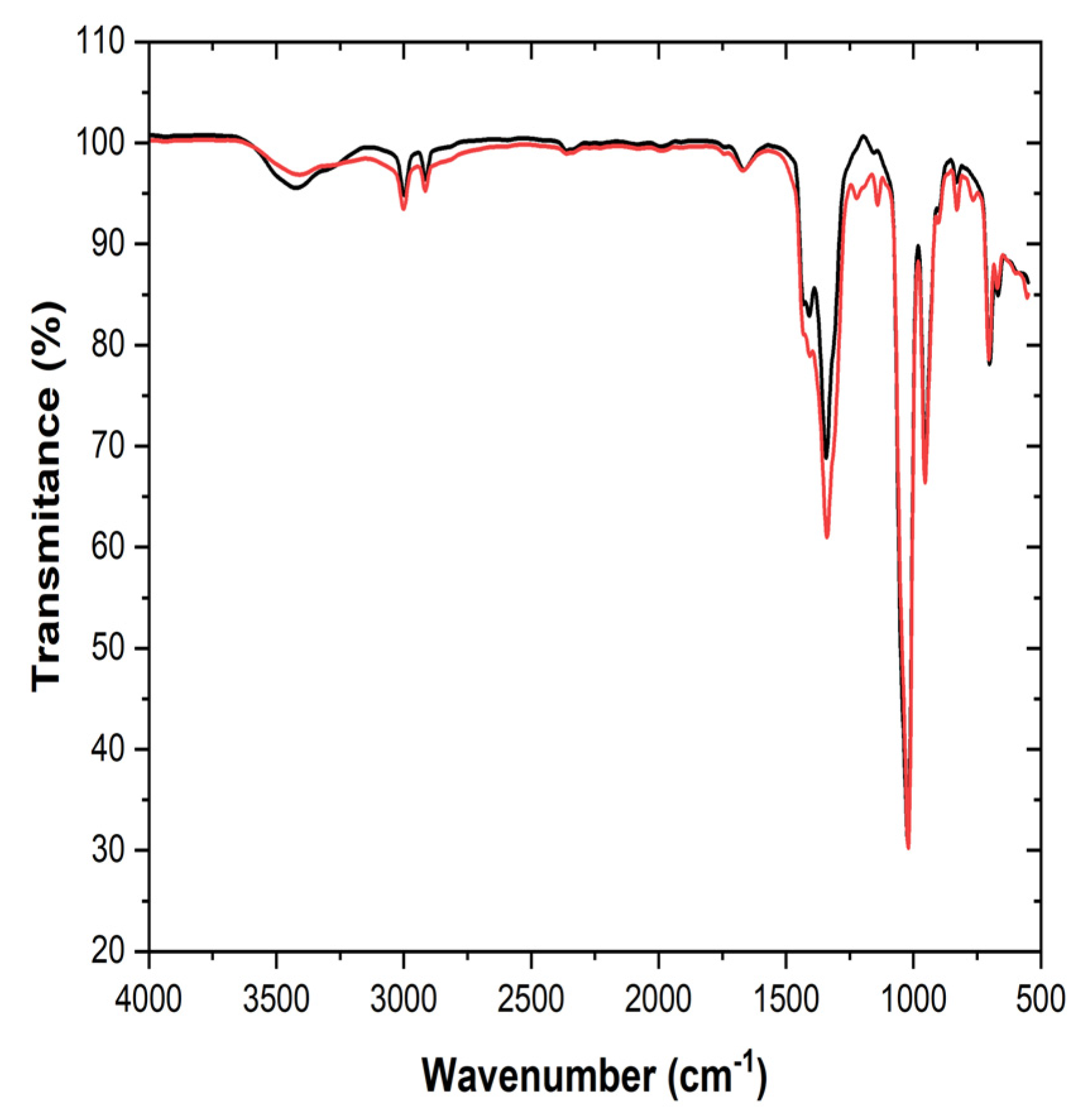
- Changes in ion–solvent interactions:
- The shift in the characteristic bands for the S=O group in DMSO (1022 cm−1 → 1020 cm−1) and the high intensity of this band after deposition indicate a strong coordination between DMSO and lithium ions (Li+).
- The broadening and modification of the bands assigned to nitrate (NO3−, 1429–1340 cm−1) suggest a reduction in the symmetry of the nitrate ion, most likely through interactions with DMSO or through the formation of new molecular structures.
- Stability of DMSO:
- The C-H bands (3001–2916 cm−1) remain unchanged, indicating that the methyl groups in DMSO are not chemically affected and are only involved in physical interactions.
- Formation of post-deposition complexes:
- The observed changes in the S=O and NO3− bands suggest the formation of a DMSO-Li+-NO3− complex, in which DMSO plays a ligand role and the nitrate becomes more asymmetric.
3.3.4. System DMSO-BMIMTFSI-LiNO3
3.4. Characterization of the Lithium Deposits
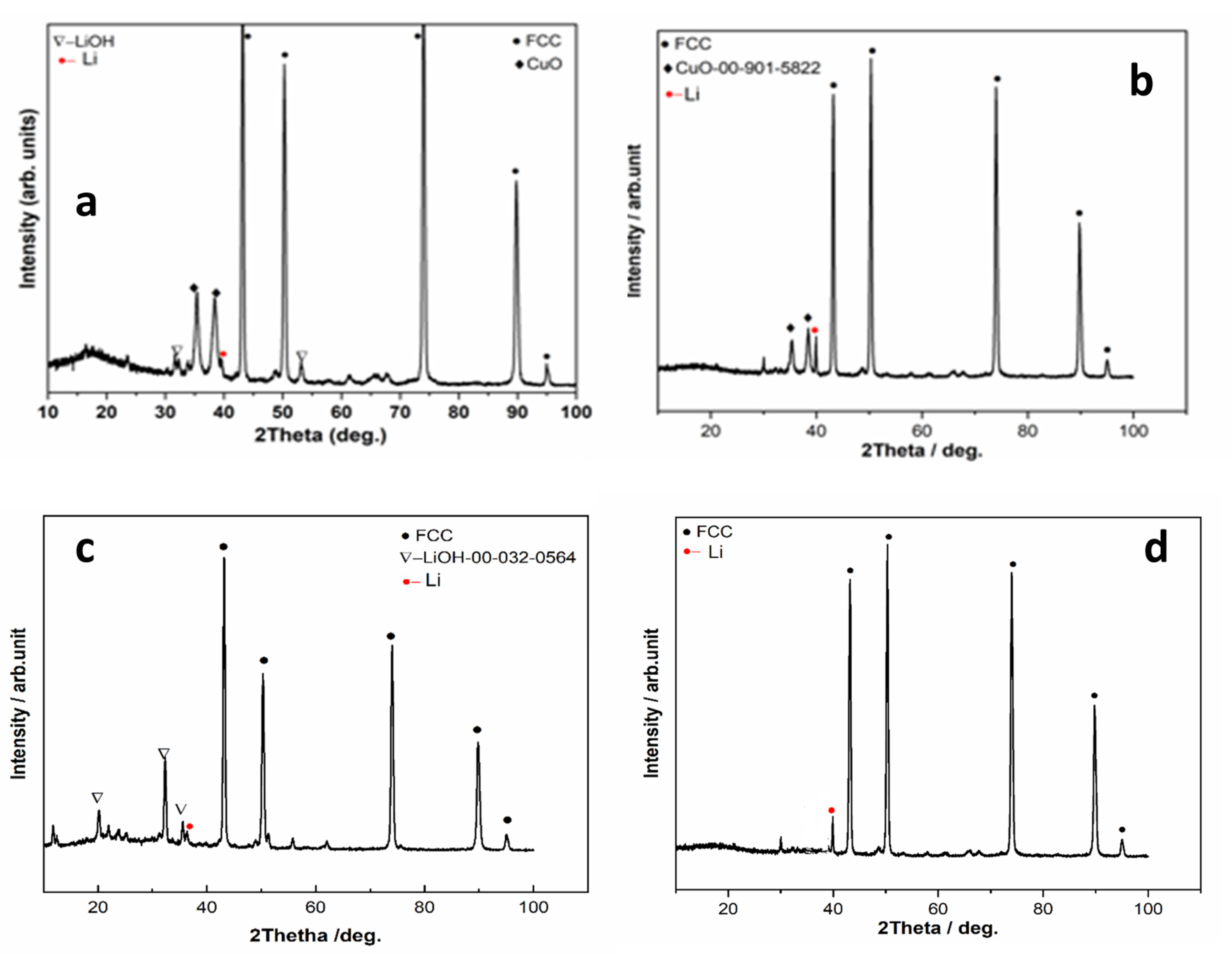
3.4.1. Chemical Analysis and XRD Measurements
3.4.2. SEM Analysis
4. Conclusions
- The developed systems were organic/organic-IL with the addition of LiNO3 and were used for the potentiostatic electrodeposition of Li on copper substrates.
- The electrodeposition process was characterized by cyclic voltammetry, and for the studied systems, the process was found to be irreversible for DMF-LiNO3 and DMA-LiNO3; diffusion-limited irreversible for DMSO-LiNO3; and irreversible or quasi-reversible for DMSO-BMIMTFSI-LiNO3.
- Cyclic voltammetry was used to prove the lithium electrodeposition in the range −3.011 → −4.495 V (vs. Pt). Under these conditions, pure lithium deposits without dendritic shapes and of micrometric and submicrometric dimensions could be obtained by electrodeposition.
- Especially because lithium shows the highest activity among all elements (being able to generate fire or oxidize quickly when it is removed from the glove box), it was quite a challenge to obtain thin films of lithium on the copper substrate.
- The obtained deposits were characterized by chemical analysis, XRD, and SEM. Diffraction patterns reveal the existence of Li and sometimes LiOH, which was confirmed by chemical analysis. SEM images generated thin lithium coatings on copper support, without dendritic shapes and of micrometric and submicrometric dimensions.
- Moreover, the newly developed electrolyte of organic/IL allowed the electrodeposition of Li under no special conditions and proved to be an excellent solvent for lithium nitrate and to have good electrochemical stability.
Author Contributions
Funding

Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fulton, L.; Schäfer, A.W.; Sperling, D. Transitions to deeply decarbonized transportation and energy systems around the world: Challenges and solutions. Transp. Res. Part. D Transp. Environ. 2024, 134, 104219. [Google Scholar] [CrossRef]
- Kasri, M.A.; Halizan, M.Z.M.; Harun, I.; Bahrudin, F.I.; Daud, N.; Aizamddin, M.F.; Shaffee, S.N.A.; Abd Rahman, N.; Shafiee, S.A.; Mahat, M.M. Addressing preliminary challenges in upscaling the recovery of lithium from spent lithium-ion batteries by the electrochemical method: A review. RSC Adv. 2024, 14, 15515. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Zhou, J.; Lu, P.; Yin, J.; Wang, Y.; Fan, Z. Electrochemical lithium extraction from aqueous sources. Matter 2022, 5, 1760–1791. [Google Scholar] [CrossRef]
- Chen, R. Ionic liquids-mediate recovery of metals from spent batteries. J. Ion. Liq. 2023, 3, 1000070. [Google Scholar]
- Tang, W.; Ma, J.; Zhang, X.; Li, Y.; Meng, S.; Zhang, Y.; Dong, H.; Liu, R.; Gao, R.; Feng, M. Interfacial strategies towards highly stable Li-metal anode of liquid-based Li-metal batteries. Energy Storage Mater. 2024, 64, 103084. [Google Scholar] [CrossRef]
- Qian, J.; Xu, W.; Bhattacharya, P.; Engelhard, M.; Henderson, W.A.; Zhang, Y.; Zhang, J.-G. Dendrite-free Li deposition using trace amounts of water as an electrolyte additive. Nano Energy 2015, 15, 135–144. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, H.; Qi, S.; Wu, D.; Huang, J.; Li, X.; Wang, C.; Ma, J. Structural regulation chemistry of lithium-ion solvation for lithium batteries. EcoMat 2022, 4, e12200. [Google Scholar] [CrossRef]
- Takei, T. Electrolytic deposition of lithium from non-aqueous solutions. J. Appl. Electrochem. 1979, 9, 587–593. [Google Scholar] [CrossRef]
- Lin, X.; Khosravinia, K.; Hu, X.; Li, J.; Lu, W. Lithium Plating Mechanism, Detection, and Mitigation in Lithium-Ion Batteries. Progress. Energy Combust. Sci. 2021, 87, 100953. [Google Scholar] [CrossRef]
- Yamada, H.; Yoshi, K.; Asahi, M.; Chiku, M.; Kitazumi, Y. Cyclic Voltametry-Part.1 Fundamentals. Electrochemistry 2022, 90, 102005. [Google Scholar] [CrossRef]
- Krtil, P.; Kavan, L.; Hoskovcová, I.; Kratochvilová, K. Anodic oxidation of dimethyl sulfoxide based electrolyte solutions: Anin situ FTIR study. J. Appl. Electrochem. 1996, 26, 523–527. [Google Scholar] [CrossRef]
- Infrared Spectroscopy: A Quick Primer on Interpreting Spectra. Available online: https://www.masterorganicchemistry.com/2016/11/23/quick_analysis_of_ir_spectra/ (accessed on 10 April 2025).
- Jascindra, R.; Hills, A.E.; Cerasoli, E.; Rakowska, P.D.; Ryadnov, M.G. FTIR markers of methionine oxidation for early detection of oxidized protein therapeutics. Eur. Biophys. J. 2011, 40, 339–345. [Google Scholar]
- Mozhshukhina, N.; Mendez de Leo, L.P.; Calvo, E.J. Infrared Spectroscopy Studies on Stability of Dimethyl Sulfoxide for Application in a Li–Air Battery. J. Phys. Chem. C 2013, 117, 18375–18380. [Google Scholar] [CrossRef]
- Levi, D.; Gamolsky, M.D.; Markovsky, K.; Salitra, B.; Levi, G.; Heider, E.; Oesten, U.; Schmidt, R. Capacity fading of LiCoO2 cathodes in lithium-ion batteries: The effect of cycling, storage, temperature, and surface film formation. J. Power Sources 1999, 81–82, 95–111. [Google Scholar]
- Qian, J.; Henderson, W.A.; Xu, W.; Bhattacharya, P.; Engelhard, M.; Borodin, O.; Zhang, J.G. High rate and stable cycling of lithium metal anode. Nat. Commun. 2015, 6, 6362. [Google Scholar] [CrossRef] [PubMed]
- National Bureau of Standards Monograph 25, Section 17. 1980, p. 46. Available online: https://nvlpubs.nist.gov/nistpubs/Legacy/MONO/nbsmonograph25-17.pdf (accessed on 10 April 2025).
- Rutt, O.J.; Williams, G.R.; Simon, C. Reversible lithium insertion and copper extrusion in layered oxysulfides. Chem. Commun. 2006, 2869–2871. [Google Scholar] [CrossRef] [PubMed]
- Covington, E.J.; Montgomery, D.J. Sample: At T = 20 °C Note: Li6 isotope. J. Chem. Phys. 1957, 27, 1030–1032. [Google Scholar] [CrossRef]
- Olinger, B.; Shaner, J.W. Lithium, compression and high-pressure structure. Science 1983, 219, 1071–1072. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Lithium Salt | Observations |
|---|---|---|
| DMA | Li2CO3 | Totally undissolved |
| LiCl | Dissolved extremely slowly | |
| LiNO3 | Totally dissolved | |
| DMF | Li2CO3 | Totally undissolved |
| LiCl | Undissolved | |
| LiNO3 | Totally dissolved | |
| DMSO | Li2CO3 | Most undissolved |
| LiCl | Totally undissolved | |
| LiNO3 | Totally dissolved | |
| DMSO-BMIMTFSI | Li2CO3 | Most undissolved |
| LiCl | Most undissolved | |
| LiNO3 | Totally dissolved |
| Electrolyte | EdepLi vs. QREPt (V) |
|---|---|
| DMA + LiNO3 | −3.815 |
| DMF + LiNO3 | −3.011 |
| DMSO + LiNO3 | −4.266 |
| DMSO + BMIMTFSI + LiNO3 | −4.495 |
| Solvent | Lithium Salt | Electrodeposition Conditions |
|---|---|---|
| DMA | LiNO3 | 4.2 V; 0.01 A; 298 K |
| DMF | LiNO3 | 4 V; 0.01 A; 298 K |
| DMSO | LiNO3 | 5.06 V; 0.09 A; 313–323 K; stirring |
| DMSO + BMIMTFSI | LiNO3 | 5.29 V; 0.01 A; 313 K, stirring |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olaru, M.T.; Matei, A.; Atkinson, I.; Matei, A.I.; Bacalum, E.; Iota, M.; Popescu, A.-M. Organic and Ionic Liquids Electrolyte Solutions as Versatile Media for Metallic Lithium Recovery. Materials 2025, 18, 2899. https://doi.org/10.3390/ma18122899
Olaru MT, Matei A, Atkinson I, Matei AI, Bacalum E, Iota M, Popescu A-M. Organic and Ionic Liquids Electrolyte Solutions as Versatile Media for Metallic Lithium Recovery. Materials. 2025; 18(12):2899. https://doi.org/10.3390/ma18122899
Chicago/Turabian StyleOlaru, Mihai Tudor, Alexandru Matei, Irina Atkinson, Adelina Ionela Matei, Elena Bacalum, Miruna Iota, and Ana-Maria Popescu. 2025. "Organic and Ionic Liquids Electrolyte Solutions as Versatile Media for Metallic Lithium Recovery" Materials 18, no. 12: 2899. https://doi.org/10.3390/ma18122899
APA StyleOlaru, M. T., Matei, A., Atkinson, I., Matei, A. I., Bacalum, E., Iota, M., & Popescu, A.-M. (2025). Organic and Ionic Liquids Electrolyte Solutions as Versatile Media for Metallic Lithium Recovery. Materials, 18(12), 2899. https://doi.org/10.3390/ma18122899