
Boosting the Performance of Electrocatalytic NO Reduction to NH3 by Decorating WS2 with Single Transition Metal Atoms: A DFT Study


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
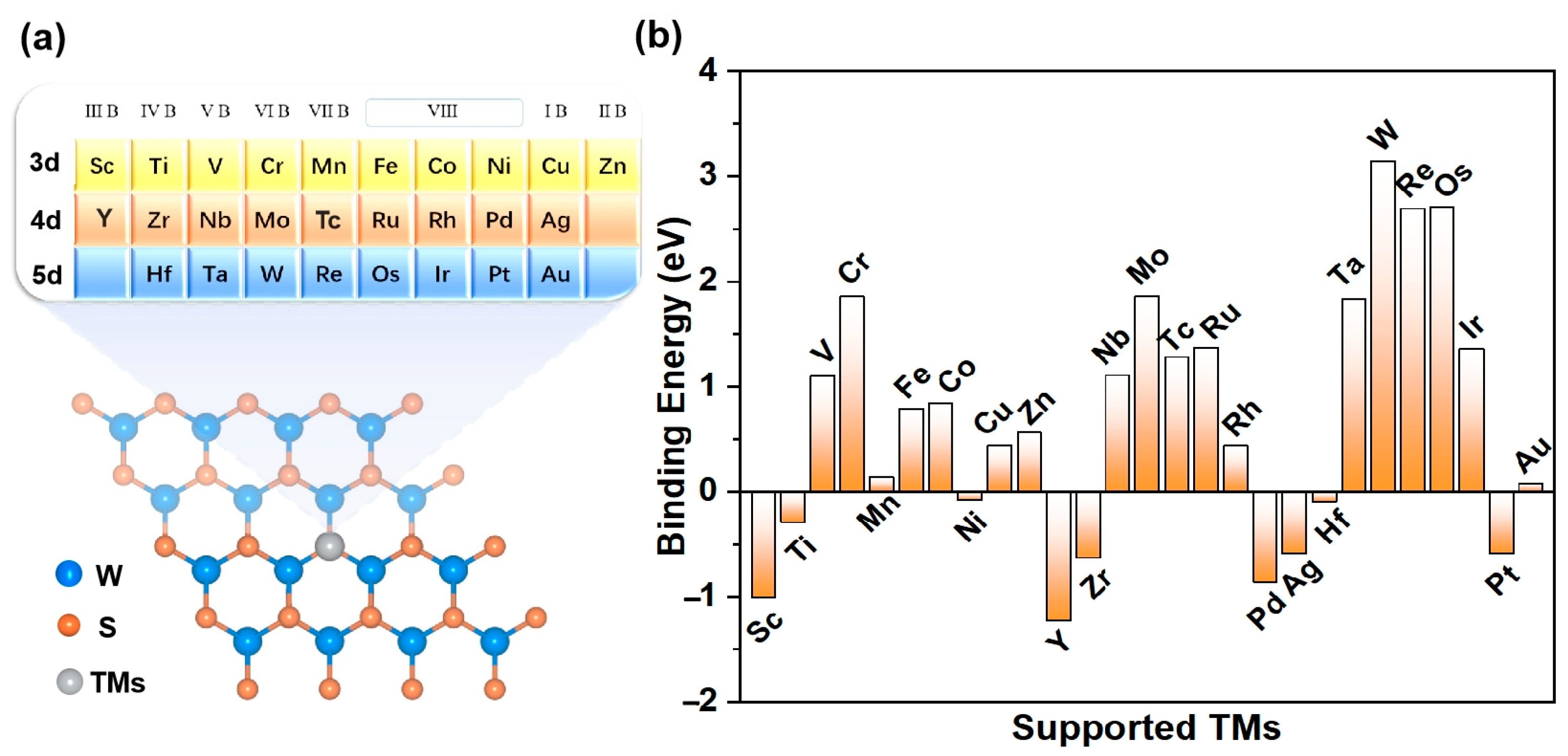
3.1. Stability of TM@WS2
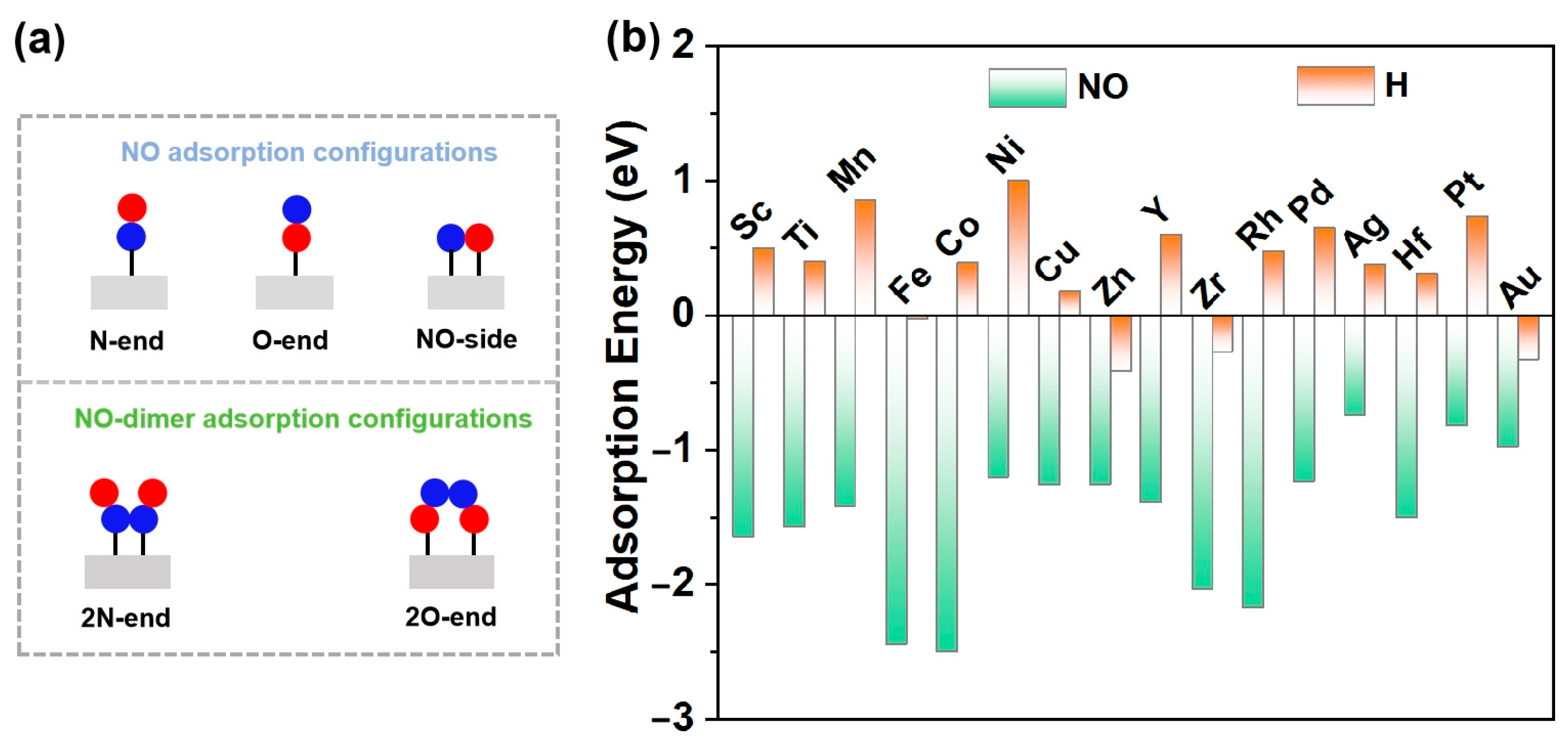
3.2. NO Adsorption
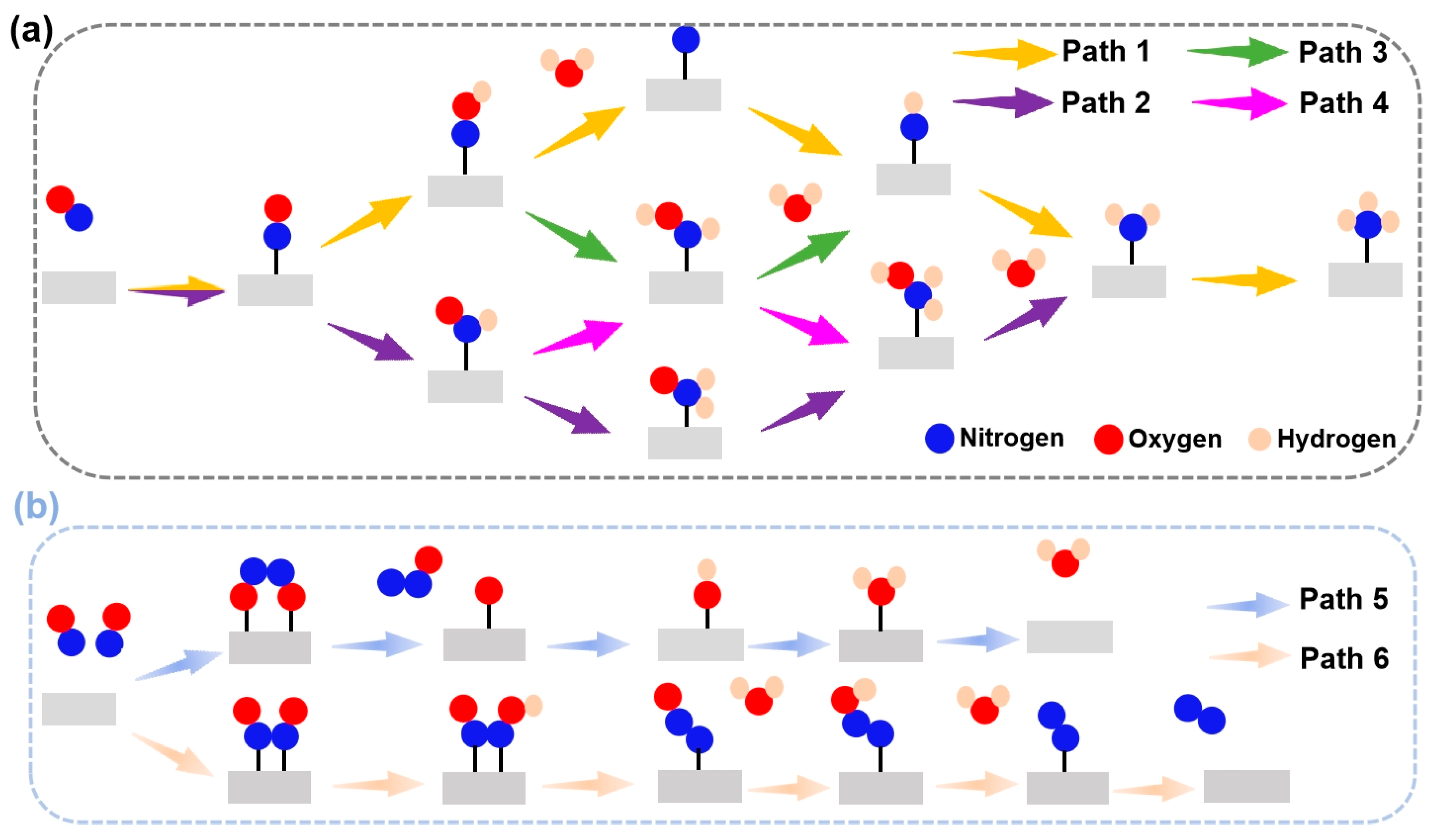
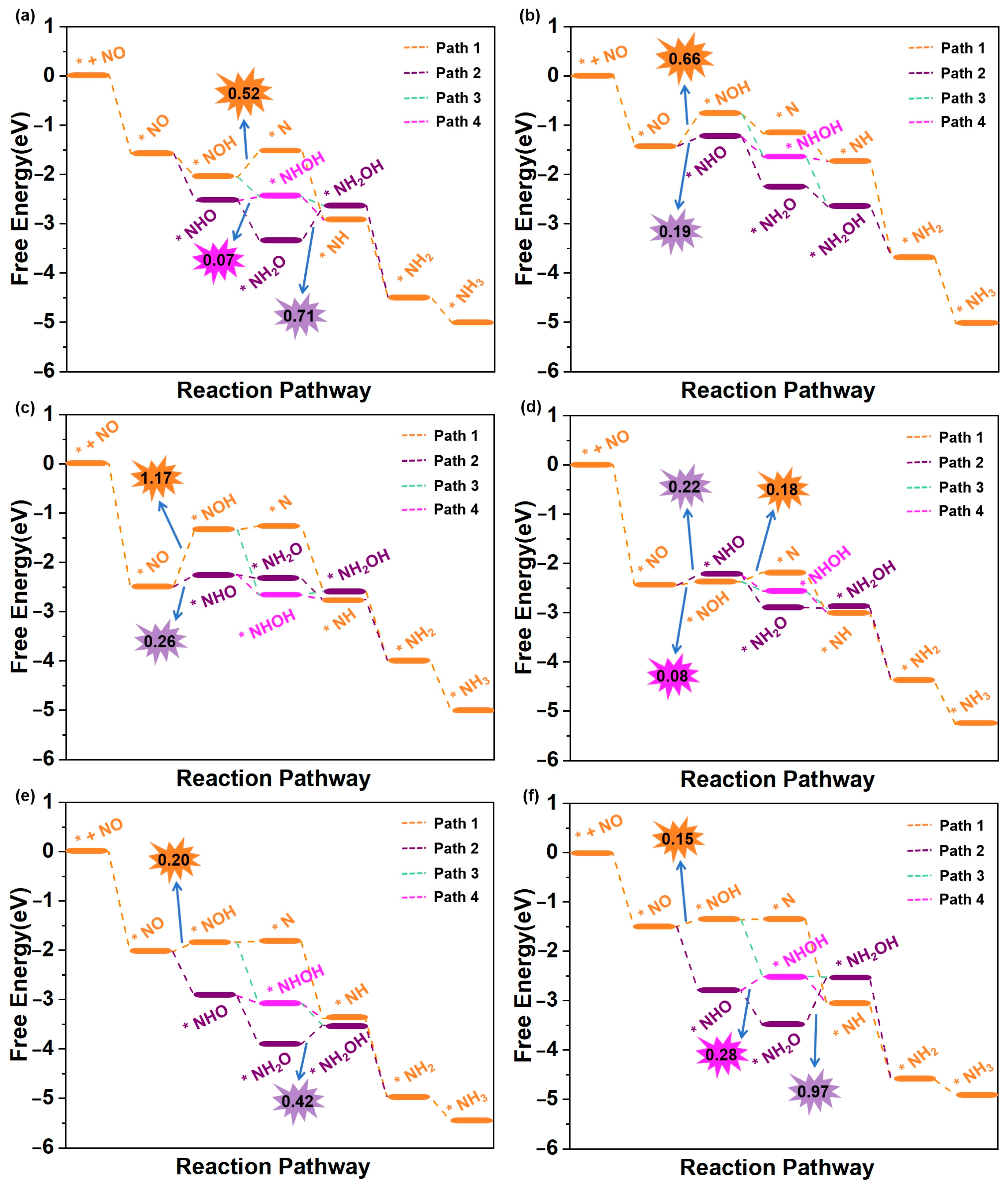
3.3. eNORR Mechanism and Evaluation of Activity
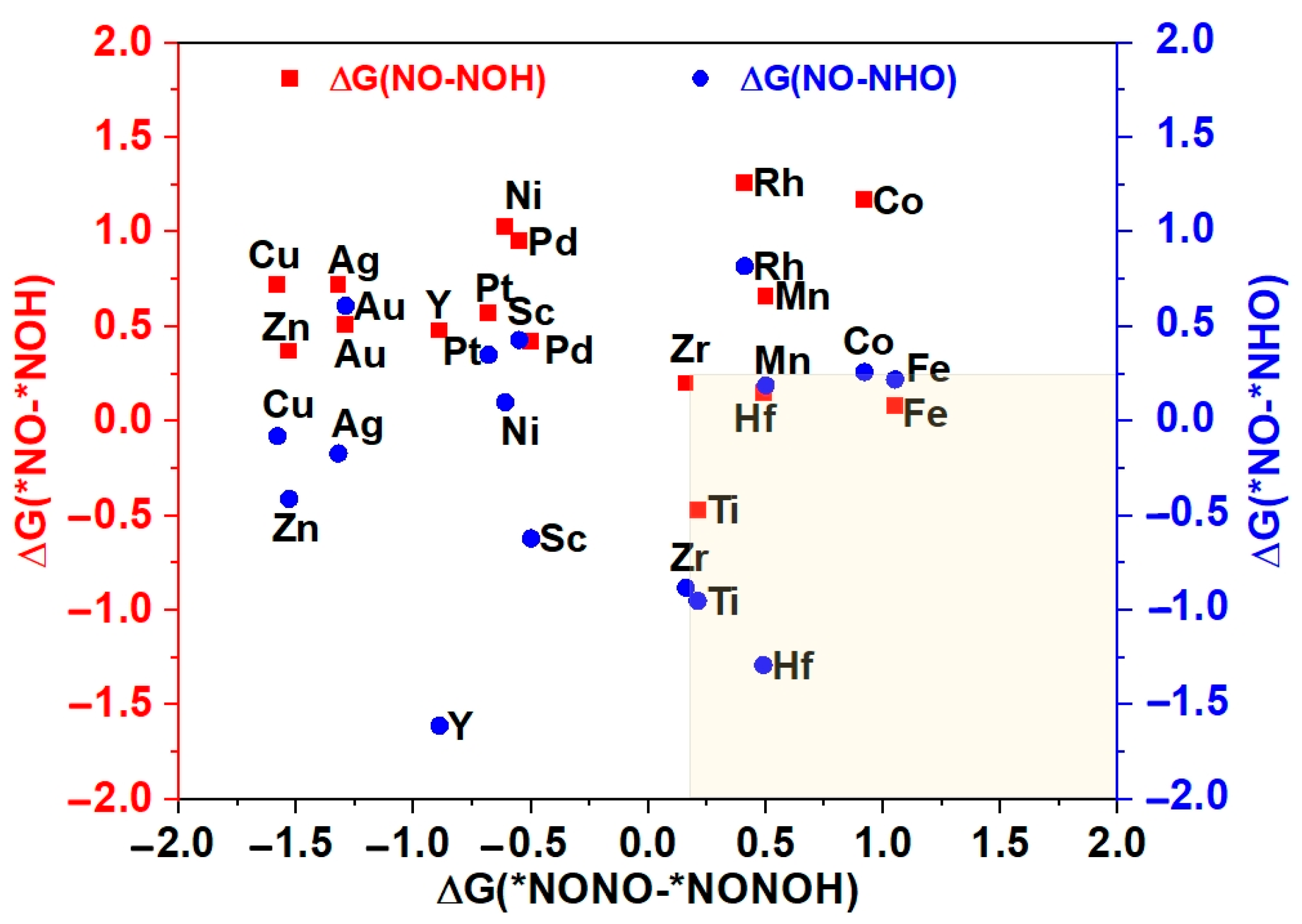
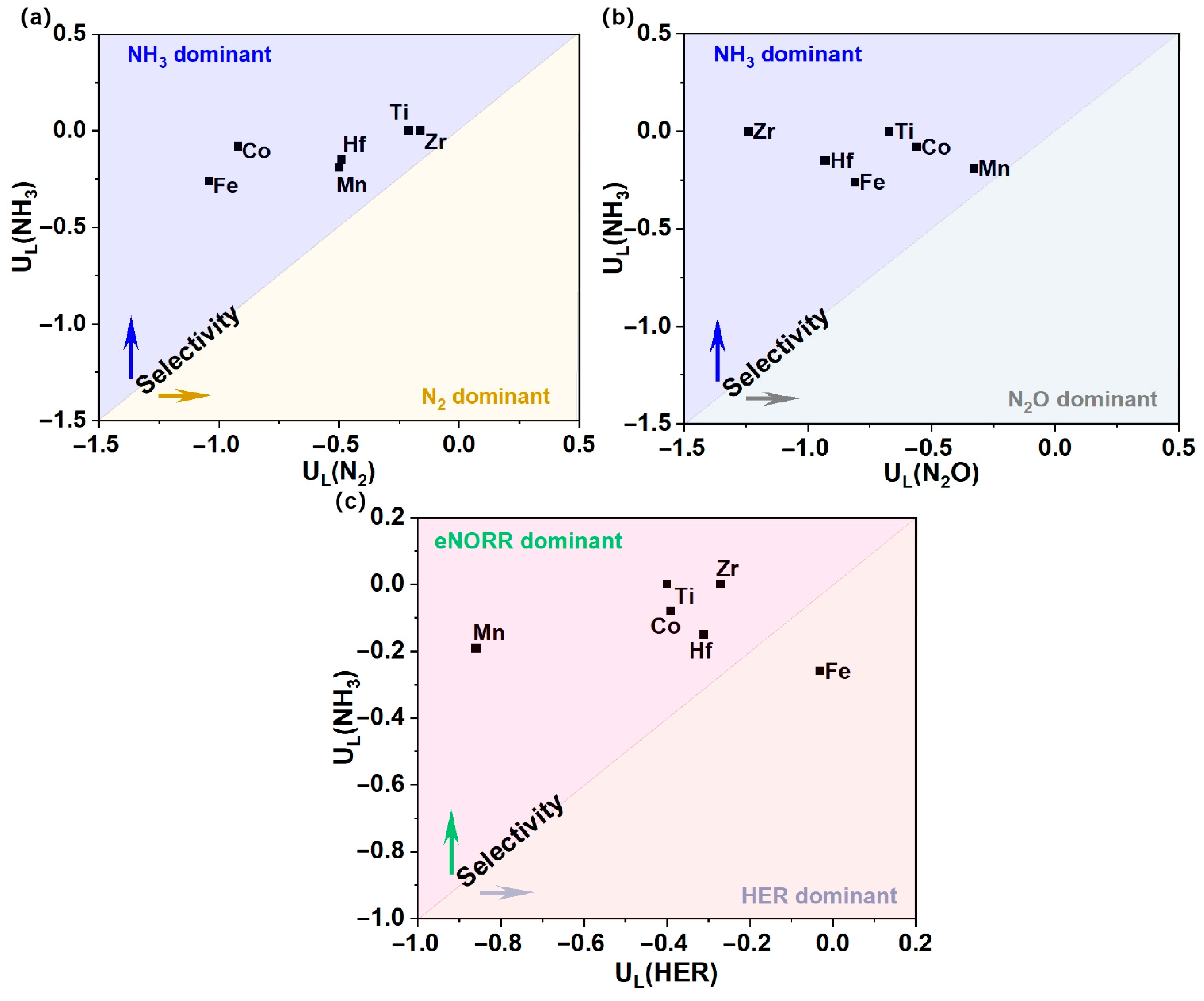
3.4. Selectivity Analysis
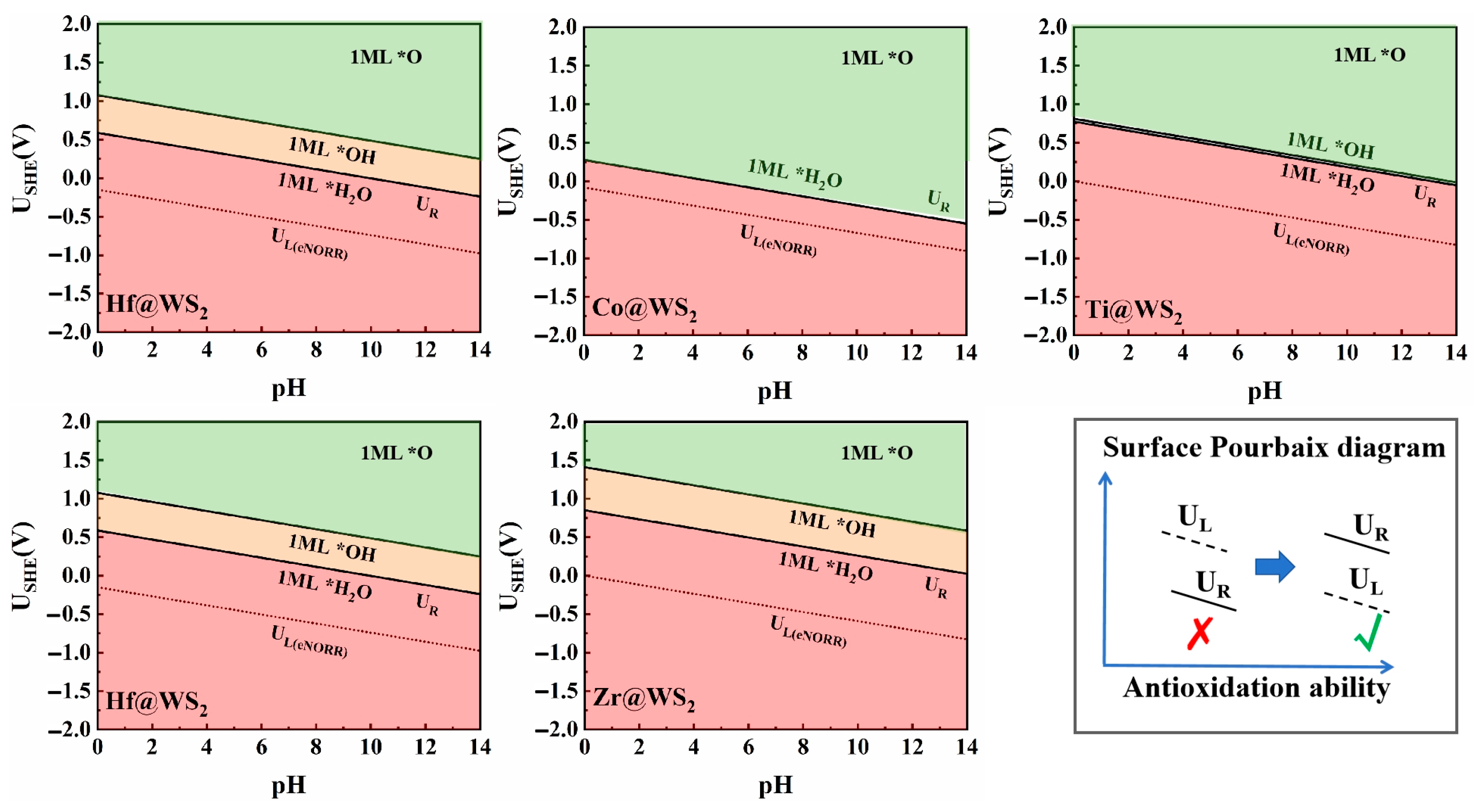
3.5. Pourbaix Diagram Analysis
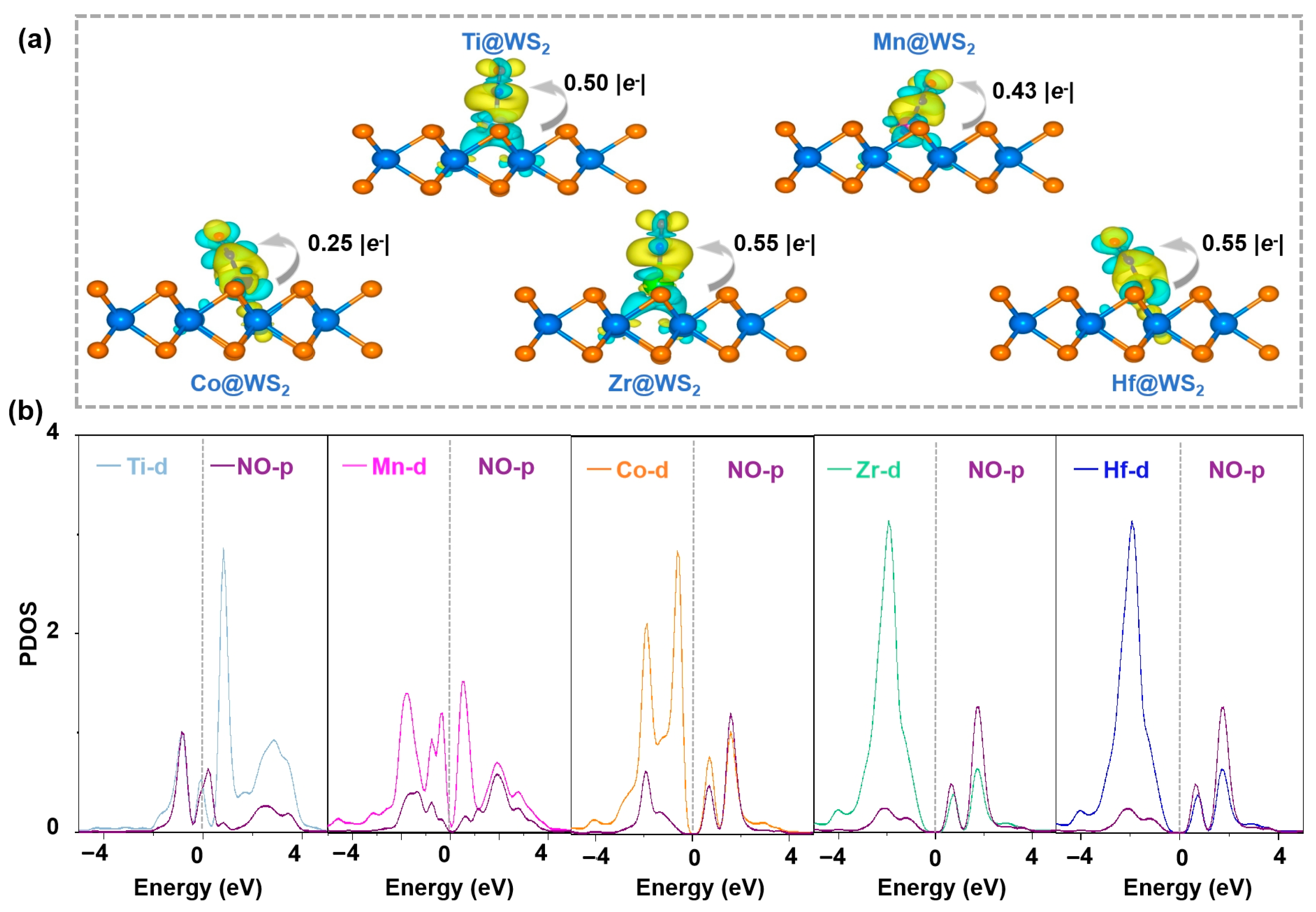
3.6. Revealing the Origin of Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| eNORR | Electrocatalytic nitric oxide reduction |
| TM | Transition metal |
| HER | Hydrogen evolution reaction |
| eNO3RR | Electrocatalytic nitrate reduction |
| eNRR | Electrocatalytic N2 reduction reaction |
| SACs | Single-atom catalysts |
| Eb | Binding energies |
References
- Christensen, C.H.; Johannessen, T.; Sørensen, R.Z.; Nørskov, J.K. Towards an ammonia-mediated hydrogen economy? Catal. Today 2006, 111, 140–144. [Google Scholar] [CrossRef]
- Soloveichik, G. Electrochemical synthesis of ammonia as a potential alternative to the Haber–Bosch process. Nat. Catal. 2019, 2, 377–380. [Google Scholar] [CrossRef]
- MacFarlane, D.R.; Cherepanov, P.V.; Choi, J.; Suryanto, B.H.R.; Hodgetts, R.Y.; Bakker, J.M.; Ferrero Vallana, F.M.; Simonov, A.N. A Roadmap to the Ammonia Economy. Joule 2020, 4, 1186–1205. [Google Scholar] [CrossRef]
- Lin, Q.F.; Jiang, Y.M.; Liu, C.Z.; Chen, L.W.; Zhang, W.J.; Ding, J.; Li, J.G. Instantaneous hydrogen production from ammonia by non-thermal arc plasma combining with catalyst. Energy Rep. 2021, 7, 4064–4070. [Google Scholar] [CrossRef]
- Wang, M.; Khan, M.A.; Mohsin, I.; Wicks, J.; Ip, A.H.; Sumon, K.Z.; Dinh, C.-T.; Sargent, E.H.; Gates, I.D.; Kibria, M.G. Can sustainable ammonia synthesis pathways compete with fossil-fuel based Haber–Bosch processes? Energy Environ. Sci. 2021, 14, 2535–2548. [Google Scholar] [CrossRef]
- Mohan, N.G.; Ramanujam, K. Electrocatalysts for ammonia synthesis: How close are we to the Haber-Bosch process? Curr. Opin. Electrochem. 2024, 45, 101520. [Google Scholar] [CrossRef]
- Liu, H.-M.; Han, S.-H.; Zhao, Y.; Zhu, Y.-Y.; Tian, X.-L.; Zeng, J.-H.; Jiang, J.-X.; Xia, B.Y.; Chen, Y. Surfactant-free atomically ultrathin rhodium nanosheet nanoassemblies for efficient nitrogen electroreduction. J. Mater. Chem. A 2018, 6, 3211–3217. [Google Scholar] [CrossRef]
- Zeng, Y.; Priest, C.; Wang, G.; Wu, G. Restoring the Nitrogen Cycle by Electrochemical Reduction of Nitrate: Progress and Prospects. Small Methods 2020, 4, 2000672. [Google Scholar] [CrossRef]
- Liu, D.; Qiao, L.; Peng, S.; Bai, H.; Liu, C.; Ip, W.F.; Lo, K.H.; Liu, H.; Ng, K.W.; Wang, S.; et al. Recent Advances in Electrocatalysts for Efficient Nitrate Reduction to Ammonia. Adv. Funct. Mater. 2023, 33, 2303480. [Google Scholar] [CrossRef]
- Liang, X.; Zhu, H.; Yang, X.; Xue, S.; Liang, Z.; Ren, X.; Liu, A.; Wu, G. Recent Advances in Designing Efficient Electrocatalysts for Electrochemical Nitrate Reduction to Ammonia. Small Struct. 2022, 4, 2200202. [Google Scholar] [CrossRef]
- Song, Z.; Qin, L.; Liu, Y.; Zhong, Y.; Guo, Q.; Geng, Z.; Zeng, J. Efficient Electroreduction of Nitrate to Ammonia with CuPd Nanoalloy Catalysts. ChemSusChem 2023, 16, e202300202. [Google Scholar] [CrossRef]
- Qing, G.; Ghazfar, R.; Jackowski, S.T.; Habibzadeh, F.; Ashtiani, M.M.; Chen, C.P.; Smith, M.R., III; Hamann, T.W. Recent Advances and Challenges of Electrocatalytic N2 Reduction to Ammonia. Chem. Rev. 2020, 120, 5437–5516. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Mi, Y.; Bao, H.; Liu, Y.; Qi, D.; Qiu, Y.; Zhuo, L.; Zhao, S.; Sun, J.; Tang, X.; et al. Ambient electrosynthesis of ammonia with efficient denitration. Nano Energy 2020, 78, 105321. [Google Scholar] [CrossRef]
- Miao, R.; Chen, D.; Guo, Z.; Zhou, Y.; Chen, C.; Wang, S. Recent advances in electrocatalytic upgrading of nitric oxide and beyond. Appl. Catal. B-Environ. 2024, 344, 123662. [Google Scholar] [CrossRef]
- Wu, Z.Y.; Karamad, M.; Yong, X.; Huang, Q.; Cullen, D.A.; Zhu, P.; Xia, C.; Xiao, Q.; Shakouri, M.; Chen, F.Y.; et al. Electrochemical ammonia synthesis via nitrate reduction on Fe single atom catalyst. Nat. Commun. 2021, 12, 2870. [Google Scholar] [CrossRef]
- Chen, D.; Yin, D.; Zhang, S.; Yip, S.; Ho, J.C. Nitrate electroreduction: Recent development in mechanistic understanding and electrocatalyst design. Mater. Today Energy 2024, 44, 101610. [Google Scholar] [CrossRef]
- Jiang, H.; Chen, G.F.; Savateev, O.; Xue, J.; Ding, L.X.; Liang, Z.; Antonietti, M.; Wang, H. Enabled Efficient Ammonia Synthesis and Energy Supply in a Zinc-Nitrate Battery System by Separating Nitrate Reduction Process into Two Stages. Angew. Chem. Int. Ed. 2023, 62, e202218717. [Google Scholar] [CrossRef]
- Liu, H.; Xiang, K.; Yang, B.; Xie, X.; Wang, D.; Zhang, C.; Liu, Z.; Yang, S.; Liu, C.; Zou, J.; et al. The electrochemical selective reduction of NO using CoSe2@CNTs hybrid. Environ. Sci. Pollut. Res. Int. 2017, 24, 14249–14258. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, H.; Haller, G.; Li, Y. Recent advances in the selective catalytic reduction of NOx with NH3 on Cu-Chabazite catalysts. Appl. Catal. B Environ. 2017, 202, 346–354. [Google Scholar] [CrossRef]
- Tursun, M.; Wu, C. Vacancy-triggered and dopant-assisted NO electrocatalytic reduction over MoS2. Phys. Chem. Chem. Phys. 2021, 23, 19872–19883. [Google Scholar] [CrossRef]
- Katsounaros, I.; Figueiredo, M.C.; Chen, X.; Calle-Vallejo, F.; Koper, M.T.M. Structure- and Coverage-Sensitive Mechanism of NO Reduction on Platinum Electrodes. ACS Catal. 2017, 7, 4660–4667. [Google Scholar] [CrossRef]
- Shi, J.; Wang, C.; Yang, R.; Chen, F.; Meng, N.; Yu, Y.; Zhang, B. Promoting nitric oxide electroreduction to ammonia over electron-rich Cu modulated by Ru doping. Sci. China Chem. 2021, 64, 1493–1497. [Google Scholar] [CrossRef]
- Tursun, M.; Wu, C. NO Electroreduction by Transition Metal Dichalcogenides with Chalcogen Vacancies. ChemElectroChem 2021, 8, 3113–3122. [Google Scholar] [CrossRef]
- Ko, B.H.; Hasa, B.; Shin, H.; Zhao, Y.; Jiao, F. Electrochemical Reduction of Gaseous Nitrogen Oxides on Transition Metals at Ambient Conditions. J. Am. Chem. Soc. 2022, 144, 1258–1266. [Google Scholar] [CrossRef]
- Tursun, M.; Wu, C. Defective 1T’-MoX2 (X = S, Se, Te) monolayers for electrocatalytic ammonia synthesis: Steric and electronic effects on the catalytic activity. Fuel 2023, 342, 127779. [Google Scholar] [CrossRef]
- Clayborne, A.; Chun, H.-J.; Rankin, R.B.; Greeley, J. Elucidation of Pathways for NO Electroreduction on Pt(111) from First Principles. Angew. Chem. Int. Ed. 2015, 54, 8255–8258. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Jiang, Y.-X.; Tian, N.; Zhou, Z.-Y.; Sun, S.-G. Electrocatalytic reduction of nitric oxide on Pt nanocrystals of different shape in sulfuric acid solutions. Electrochim. Acta 2010, 55, 8273–8279. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, J.; Wang, J.; Cabrera, C.R.; Chen, Z. A Co–N4 moiety embedded into graphene as an efficient single-atom-catalyst for NO electrochemical reduction: A computational study. J. Mater. Chem. A 2018, 6, 7547–7556. [Google Scholar] [CrossRef]
- Li, H.; Wu, D.; Wu, J.; Lv, W.; Duan, Z.; Ma, D. Graphene-based iron single-atom catalysts for electrocatalytic nitric oxide reduction: A first-principles study. Nanoscale 2024, 16, 7058–7067. [Google Scholar] [CrossRef]
- Wang, J.; Li, K.; Hao, Q.; Liu, D.; Zhang, X. Electroreduction NO to NH3 over single metal atom anchored on pyrrole type defective graphene: A DFT study. Chin. Chem. Lett. 2023, 34, 107567. [Google Scholar] [CrossRef]
- Wu, Q.; Huang, B.; Dai, Y.; Heine, T.; Ma, Y. Main-group metal elements as promising active centers for single-atom catalyst toward nitric oxide reduction reaction. Npj 2d Mater. Appl. 2022, 6, 52. [Google Scholar] [CrossRef]
- Tursun, M.; Wu, C. Single Transition Metal Atoms Anchored on Defective MoS2 Monolayers for the Electrocatalytic Reduction of Nitric Oxide into Ammonia and Hydroxylamine. Inorg. Chem. 2022, 61, 17448–17458. [Google Scholar] [CrossRef] [PubMed]
- Ruan, W.; Yang, C.; Hu, J.; Lin, W.; Guo, X.; Ding, K. Investigation of a Single Atom Iron Catalyst for the Electrocatalytic Reduction of Nitric Oxide to Hydroxylamine: A DFT Study. Langmuir 2024, 40, 24062–24073. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zheng, J.; Yao, Z.; Deng, S.; Pan, Z.; Wang, S.; Wang, J. DFT Investigation of Single Metal Atom-Doped 2D MA2Z4 Materials for NO Electrocatalytic Reduction to NH3. J. Phys. Chem. C 2022, 126, 17598–17607. [Google Scholar] [CrossRef]
- Xiao, X.; Cao, Y.; Hu, L. First-principles study on single-layer electronic structure of Fe-doped MoS2 and the reduction of NO on the doped surface. Comput. Theor. Chem. 2025, 1248, 115172. [Google Scholar] [CrossRef]
- Wu, Y.-W.; Wang, H.-W.; Wu, Z.-L.; Zhang, X.; Dong, Y.; Hu, Z.; Lv, Y.; Zhou, X.-Y.; Zhao, L.; Zhang, B.; et al. Mechanism of NO electrocatalytic reduction over the MoS2-based single atom catalyst: A DFT investigation. Sep. Purif. Technol. 2025, 366, 132813. [Google Scholar] [CrossRef]
- Lin, L.; Pang, D.; Shi, P.; Xie, K.; Su, L.; Zhang, Z. First-principles study of TM supported SnSe2 monolayer as an efficient electrocatalyst for NOER. Mol. Catal. 2022, 533, 112789. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, N.; Wang, F.; Kang, J.; Chu, K. Main-group indium single-atom catalysts for electrocatalytic NO reduction to NH3. J. Mater. Chem. A 2023, 11, 6814–6819. [Google Scholar] [CrossRef]
- Venkateswara Rao Nulakani, N.; Surya Kumar Choutipalli, V.; Akbar Ali, M. Efficient electrocatalytic reduction of nitric oxide (NO) to ammonia (NH3) on metal-free B4@g-C3N4 nanosheet. Appl. Surf. Sci. 2025, 680, 161470. [Google Scholar] [CrossRef]
- Guo, W.; Tang, X.; Liao, H.; Peng, J.; Lian, X. Theoretical screening of single-metal atom deposited on 2D BC3N2 monolayers for NO electrocatalytic reduction to NH3. Appl. Surf. Sci. 2025, 690, 162605. [Google Scholar] [CrossRef]
- Xiao, Y.; Shen, C.; Zhang, W.B. Screening and prediction of metal-doped α-borophene monolayer for nitric oxide elimination. Mater. Today Chem. 2022, 25, 100958. [Google Scholar] [CrossRef]
- Yang, L.; Fan, J.; Zhu, W. Single atom decorated wavy antimony nitride for nitric oxide degradation: A first-principles and machine learning study. Fuel 2025, 380, 133219. [Google Scholar] [CrossRef]
- Wang, J.; Sun, C.; Sheng, L.; Zhuo, Z.; Li, S.; Wang, J.; Wang, W.; Sun, J.; Yang, J.; Xu, K.; et al. Unveiling the electrocatalytic potential of main-group metal-embedded BC3 monolayer for highly efficient NO reduction to NH3. Chin. Chem. Lett. 2025, in press. [CrossRef]
- Niu, H.; Zhang, Z.; Wang, X.; Wan, X.; Kuai, C.; Guo, Y. A Feasible Strategy for Identifying Single-Atom Catalysts Toward Electrochemical NO-to-NH3 Conversion. Small 2021, 17, e2102396. [Google Scholar] [CrossRef]
- He, C.-Z.; Zhang, Y.-X.; Wang, J.; Fu, L. Anchor single atom in h-BN assist NO synthesis NH3: A computational view. Rare Met. 2022, 41, 3456–3465. [Google Scholar] [CrossRef]
- Sun, P.F.; Wang, W.L.; Zhao, X.; Dang, J.S. Defective h-BN sheet embedded atomic metals as highly active and selective electrocatalysts for NH3 fabrication via NO reduction. Phys. Chem. Chem. Phys. 2020, 22, 22627–22634. [Google Scholar] [CrossRef]
- Fan, J.; Yang, L.; Zhu, W. Transition metal-anchored BN tubes as single-atom catalysts for NO reduction reaction: A study of DFT and deep learning. Fuel 2025, 386, 134302. [Google Scholar] [CrossRef]
- Wu, J.; Yu, Y.X. A theoretical descriptor for screening efficient NO reduction electrocatalysts from transition-metal atoms on N-doped BP monolayer. J. Colloid. Interface Sci. 2022, 623, 432–444. [Google Scholar] [CrossRef]
- Zang, Y.; Wu, Q.; Wang, S.; Huang, B.; Dai, Y.; Ma, Y. Activating dual atomic electrocatalysts for the nitric oxide reduction reaction through the P/S element. Mater. Horiz. 2023, 10, 2160–2168. [Google Scholar] [CrossRef]
- Liu, S.; Xing, G.; Liu, J.-Y. Computational screening of single-atom catalysts for direct electrochemical NH3 synthesis from NO on defective boron phosphide monolayer. Appl. Surf. Sci. 2023, 611, 155764. [Google Scholar] [CrossRef]
- Tong, T.; Linghu, Y.; Wu, G.; Wang, C.; Wu, C. Nitric oxide electrochemical reduction reaction on transition metal-doped MoSi2N4 monolayers. Phys. Chem. Chem. Phys. 2022, 24, 18943–18951. [Google Scholar] [CrossRef]
- Wang, X.; Wu, J.; Zhang, Y.; Sun, Y.; Ma, K.; Xie, Y.; Zheng, W.; Tian, Z.; Kang, Z.; Zhang, Y. Vacancy Defects in 2D Transition Metal Dichalcogenide Electrocatalysts: From Aggregated to Atomic Configuration. Adv. Mater. 2023, 35, e2206576. [Google Scholar] [CrossRef]
- Zheng, J.; Lebedev, K.; Wu, S.; Huang, C.; Ayvali, T.; Wu, T.S.; Li, Y.; Ho, P.L.; Soo, Y.L.; Kirkland, A.; et al. High Loading of Transition Metal Single Atoms on Chalcogenide Catalysts. J. Am. Chem. Soc. 2021, 143, 7979–7990. [Google Scholar] [CrossRef] [PubMed]
- Tursun, M.; Abdukayum, A.; Wu, C.; Wang, C. Screening WS2−based single−atom catalysts for electrocatalytic nitrate reduction to ammonia. Int. J. Hydrogen Energy 2024, 73, 183–190. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Norskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Farberow, C.A.; Dumesic, J.A.; Mavrikakis, M. Density Functional Theory Calculations and Analysis of Reaction Pathways for Reduction of Nitric Oxide by Hydrogen on Pt(111). ACS Catal. 2014, 4, 3307–3319. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Mathew, K.; Sundararaman, R.; Letchworth-Weaver, K.; Arias, T.A.; Hennig, R.G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 2014, 140, 084106. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhu, Z.; Chen, J.; Xia, L.; Gan, L.; Zhou, Y. Evaluating the efficiency of single-double atom catalysts in electrochemical NH3 production from NO based on CN monolayers. J. Mater. Chem. A 2024, 12, 14035–14044. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, Y.; Liu, W.; Yang, D.; Zhou, G.; Yang, Z. Exploring the Catalytic Performance of Oxygen-Coordinated Single-Atom Catalysts for Nitric Oxide Electroreduction. J. Phys. Chem. C 2025, 129, 3522–3530. [Google Scholar] [CrossRef]
- Guo, X.; Lin, S.; Gu, J.; Zhang, S.; Chen, Z.; Huang, S. Establishing a Theoretical Landscape for Identifying Basal Plane Active 2D Metal Borides (MBenes) toward Nitrogen Electroreduction. Adv. Funct. Mater. 2020, 31, 2008056. [Google Scholar] [CrossRef]
- Wu, Q.; Wei, W.; Lv, X.; Wang, Y.; Huang, B.; Dai, Y. Cu@g-C3N4: An Efficient Single-Atom Electrocatalyst for NO Electrochemical Reduction with Suppressed Hydrogen Evolution. J. Phys. Chem. C 2019, 123, 31043–31049. [Google Scholar] [CrossRef]
- Lin, L.; Yan, L.; Fu, L.; He, C.; Xie, K.; Zhu, L.; Sun, J.; Zhang, Z. First principle investigation of W/P3C sheet as an efficient single atom electrocatalyst for N2 and NO electrochemical reaction with suppressed hydrogen evolution. Fuel 2022, 308, 122068. [Google Scholar] [CrossRef]
- Liu, L.; Zuo, Z.J.; Du, Y.; Wu, T.; Wu, J.; Gao, J.; Mu, T.; Zhang, Y.C.; Zhu, X.D. Role of synergies of Cu/Fe3O4 electrocatalyst for nitric oxide reduction to ammonia. J. Colloid Interface Sci. 2025, 691, 137376. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tursun, M.; Abduryim, A.; Wu, C. Boosting the Performance of Electrocatalytic NO Reduction to NH3 by Decorating WS2 with Single Transition Metal Atoms: A DFT Study. Materials 2025, 18, 2341. https://doi.org/10.3390/ma18102341
Tursun M, Abduryim A, Wu C. Boosting the Performance of Electrocatalytic NO Reduction to NH3 by Decorating WS2 with Single Transition Metal Atoms: A DFT Study. Materials. 2025; 18(10):2341. https://doi.org/10.3390/ma18102341
Chicago/Turabian StyleTursun, Mamutjan, Ayxamgul Abduryim, and Chao Wu. 2025. "Boosting the Performance of Electrocatalytic NO Reduction to NH3 by Decorating WS2 with Single Transition Metal Atoms: A DFT Study" Materials 18, no. 10: 2341. https://doi.org/10.3390/ma18102341
APA StyleTursun, M., Abduryim, A., & Wu, C. (2025). Boosting the Performance of Electrocatalytic NO Reduction to NH3 by Decorating WS2 with Single Transition Metal Atoms: A DFT Study. Materials, 18(10), 2341. https://doi.org/10.3390/ma18102341