Theoretical Insights into Hydrogen Production from Formic Acid Catalyzed by Pt-Group Single-Atom Catalysts

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
3. Results and Discussion
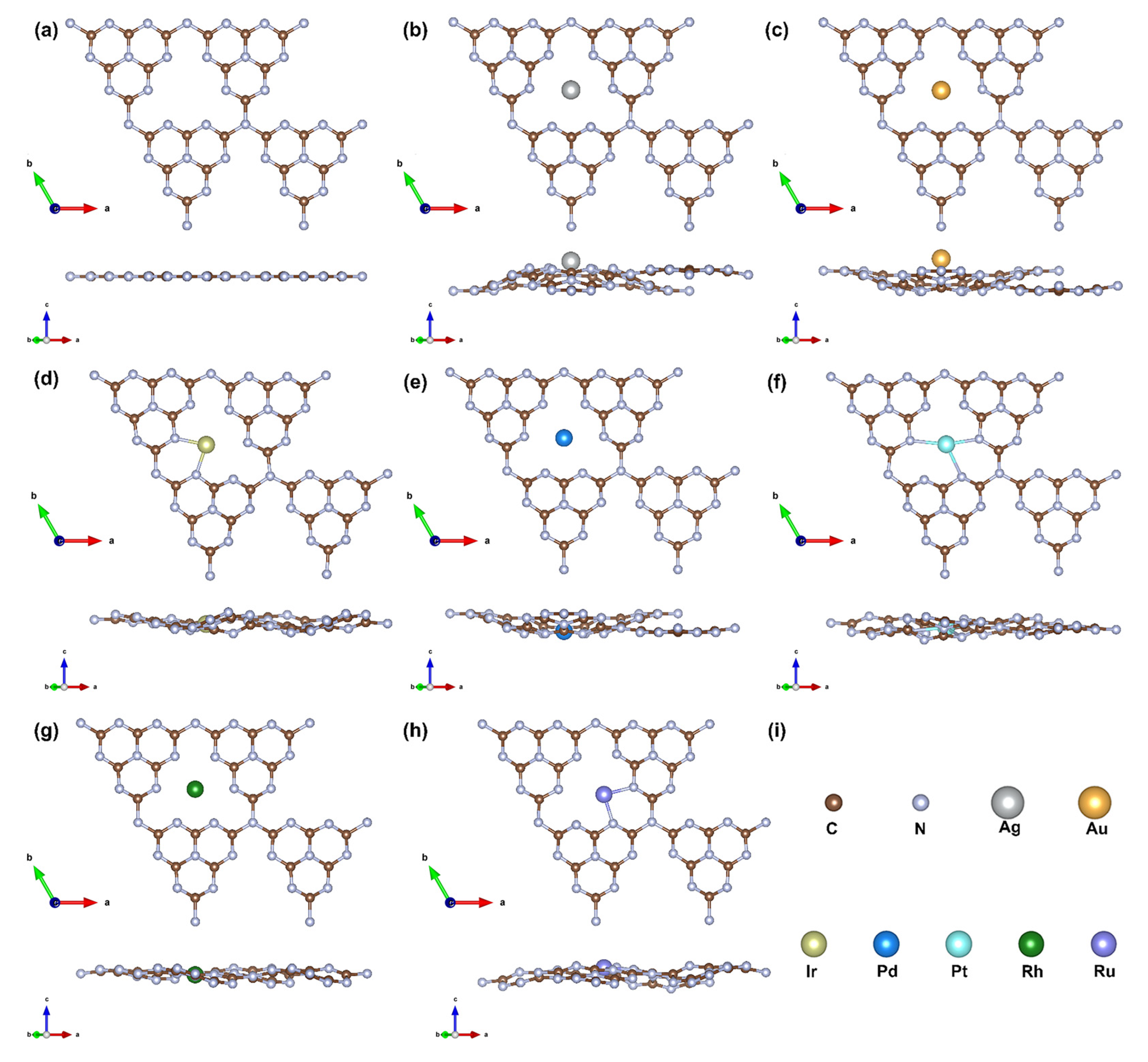
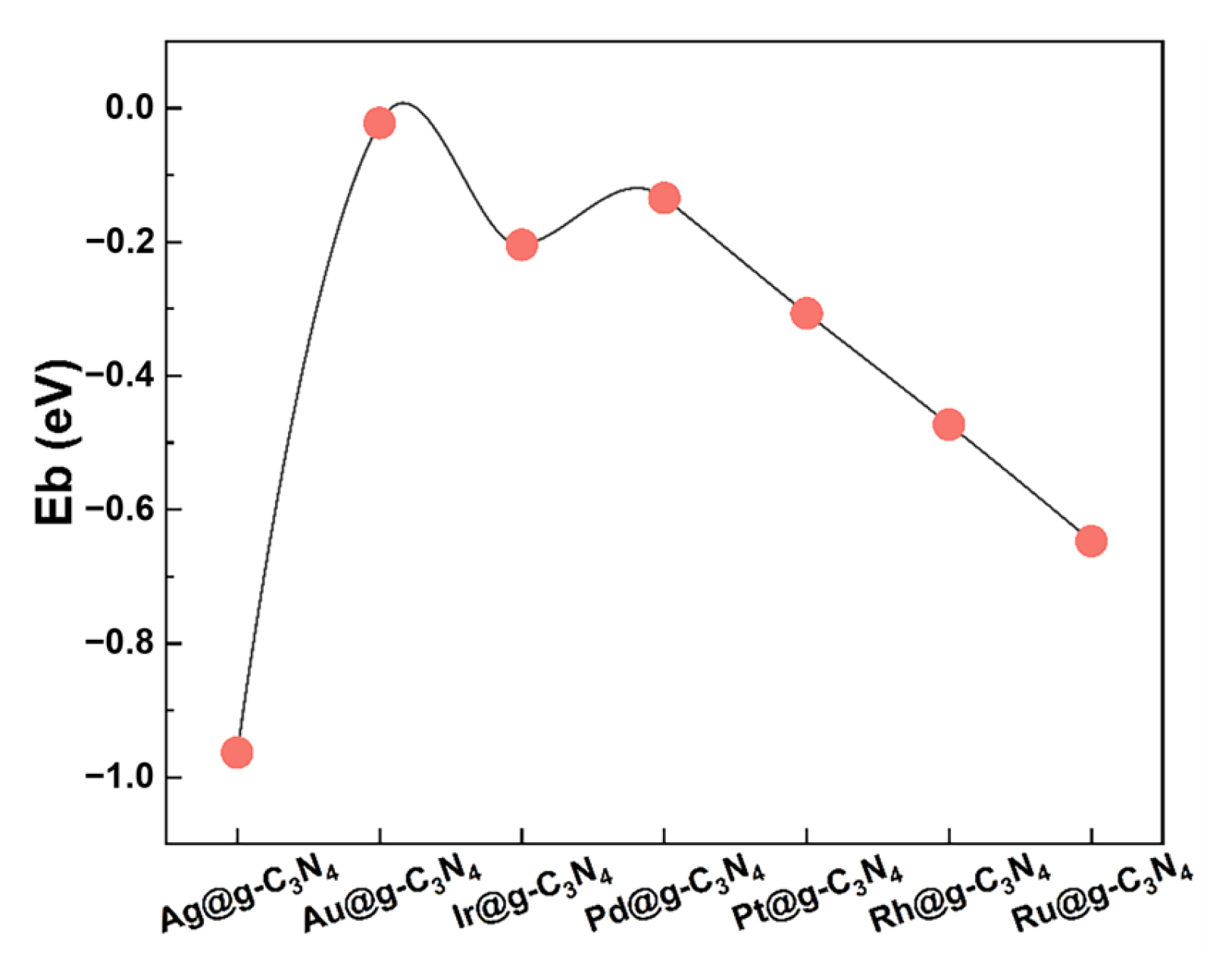
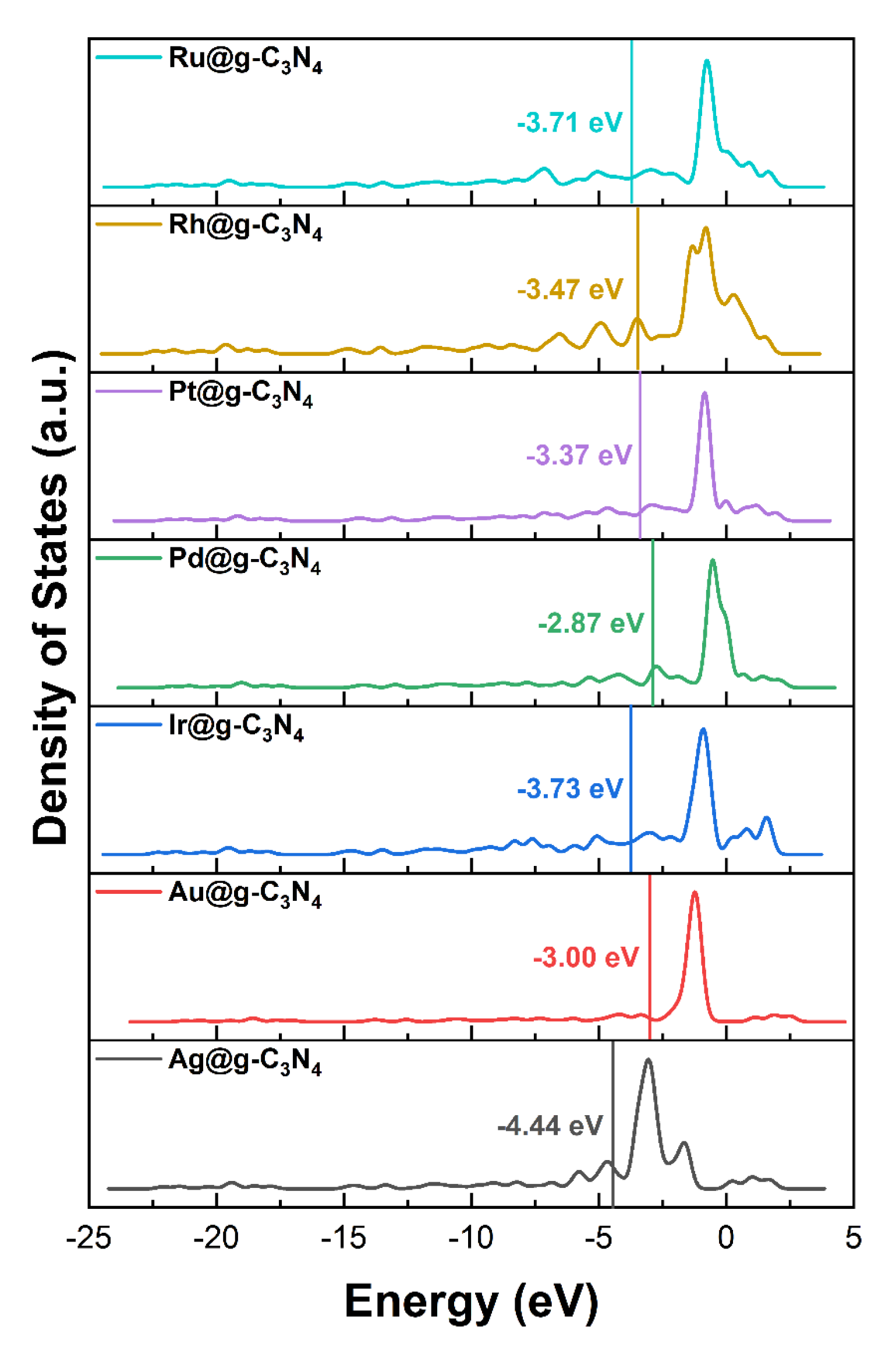
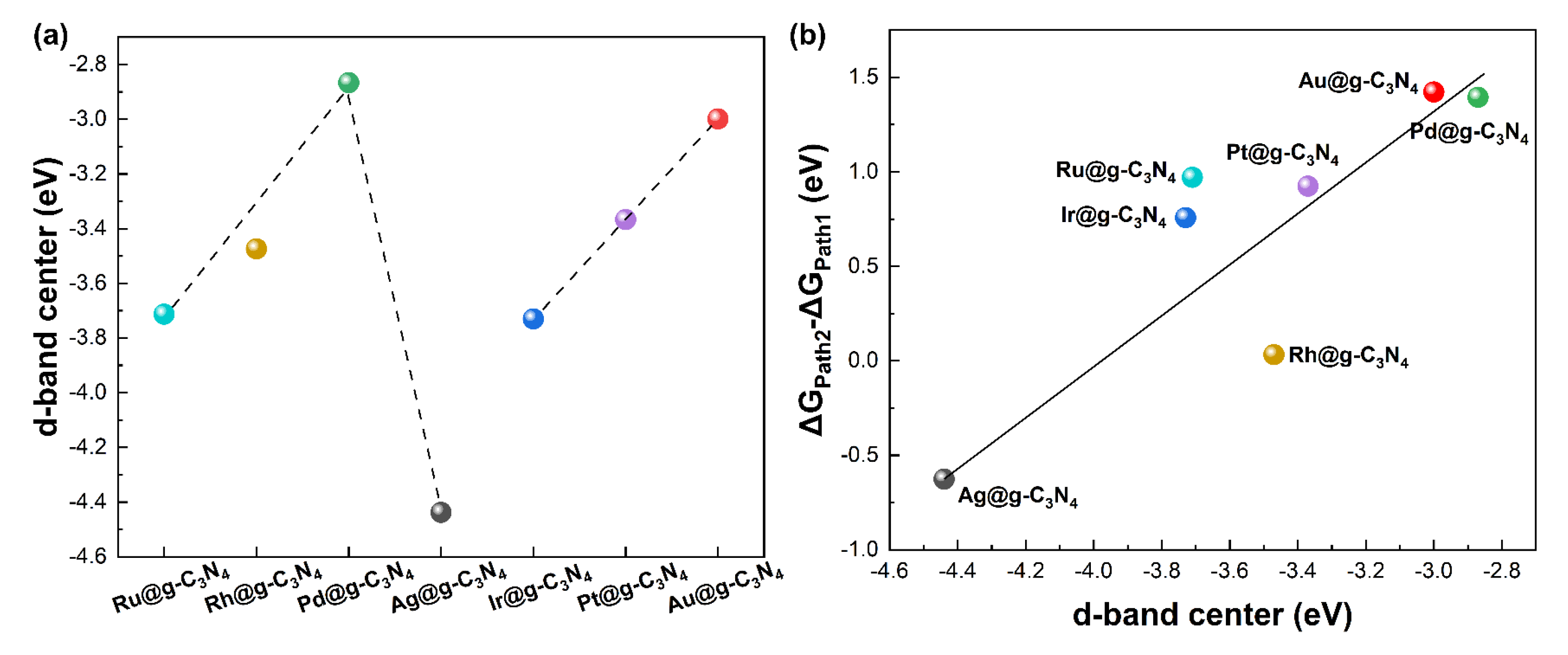
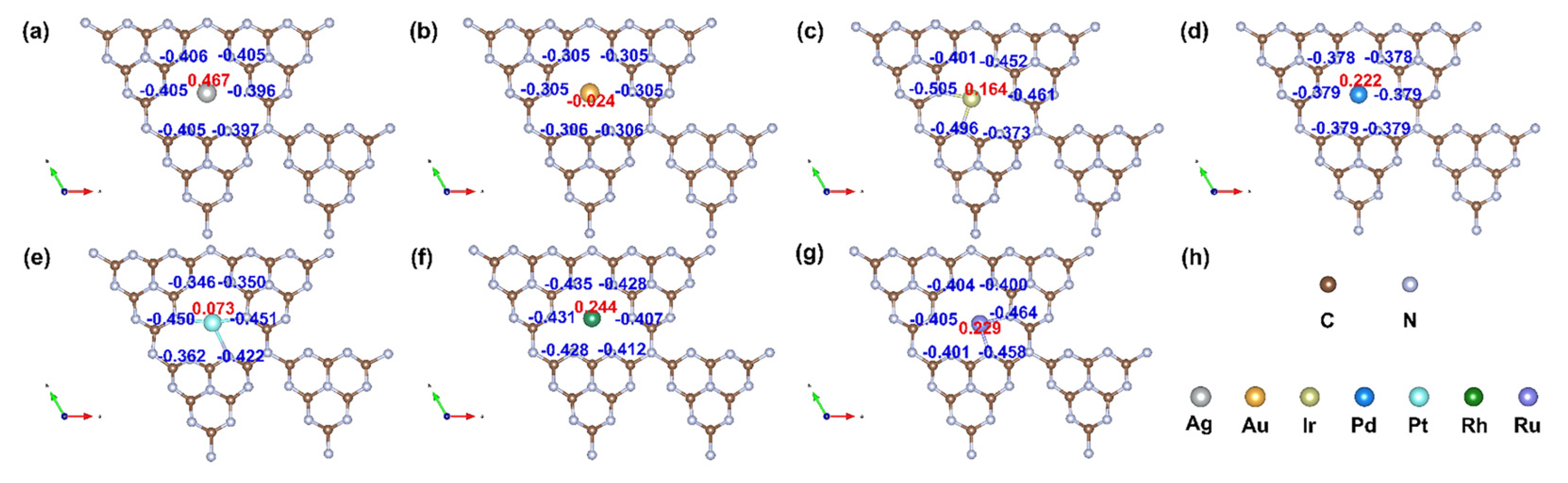
3.1. Structure Stability and Metal–Support Interactions
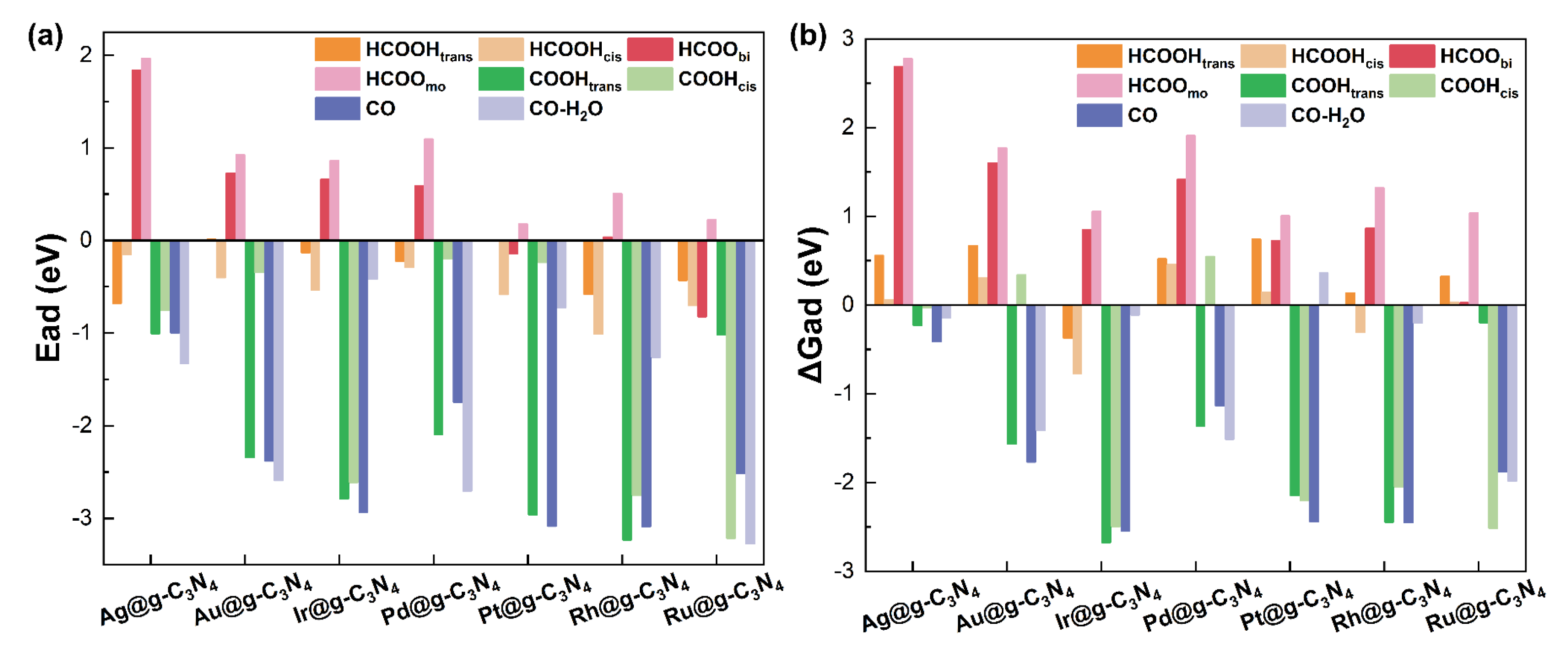
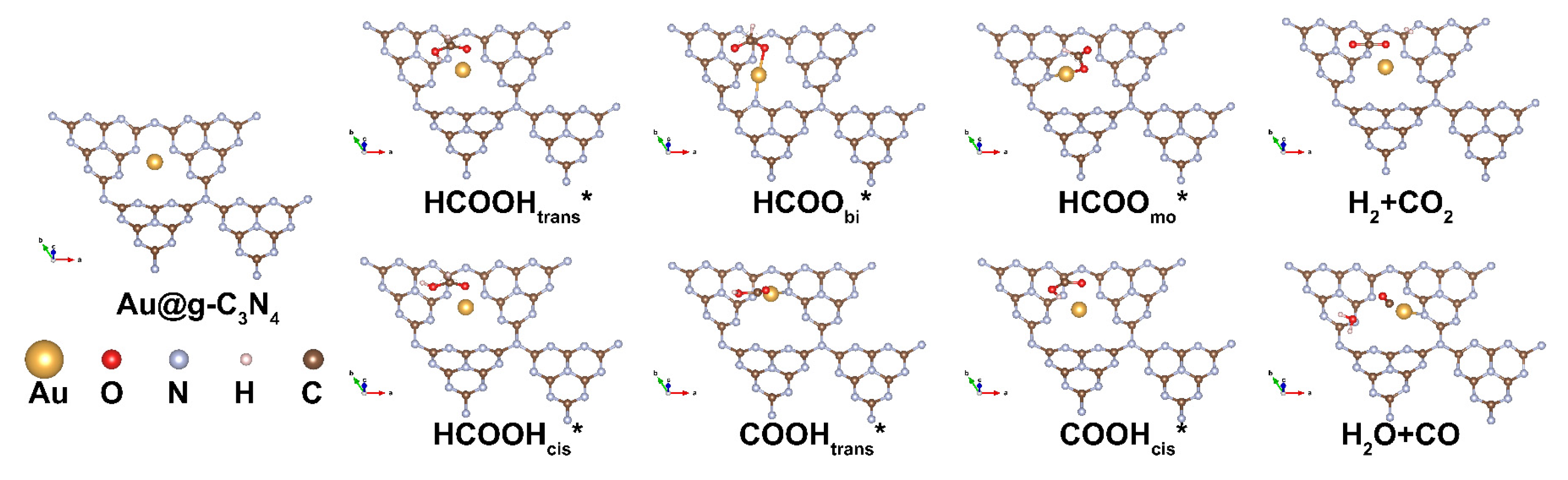
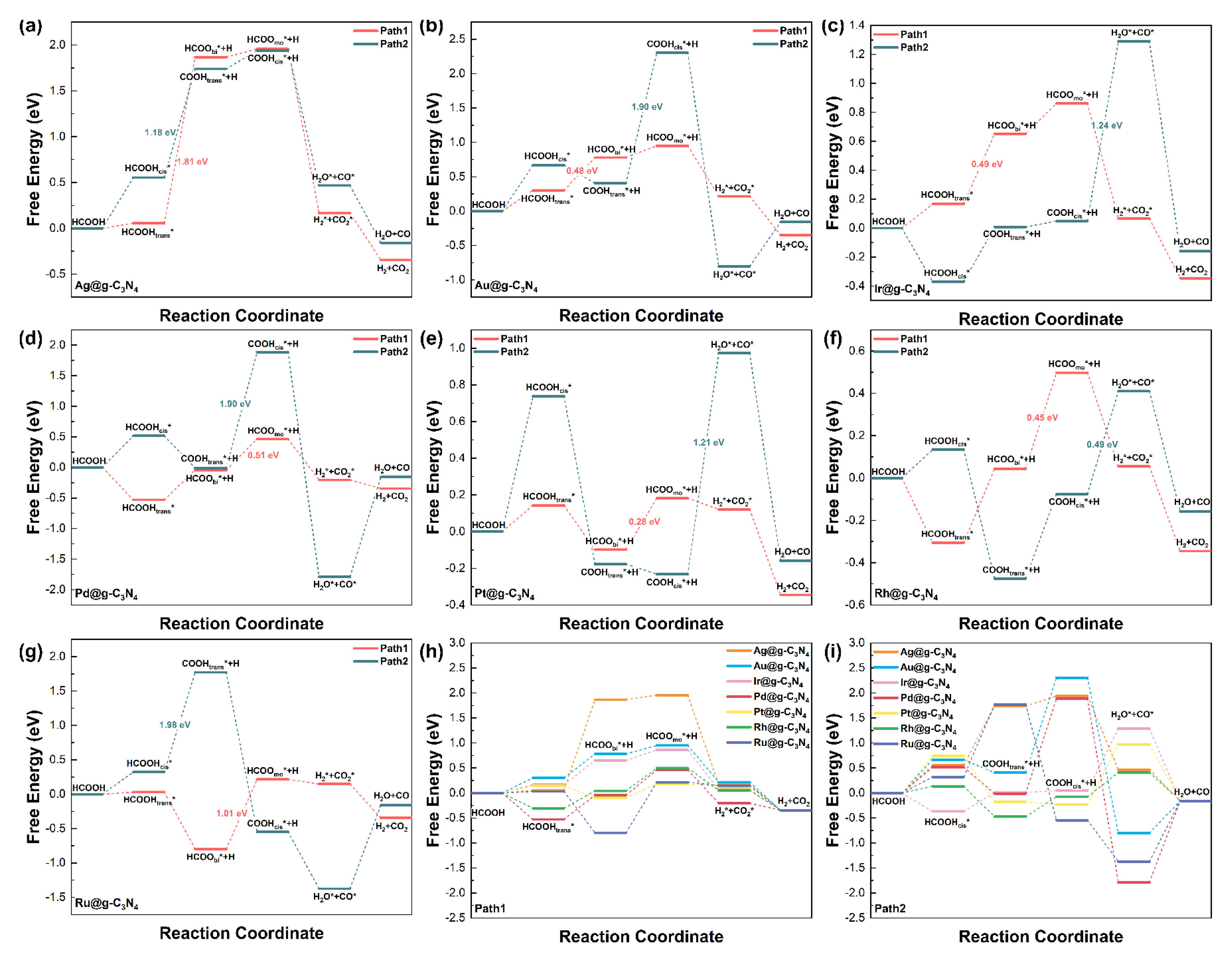
3.2. Adsorption Behavior and Pathways for FAD
3.3. Catalytic Activity and Selectivity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DFT | Density Functional Theory |
| SACs | Single Atom Catalysts |
| FAD | Formic Acid Dehydrogenation |
| g-C3N4 | graphitic carbon nitride |
References
- Wang, C.R.; Stansberry, J.M.; Mukundan, R.; Chang, H.-M.J.; Kulkarni, D.; Park, A.M.; Plymill, A.B.; Firas, N.M.; Liu, C.P.; Lang, J.T.; et al. Proton exchange membrane (PEM) water electrolysis: Cell-level considerations for gigawatt-scale deployment. Chem. Rev. 2025, 125, 1257–1302. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Ullah, A.; Samreen, A.; Qasim, M.; Nawaz, K.; Ahmad, W.; Alnaser, A.; Kannan, A.M.; Egilmez, M. Hydrogen production, storage, transportation and utilization for energy sector: A current status review. J. Energy Storage 2024, 101, 113733. [Google Scholar] [CrossRef]
- Squadrito, G.; Maggio, G.; Nicita, A. The green hydrogen revolution. Renew. Energy 2023, 216, 119041. [Google Scholar] [CrossRef]
- Zhu, Q.-L.; Xu, Q. Liquid organic and inorganic chemical hydrides for high-capacity hydrogen storage. Energy Environ. Sci. 2015, 8, 478–512. [Google Scholar] [CrossRef]
- Yadav, M.; Xu, Q. Liquid-phase chemical hydrogen storage materials. Energy Environ. Sci. 2012, 5, 9698. [Google Scholar] [CrossRef]
- Chen, B.W.J.; Mavrikakis, M. Formic acid: A hydrogen-bonding cocatalyst for formate decomposition. ACS Catal. 2020, 10, 10812–10825. [Google Scholar] [CrossRef]
- Martin, C.; Quintanilla, A.; Vega, G.; Casas, J.A. Formic acid-to-hydrogen on pd/AC catalysts: Kinetic study with catalytic deactivation. Appl. Catal. B 2022, 317, 121802. [Google Scholar] [CrossRef]
- Dai, H.; Xia, B.; Wen, L.; Du, C.; Su, J.; Luo, W.; Cheng, G. Synergistic catalysis of AgPd@ZIF-8 on dehydrogenation of formic acid. Appl. Catal. B 2015, 165, 57–62. [Google Scholar] [CrossRef]
- Choi, B.-S.; Song, J.; Song, M.; Goo, B.S.; Lee, Y.W.; Kim, Y.; Yang, H.; Han, S.W. Core–shell engineering of pd–Ag bimetallic catalysts for efficient hydrogen production from formic acid decomposition. ACS Catal. 2019, 9, 819–826. [Google Scholar] [CrossRef]
- Coffey, R.S. A catalyst for the homogeneous hydrogenation of aldehydes under mild conditions. Chem. Commun. 1967, 18, 923a. [Google Scholar] [CrossRef]
- Boddien, A.; Junge, H. Acidic ideas for hydrogen storage. Nat. Nanotechnol. 2011, 6, 265–266. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhou, X.; Yin, M.; Liu, C.; Xing, W. Novel PdAu@Au/C core−shell catalyst: Superior activity and selectivity in formic acid decomposition for hydrogen generation. Chem. Mater. 2010, 22, 5122–5128. [Google Scholar] [CrossRef]
- Yin, R.; Jiang, B.; Guo, H. Mechanism and dynamics of CO2 formation in formic acid decomposition on Pt surfaces. ACS Catal. 2022, 12, 6486–6494. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, N.; Xu, Q.; Yu, J. Nanopore-supported metal nanocatalysts for efficient hydrogen generation from liquid-phase chemical hydrogen storage materials. Adv. Mater. 2020, 32, 2001818. [Google Scholar] [CrossRef]
- Li, Z.; Yang, X.; Tsumori, N.; Liu, Z.; Himeda, Y.; Autrey, T.; Xu, Q. Tandem nitrogen functionalization of porous carbon: Toward immobilizing highly active palladium nanoclusters for dehydrogenation of formic acid. ACS Catal. 2017, 7, 2720–2724. [Google Scholar] [CrossRef]
- Yu, W.-Y.; Mullen, G.M.; Flaherty, D.W.; Mullins, C.B. Selective hydrogen production from formic acid decomposition on pd–Au bimetallic surfaces. J. Am. Chem. Soc. 2014, 136, 11070–11078. [Google Scholar] [CrossRef]
- Onishi, N.; Iguchi, M.; Yang, X.; Kanega, R.; Kawanami, H.; Xu, Q.; Himeda, Y. Development of effective catalysts for hydrogen storage technology using formic acid. Adv. Energy Mater. 2019, 9, 1801275. [Google Scholar] [CrossRef]
- Nie, R.; Tao, Y.; Nie, Y.; Lu, T.; Wang, J.; Zhang, Y.; Lu, X.; Xu, C.C. Recent Advances in Catalytic Transfer Hydrogenation with Formic Acid over Heterogeneous Transition Metal Catalysts. ACS Catal. 2021, 11, 1071–1095. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Zacharska, M.; Shlyakhova, E.V.; Chuvilin, A.L.; Guo, Y.; Beloshapkin, S.; Okotrub, A.V.; Bulusheva, L.G. Single isolated Pd2+ cations supported on N-doped carbon as active sites for hydrogen production from formic acid decomposition. ACS Catal. 2016, 6, 681–691. [Google Scholar] [CrossRef]
- Han, L.; Zhang, L.; Wu, H.; Zu, H.; Cui, P.; Guo, J.; Guo, R.; Ye, J.; Zhu, J.; Zheng, X.; et al. Anchoring Pt single atoms on Te nanowires for plasmon-enhanced dehydrogenation of formic acid at room temperature. Adv. Sci. 2019, 6, 1900006. [Google Scholar] [CrossRef]
- Rocha, G.F.S.R.; da Silva, M.A.R.; Rogolino, A.; Diab, G.A.A.; Noleto, L.F.G.; Antonietti, M.; Teixeira, I.F. Carbon nitride based materials: More than just a support for single-atom catalysis. Chem. Soc. Rev. 2023, 52, 4878–4932. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Su, P.; Chen, Y.; Zhu, B.; Zhang, S.; Huang, W. g-C3N4 supported metal (pd, Ag, Pt) catalysts for hydrogen-production from formic acid. New J. Chem. 2018, 42, 9449–9454. [Google Scholar] [CrossRef]
- Lippert, G.; Hutter, J.; Parrinello, M. A hybrid gaussian and plane wave density functional scheme. Mol. Phys. 1997, 92, 477–487. [Google Scholar] [CrossRef]
- Kühne, T.D.; Iannuzzi, M.; Del Ben, M.; Rybkin, V.V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R.Z.; Schütt, O.; Schiffmann, F.; et al. CP2K: An electronic structure and molecular dynamics software package—Quickstep: Efficient and accurate electronic structure calculations. J. Chem. Phys. 2020, 152, 194103. [Google Scholar] [CrossRef] [PubMed]
- VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef]
- Krack, M. Pseudopotentials for H to Kr optimized for gradient-corrected exchange-correlation functionals. Theor. Chem. Acc. 2005, 114, 145–152. [Google Scholar] [CrossRef]
- Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Shermo: A general code for calculating molecular thermochemistry properties. Comput. Theor. Chem. 2021, 1200, 113249. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Jiao, Y.; Zhu, Y.; Cai, Q.; Vasileff, A.; Li, L.H.; Han, Y.; Chen, Y.; Qiao, S.-Z. Molecule-level g-C3N4 coordinated transition metals as a new class of electrocatalysts for oxygen electrode reactions. J. Am. Chem. Soc. 2017, 139, 3336–3339. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Periyasamy, G.; Pandey, B.; Pati, S.K. Computational studies on magnetism and the optical properties of transition metal embedded graphitic carbon nitride sheets. J. Mater. Chem. C 2014, 2, 7943–7951. [Google Scholar] [CrossRef]
- Jin, T.; Guo, L.; Tang, Q.; Wang, J.; Pan, B.; Wang, C.; Li, Z.; Chen, F. Spontaneous formate oxidation on the 2D surface metal fluoride interface reconstructed from the AgPdF surface. J. Phys. Chem. C 2022, 126, 9683–9695. [Google Scholar] [CrossRef]
- Zhang, N.; Gao, Y.; Ma, L.; Wang, Y.; Huang, L.; Wei, B.; Xue, Y.; Zhu, H.; Jiang, R. Single transition metal atom anchored on g-C3N4 as an electrocatalyst for nitrogen fixation: A computational study. Int. J. Hydrogen Energy 2023, 48, 7621–7631. [Google Scholar] [CrossRef]
- Jin, T.; Chen, F.; Guo, L.; Tang, Q.; Wang, J.; Pan, B.; Wu, Y.; Yu, S. Pd4O3 subsurface oxide on pd(111) formed during oxygen adsorption-induced surface reconstruction and its activity toward formate oxidation reactions. J. Phys. Chem. C 2021, 125, 19497–19508. [Google Scholar] [CrossRef]
- Tang, Y.; Roberts, C.A.; Perkins, R.T.; Wachs, I.E. Revisiting formic acid decomposition on metallic powder catalysts: Exploding the HCOOH decomposition volcano curve. Surf. Sci. 2016, 650, 103–110. [Google Scholar] [CrossRef]
- Fingerhut, J.; Lecroart, L.; Schwarzer, M.; Hörandl, S.; Borodin, D.; Kandratsenka, A.; Kitsopoulos, T.N.; Auerbach, D.J.; Wodtke, A.M. Identification of reaction intermediates in the decomposition of formic acid on pd. Faraday Discuss. 2024, 251, 412–434. [Google Scholar] [CrossRef]
- Greiner, M.T.; Jones, T.E.; Beeg, S.; Zwiener, L.; Scherzer, M.; Girgsdies, F.; Piccinin, S.; Armbrüster, M.; Knop-Gericke, A.; Schlögl, R. Free-atom-like d states in single-atom alloy catalysts. Nature Chem 2018, 10, 1008–1015. [Google Scholar] [CrossRef]
- Yang, Y.; Peltier, C.R.; Zeng, R.; Schimmenti, R.; Li, Q.; Huang, X.; Yan, Z.; Potsi, G.; Selhorst, R.; Lu, X.; et al. Electrocatalysis in alkaline media and alkaline membrane-based energy technologies. Chem. Rev. 2022, 122, 6117–6321. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, T.; Liang, S.; Zhang, J.; Li, Y.; Bai, Y.; Wu, H.; Razanau, I.; Pan, K.; Wang, F. Theoretical Insights into Hydrogen Production from Formic Acid Catalyzed by Pt-Group Single-Atom Catalysts. Materials 2025, 18, 2328. https://doi.org/10.3390/ma18102328
Jin T, Liang S, Zhang J, Li Y, Bai Y, Wu H, Razanau I, Pan K, Wang F. Theoretical Insights into Hydrogen Production from Formic Acid Catalyzed by Pt-Group Single-Atom Catalysts. Materials. 2025; 18(10):2328. https://doi.org/10.3390/ma18102328
Chicago/Turabian StyleJin, Tao, Sen Liang, Jiahao Zhang, Yaru Li, Yukun Bai, Hangjin Wu, Ihar Razanau, Kunming Pan, and Fang Wang. 2025. "Theoretical Insights into Hydrogen Production from Formic Acid Catalyzed by Pt-Group Single-Atom Catalysts" Materials 18, no. 10: 2328. https://doi.org/10.3390/ma18102328
APA StyleJin, T., Liang, S., Zhang, J., Li, Y., Bai, Y., Wu, H., Razanau, I., Pan, K., & Wang, F. (2025). Theoretical Insights into Hydrogen Production from Formic Acid Catalyzed by Pt-Group Single-Atom Catalysts. Materials, 18(10), 2328. https://doi.org/10.3390/ma18102328