
Investigation of Compatibility Mechanisms and Diffusion Behavior of Polymer SBS-Modified Asphalt Compatibilizer Using Molecular Dynamics Simulation

Abstract
1. Introduction
2. Molecular Dynamics Model of SBSMA
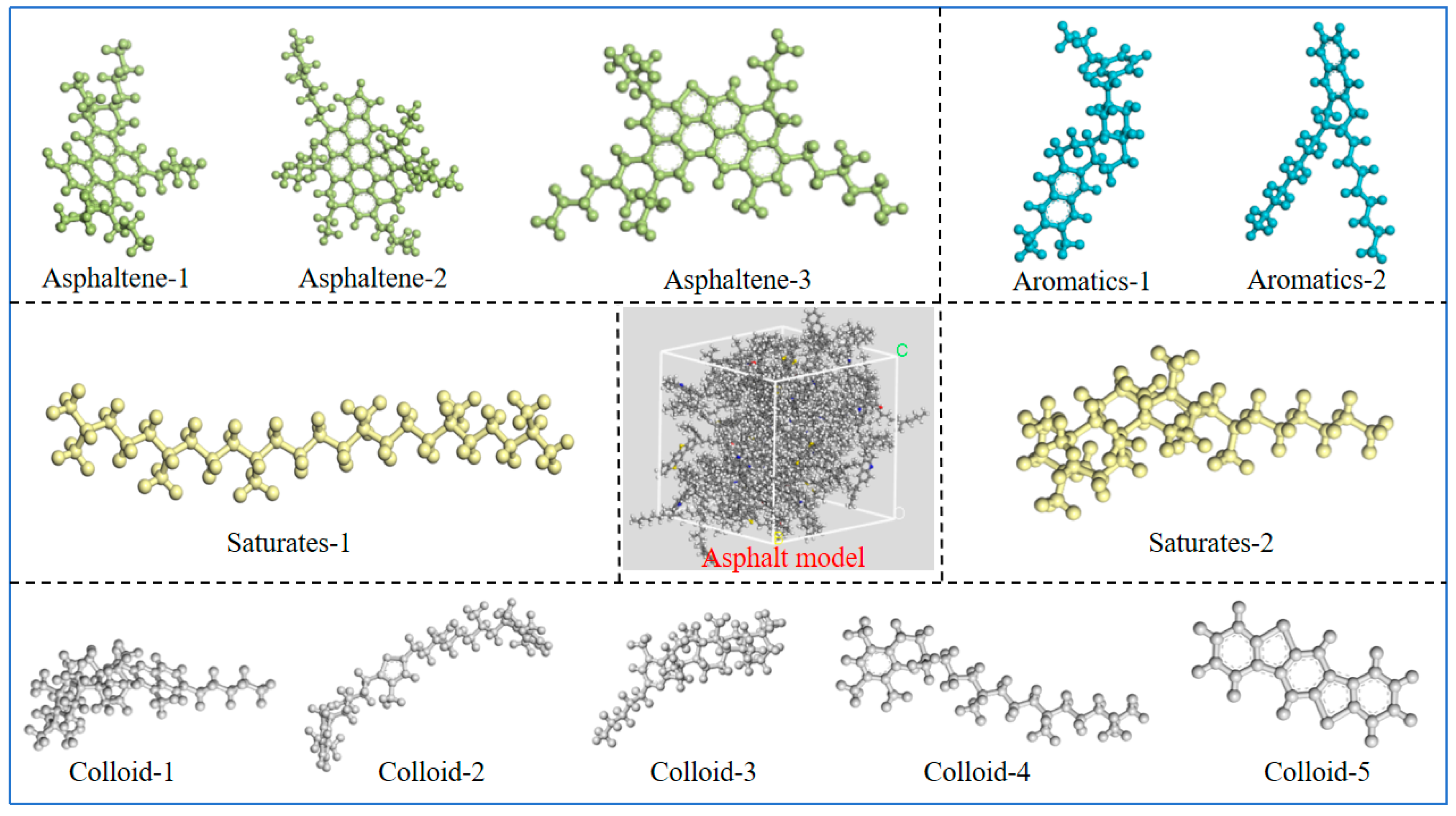
2.1. Molecular Modeling of Matrix Asphalt Binder
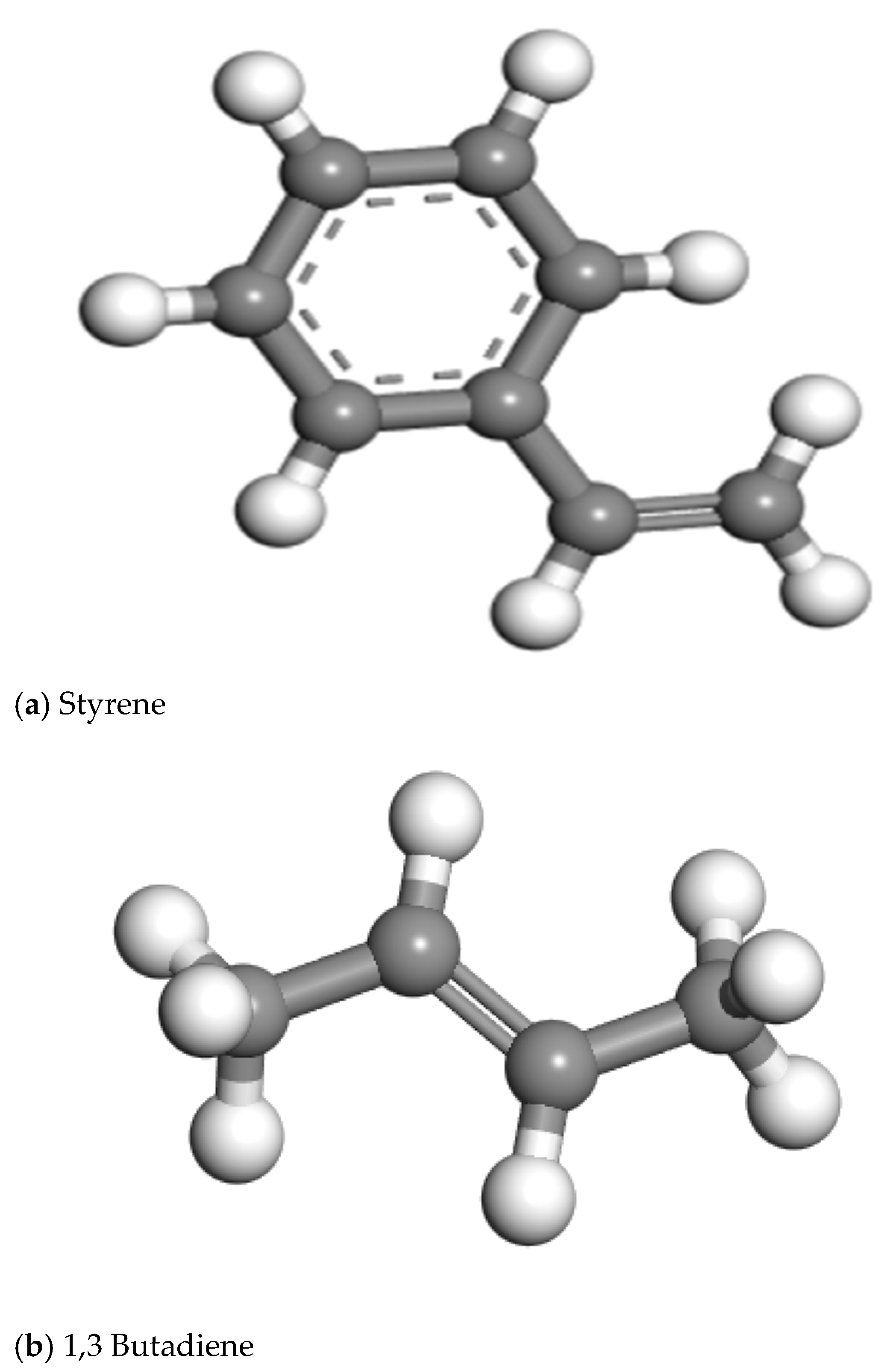
2.2. Molecular Modeling of Polymer SBS
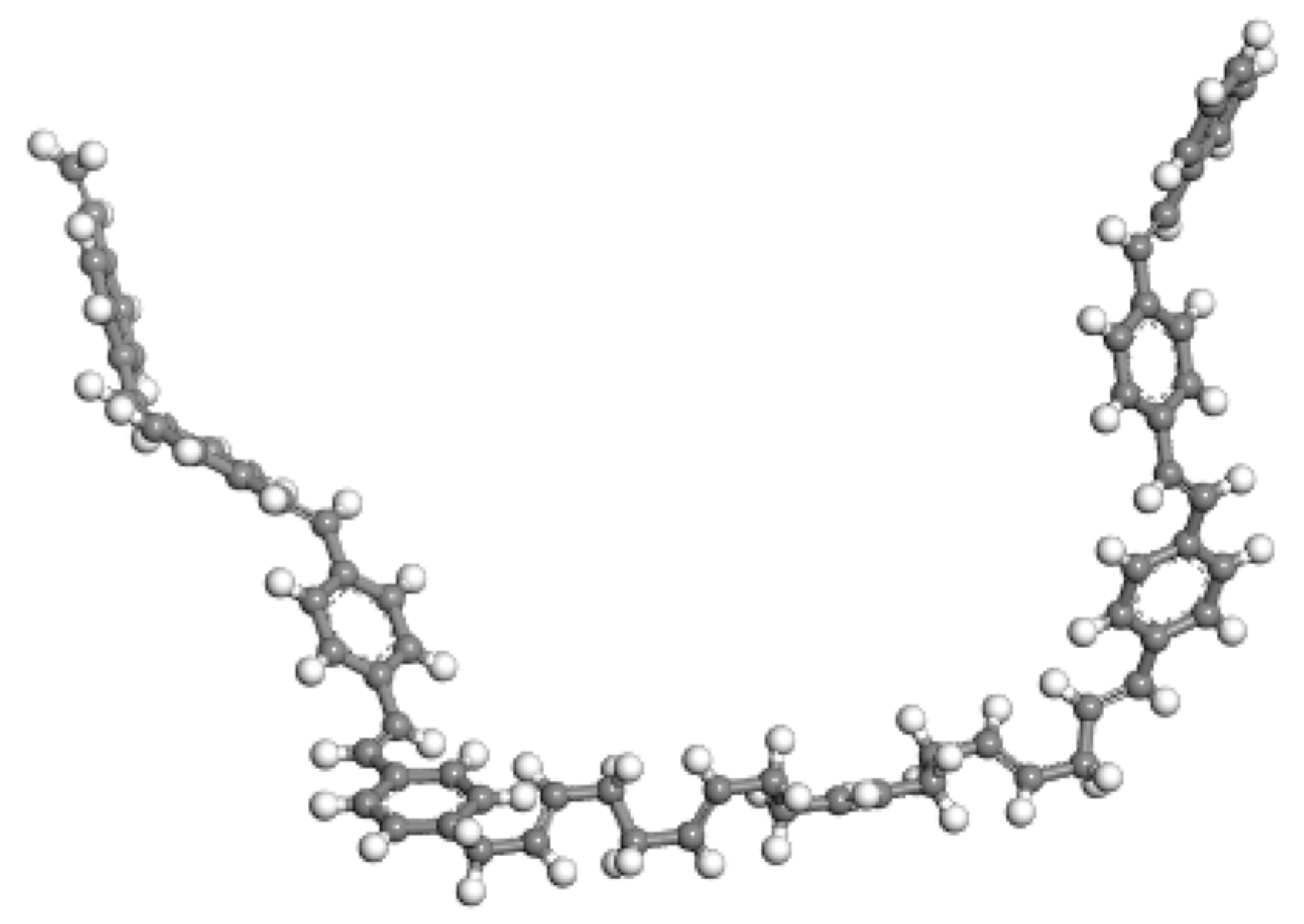
2.3. Molecular Modeling of Compatibilizer
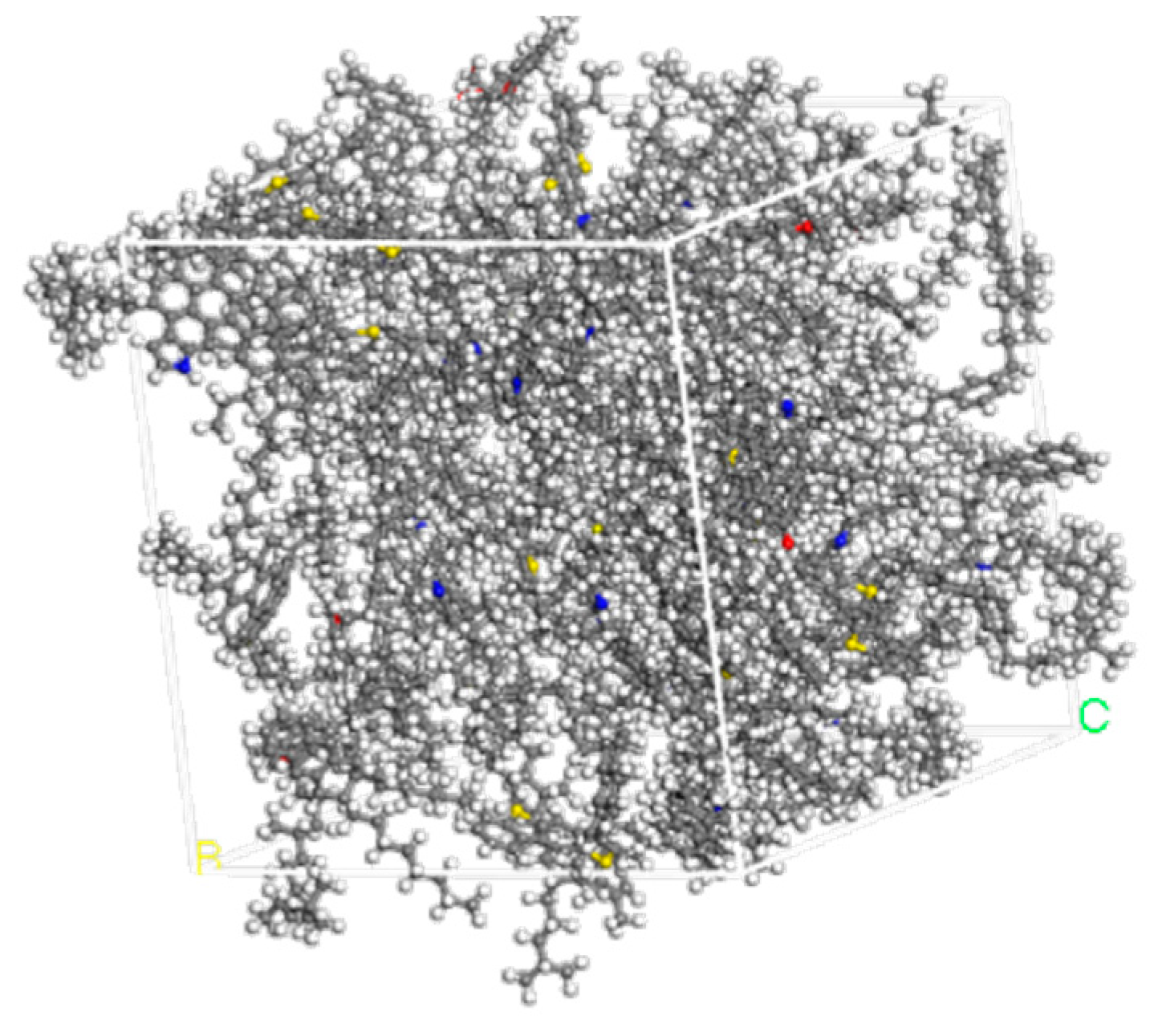
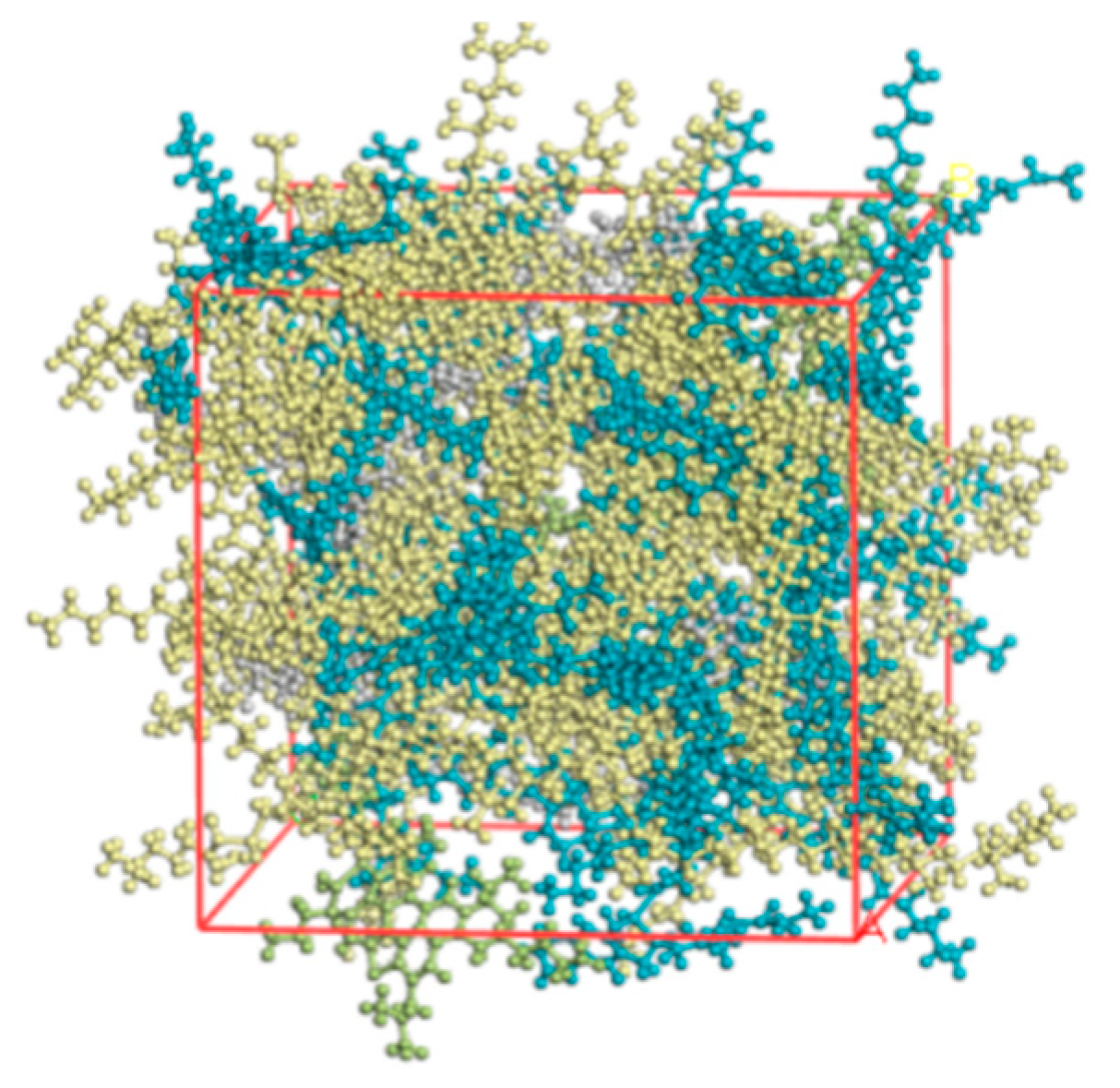
2.4. SBSMA System Model
3. Theory and Methods of Molecular Dynamics
3.1. Basic Principles and Methods
3.1.1. Molecular Dynamics Theory
- (1)
- Rationale
- (2)
- Force field
- (3)
- Periodic boundary condition
- (4)
- Synthesis
- (5)
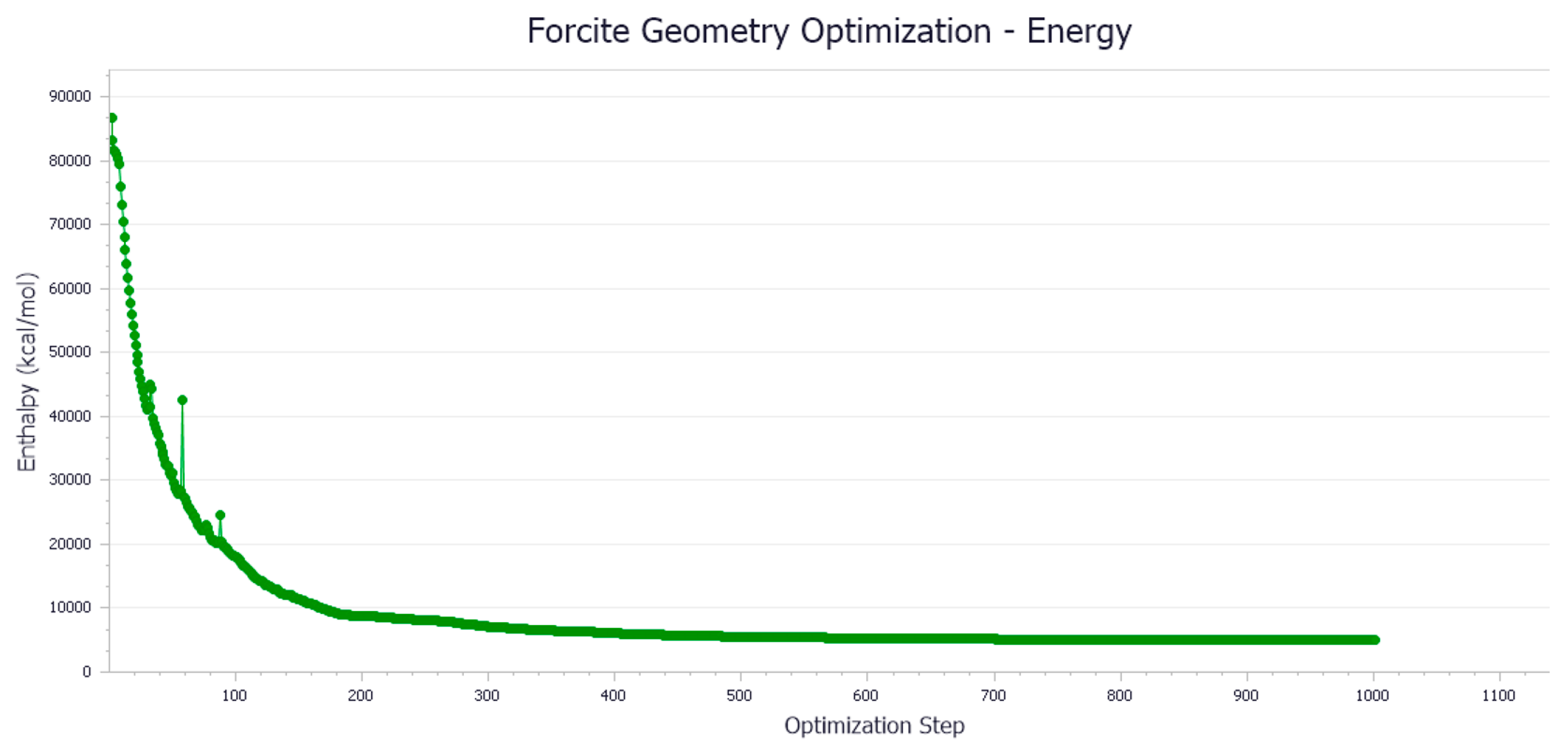
- Energy Minimization
3.1.2. Simulation Methods
- (1)
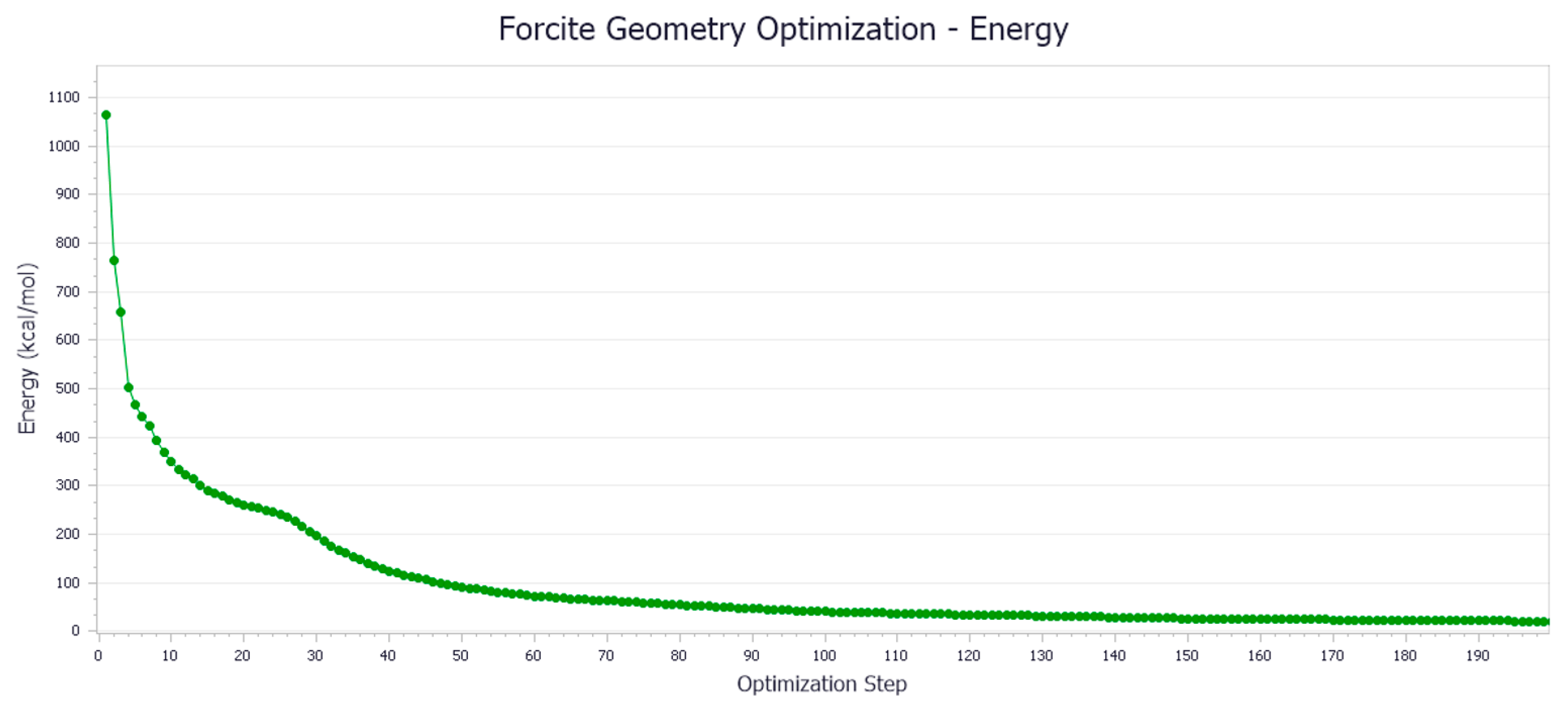
- Model Optimization
- (2)
- MD Simulation
3.1.3. Assumptions and Limitations of Molecular Dynamics Simulations
- (1)
- Coarse-Graining Approximation
- (2)
- Timescale Limitations
- (3)
- Periodic Boundary Condition Artifacts
- (4)
- Force Field Transferability
4. Results and Discussion
4.1. Model Reliability Verification
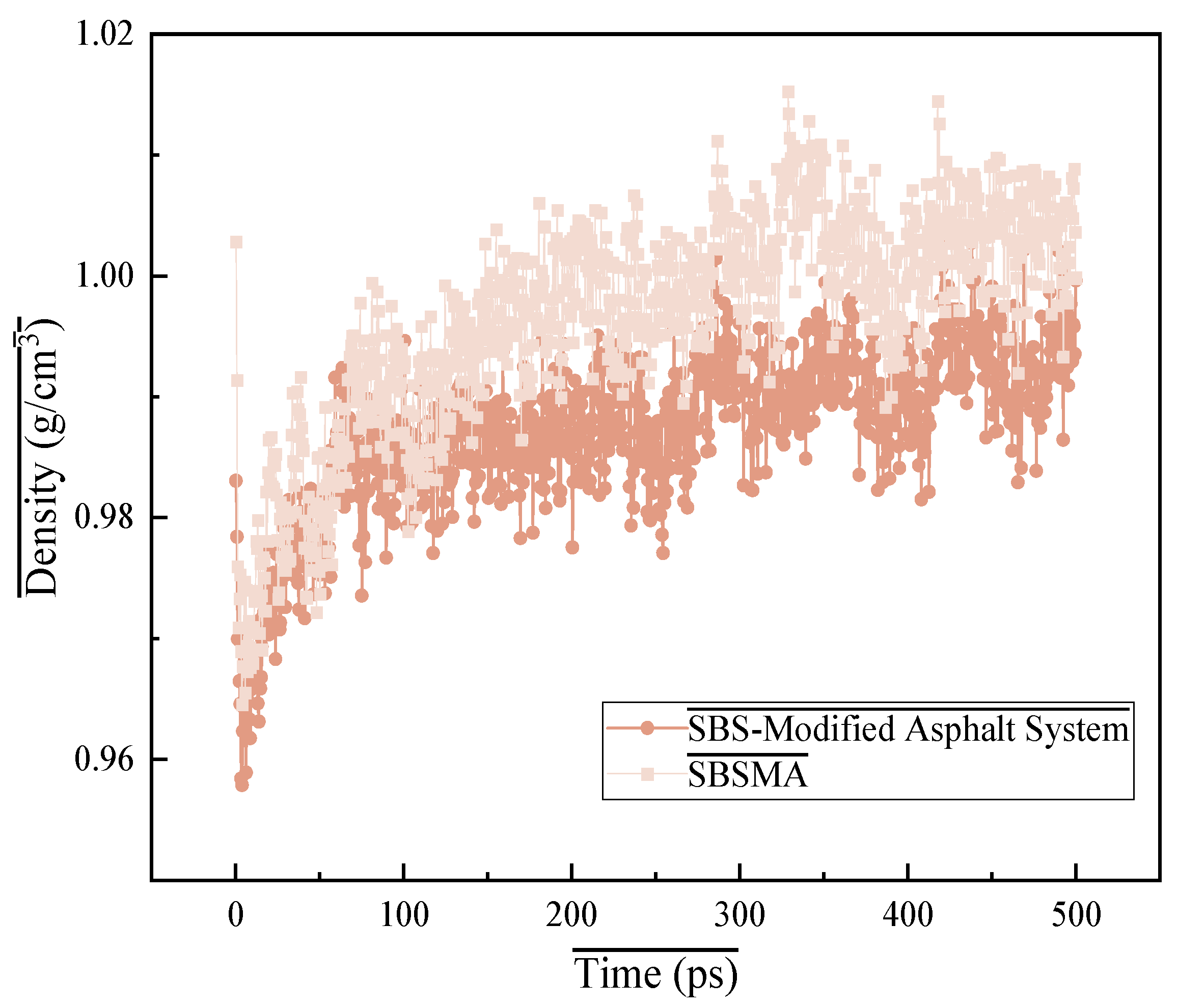
4.1.1. Density Verification
4.1.2. Verification of Glass Transition Temperature
4.2. Effect of Compatibilizer Dosage on the Compatibility of SBSMA
4.2.1. SBSMA Compatibility Characterization Parameters
- (1)
- Cohesive energy density
- (2)
- Solubility parameters
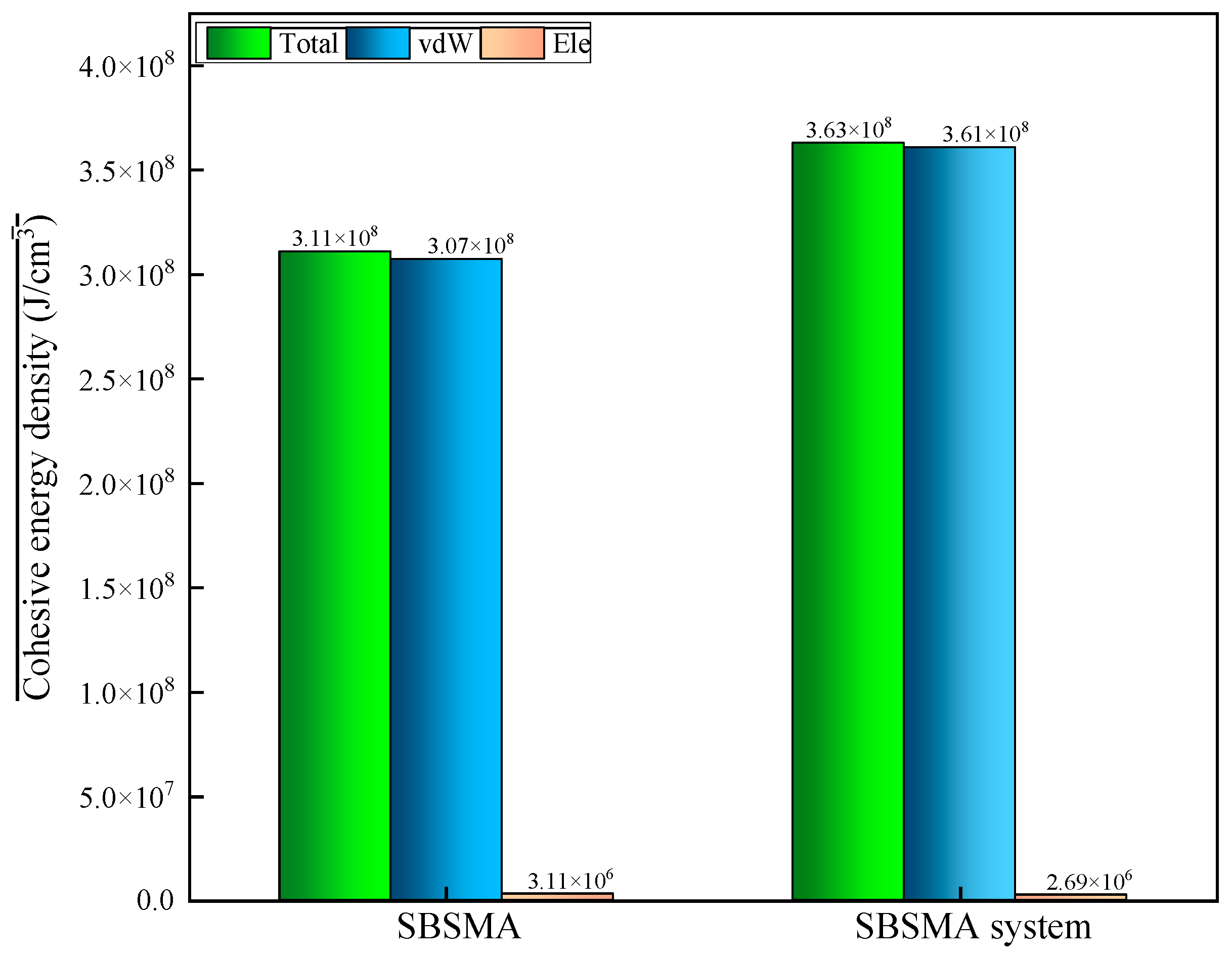
4.2.2. Changing Law of Cohesive Energy Density of SBSMA
4.2.3. Changing Law of Solubility Parameters of SBSMA
4.3. Effect of Compatibilizer Dosage on Diffusion Simulation of SBSMA
4.3.1. Diffusion Characterization Parameters of SBSMA
- (1)
- Mean Square Displacement
- (2)
- Radial distribution function
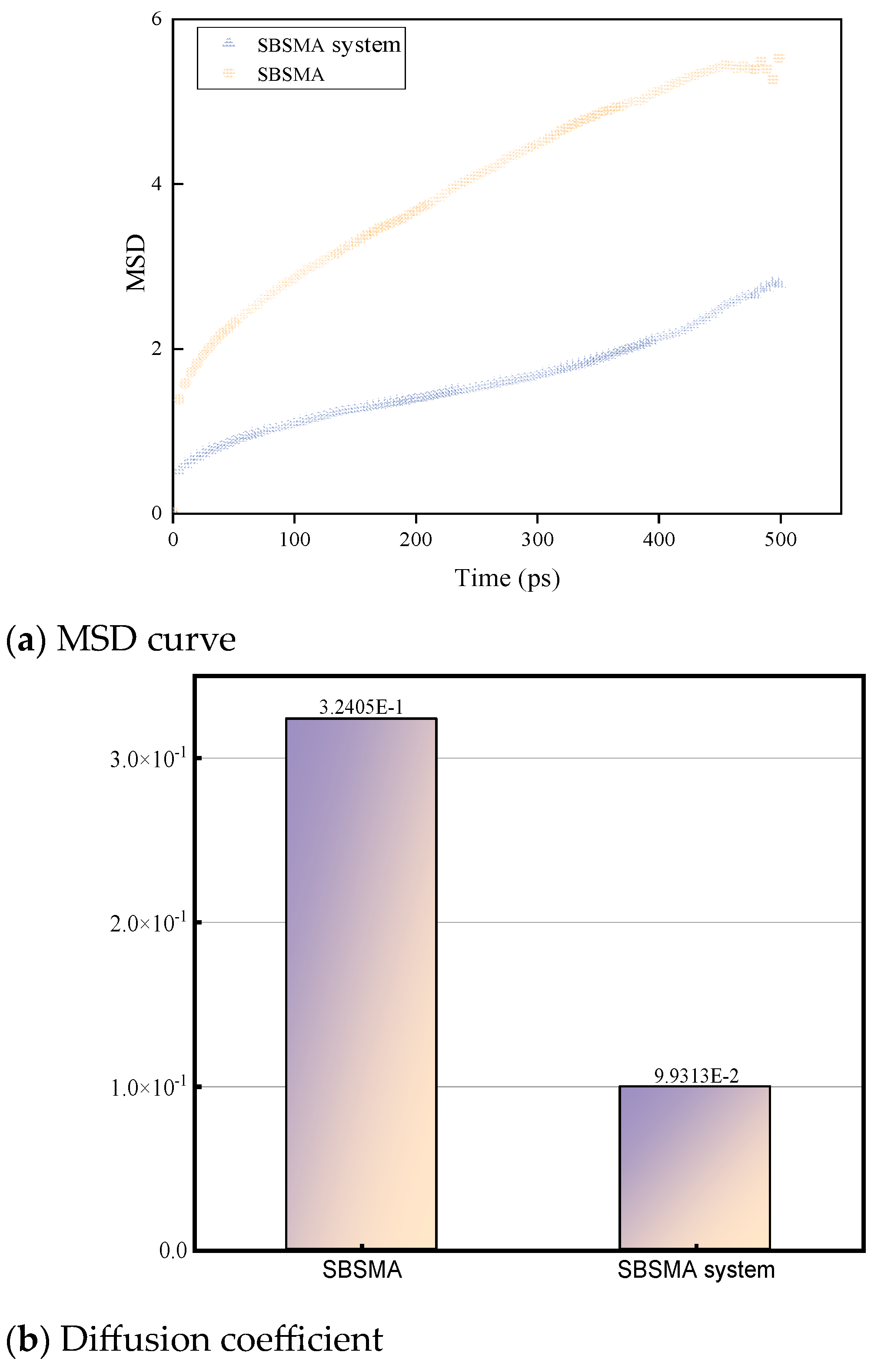
4.3.2. Changing Law of Mean Square Displacement of SBSMA
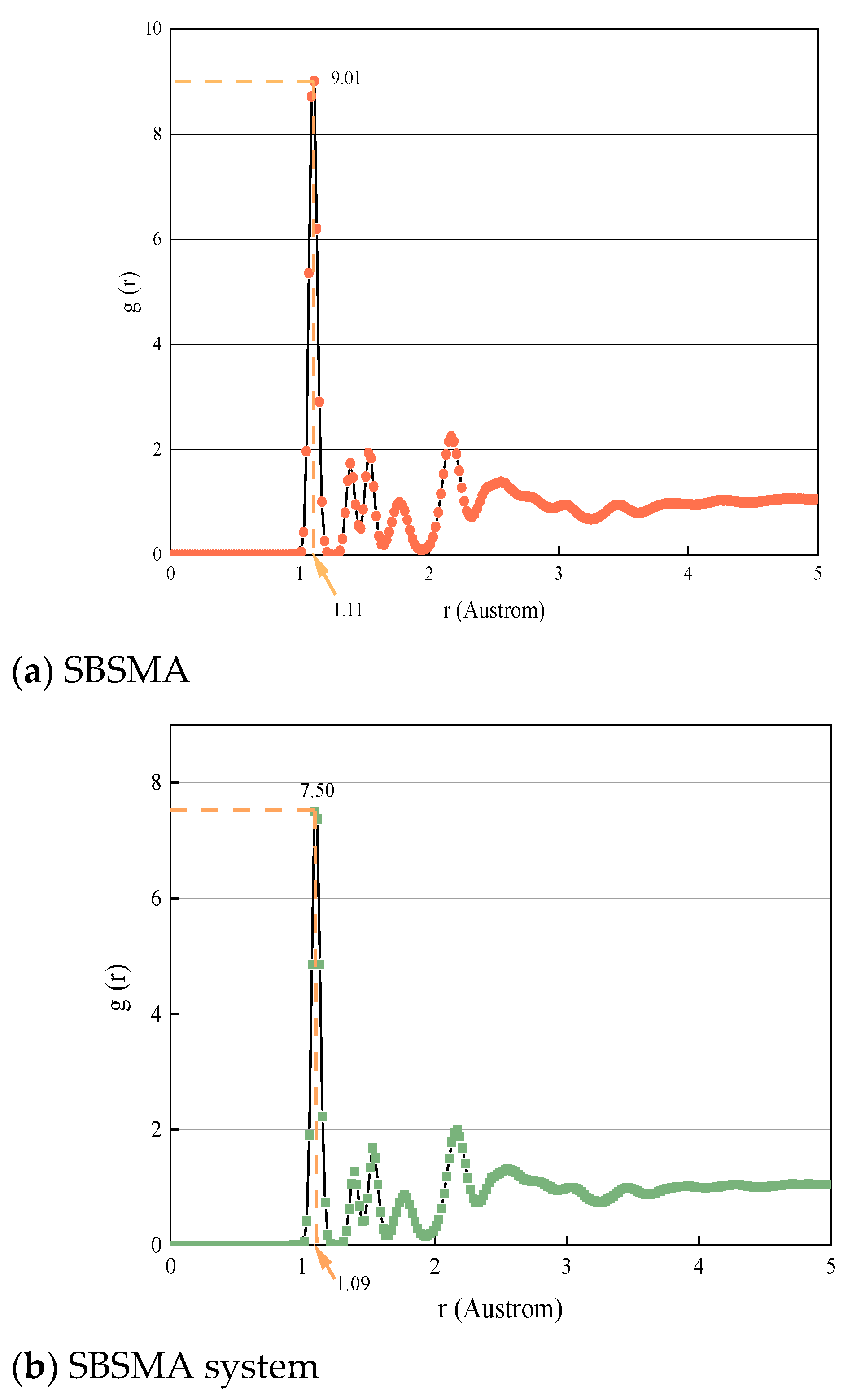
4.3.3. Changing Law of Radial Distribution Function of SBSMA
4.4. Experimental Verification
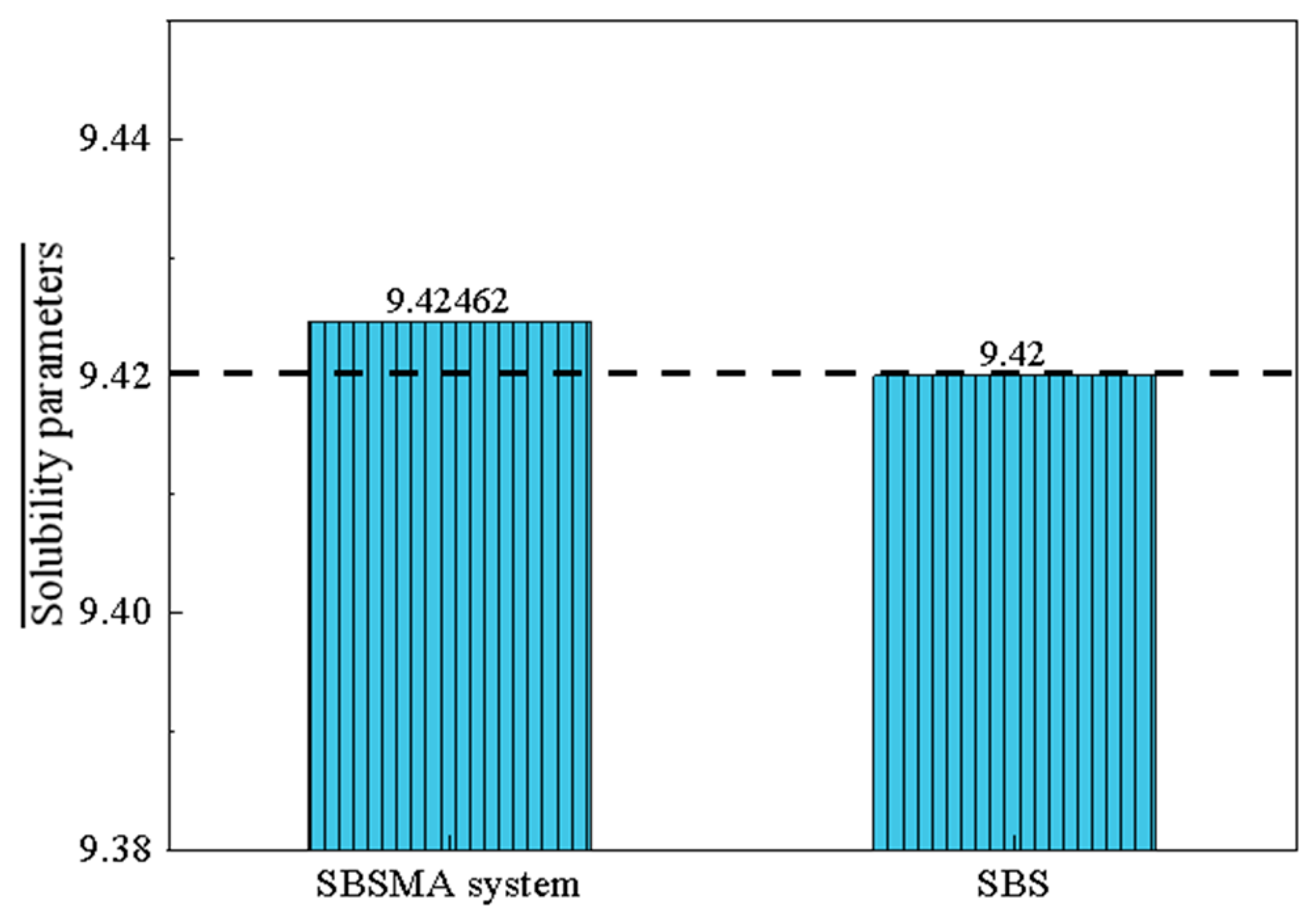
4.4.1. Analysis of Solubility Parameters of Asphalt with the Addition of Compatibilizer
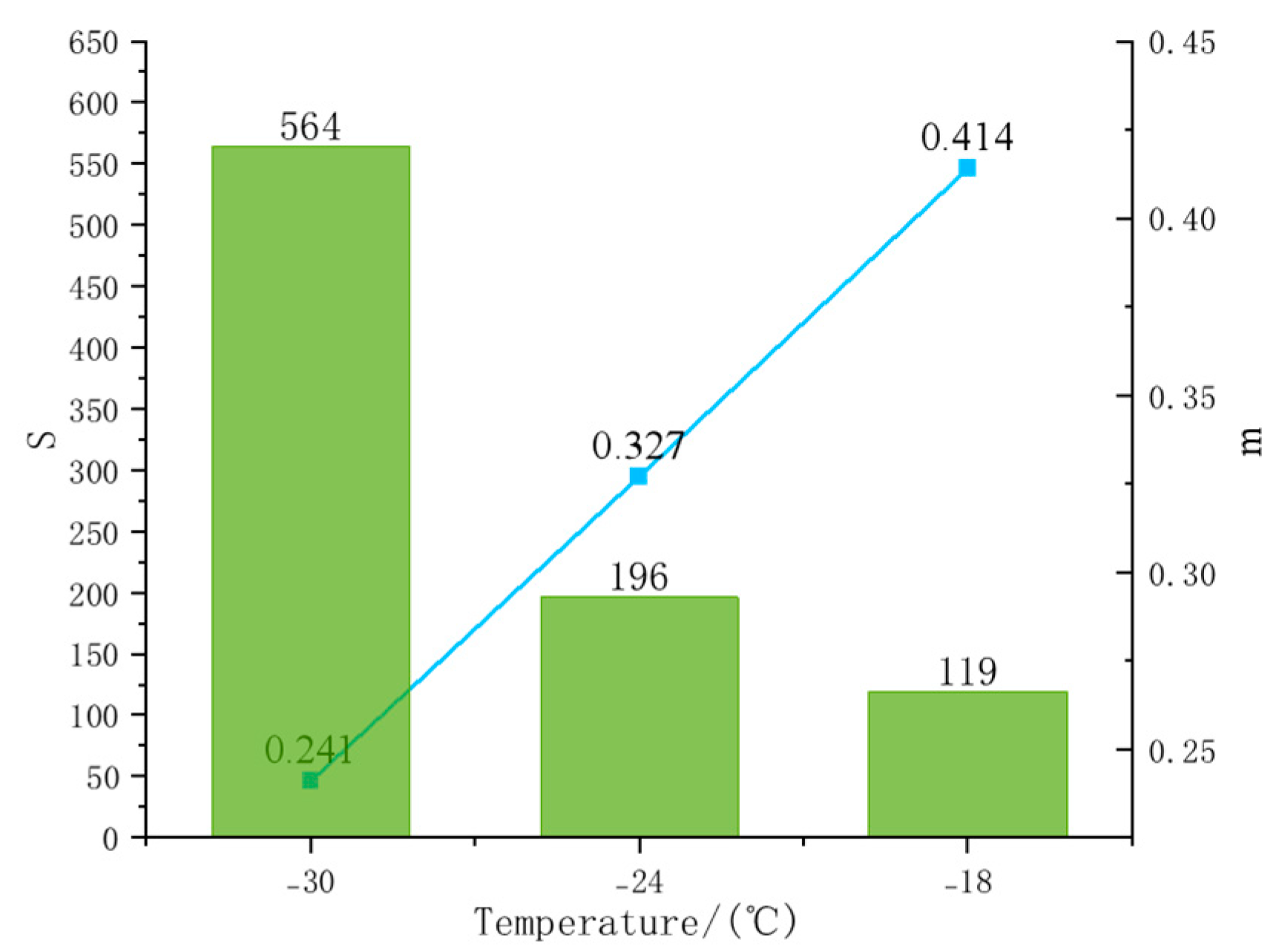
4.4.2. BBR Experimental Results and Index Analysis
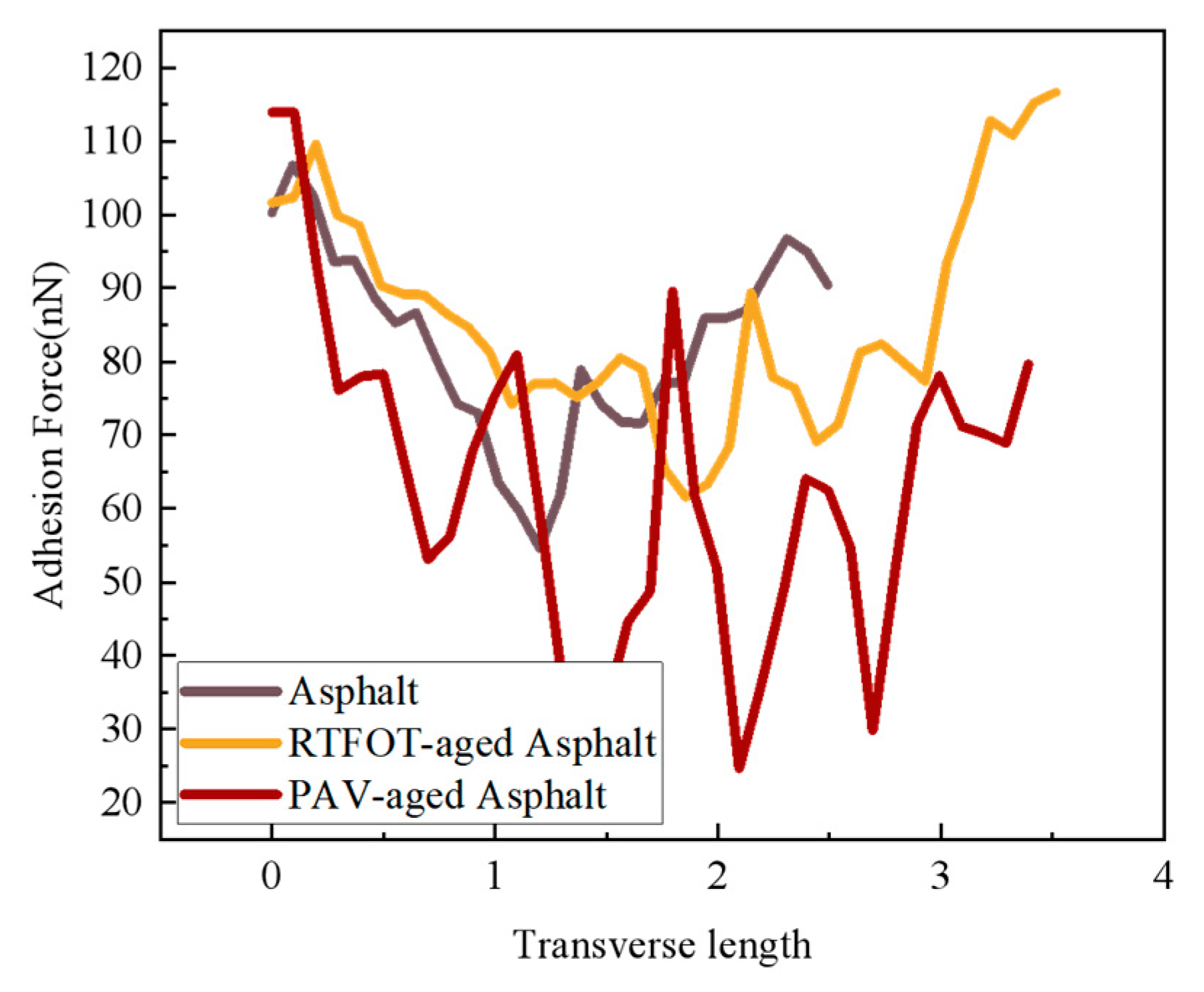
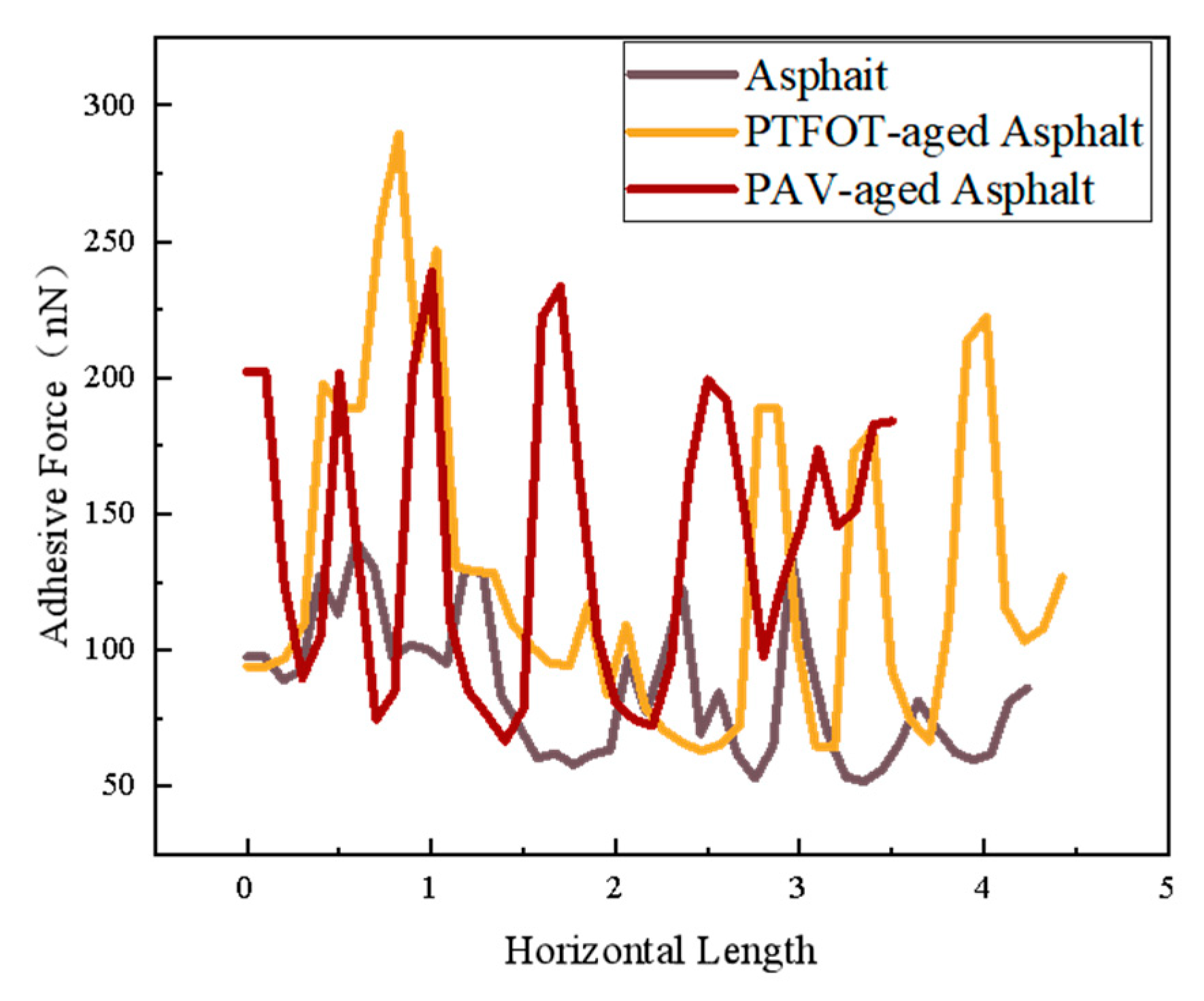
4.4.3. AFM-Based Surface Adhesion Analysis of SBS-Modified Asphalt
- (1)
- SBS-modified asphalt
- (2)
- SBS-modified asphalt with compatibilizer added
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SBS | Styrene–butadiene–styrene |
| SBSMA | Styrene–butadiene–styrene-modified asphalt |
| PS | Polystyrene |
| PB | Polybutadiene |
| Tg | Glass transition temperature |
| MD | Molecular dynamics simulation |
| MS | Materials Studio |
| SARA | Saturated, aromatic, resin, and asphaltene |
| NVT | Number of system atoms, volume, and temperature |
| NPT | Constant molecular number, external pressure, and temperature |
| CG | Coarse-graining |
| PBC | Periodic boundary conditions |
| RDF | Radial distribution function |
| DFT | Density-functional theory |
| CED | Cohesive energy density |
| SPD | Solubility parameter difference |
| MSD | Mean square displacement |
References
- Nie, X.; Li, Z.; Yao, H.; Hou, T.; Zhou, X.; Li, C. Waste bio-oil as a compatibilizer for high content SBS modified asphalt. Pet. Sci. Technol. 2020, 38, 316–322. [Google Scholar] [CrossRef]
- Li, C.; Li, Z.; Guo, T.; Chen, Y.; Liu, Q.; Wang, J.; Jin, L. Study on the performance of SBS/Polyphosphoric acid composite modified asphalt. Coatings 2024, 14, 72. [Google Scholar] [CrossRef]
- Han, M.; Li, J.; Muhammad, Y.; Yin, Y.; Yang, J.; Yang, S.; Duan, S. Studies on the secondary modification of SBS modified asphalt by the application of octadecyl amine grafted graphene nanoplatelets as modifier. Diam. Relat. Mater. 2018, 89, 140–150. [Google Scholar] [CrossRef]
- Li, Y.; Ma, R.; Wang, X.; Cheng, P.; Chen, Y. Improvement effect of different modifiers on storage stability of high content SBS modified asphalt. Case Stud. Constr. Mater. 2024, 20, e02820. [Google Scholar] [CrossRef]
- Guo, P.; Li, P.; Chang, C.; Xu, G.; Shi, X.; Bai, J.; Fang, S. Progress in the application of computer simulation technology in biomass conversion. Adv. Chem. Eng. 2020, 39, 3027–3040. [Google Scholar]
- Tang, Y.; Fu, Z.; Raos, G.; Ma, F.; Zhao, P.; Hou, Y. Molecular dynamics simulation of adhesion at the asphalt-aggregate interface: A review. Surf. Interfaces 2024, 44, 103706. [Google Scholar] [CrossRef]
- Qian, C.; Fan, W. Evaluation and characterization of properties of crumb rubber/SBS modified asphalt. Mater. Chem. Phys. 2020, 253, 123319. [Google Scholar] [CrossRef]
- Yu, C.; Hu, K.; Chen, Y.; Zhang, W.; Chen, Y.; Chang, R. Compatibility and high temperature performance of recycled polyethylene modified asphalt using molecular simulations. Mol. Simul. 2021, 47, 1037–1049. [Google Scholar] [CrossRef]
- Zhang, W.; Jia, Z.; Wang, F. Effect and prediction of aromatic oil on swelling degree of direct-to-plant SBS modifier in bitumen. Pet. Sci. Technol. 2019, 37, 1033–1040. [Google Scholar] [CrossRef]
- Yao, X.; Li, C.; Xu, T. Multi-scale studies on interfacial system compatibility between asphalt and SBS modifier using molecular dynamics simulations and experimental methods. Constr. Build. Mater. 2022, 346, 128502. [Google Scholar] [CrossRef]
- Yu, C.; Hu, K.; Chen, G.; Chang, R.; Wang, Y. Molecular dynamics simulation and microscopic observation of compatibility and interphase of composited polymer modified asphalt with carbon nanotubes. J. Zhejiang Univ. Sci. A 2021, 22, 528–546. [Google Scholar] [CrossRef]
- Li, D.D.; Greenfield, M.L. Chemical compositions of improved model asphalt systems for molecular simulations. Fuel 2014, 115, 347–356. [Google Scholar] [CrossRef]
- Liu, F.; Wang, Q.; Zhang, X.; Zhou, Z.; Wang, X. Investigation of asphalt oxidation kinetics aging mechanism using molecular dynamic simulation. Constr. Build. Mater. 2023, 377, 131159. [Google Scholar] [CrossRef]
- Yao, H.; Liu, J.; Xu, M.; Ji, J.; Dai, Q.; You, Z. Discussion on molecular dynamics (MD) simulations of the asphalt materials. Adv. Colloid Interface Sci. 2022, 299, 102565. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Chen, Y.; Fan, C.; Li, M. Molecular dynamics study on the compatibility of asphalt and rubber powder with different component contents. ACS Omega 2022, 7, 36157–36164. [Google Scholar] [CrossRef]
- Gao, Y.; Tian, W.; Li, Y.; Zhu, J.; Liao, M.; Xie, Y. Study on compatibility mechanism of plasticizer and asphalt based on molecular dynamics. Mater. Des. 2023, 228, 111827. [Google Scholar] [CrossRef]
- Liu, S.; Shan, L.; Qi, C.; Zhang, W.; Li, G.; Wang, B.; Wei, W. Effect of SBS structure on viscosity of SBS-modified asphalt based on molecular dynamics: Insights from shearing phase morphology, adsorption and swelling mechanisms. J. Mol. Liq. 2024, 393, 123567. [Google Scholar] [CrossRef]
- Li, K.; Ren, H.; Huang, W. Effect of graphene nanoplatelets (GNPs) on fatigue properties of asphalt mastics. Materials 2021, 14, 4864. [Google Scholar] [CrossRef]
- Xu, S.; Hu, C.; Yu, J.; Que, Y.; Zhou, X. Performance of mixed asphalt blended with furfural extract oil and its distinction from pure asphalt. Pet. Sci. Technol. 2017, 35, 1673–1679. [Google Scholar] [CrossRef]
- Zhang, S.; Schweitzer-Stenner, R.; Urbanc, B. Do molecular dynamics force fields capture conformational dynamics of alanine in water? J. Chem. Theory Comput. 2019, 16, 510–527. [Google Scholar] [CrossRef]
- Liu, S.; Wang, H.; Yang, J.; Luo, S.; Liu, Y.; Huang, W.; Hu, J.; Xu, G.; Min, Z. Force field benchmark of asphalt materials: Density, viscosity, glass transition temperature, diffusion coefficient, cohesive energy density and molecular structures. J. Mol. Liq. 2024, 398, 124166. [Google Scholar] [CrossRef]
- Li, B.; Han, J.; Wei, D.; Ji, H.; Yao, T.; Wang, H.; Han, J.; Zhang, Y. A molecular dynamics simulation study on the recovery performance of aged asphalt binder by waste vegetable oil rejuvenators. J. Clean. Prod. 2024, 442, 140796. [Google Scholar] [CrossRef]
- Lee, S.H. Diffusion and friction dynamics of probe molecules in liquid n-alkane systems: A molecular dynamics simulation study. J. Chem. 2019, 2019, 8134904. [Google Scholar] [CrossRef]
- Wang, M.; Lü, S.; Wu, S.; Chen, X.; Guo, W. Rejuvenation behaviors of recovery-annealed Cu–Zr metallic glass with different thermal treatment conditions: A molecular dynamics study. J. Mater. Res. Technol. 2022, 20, 3355–3362. [Google Scholar] [CrossRef]
- Han, J.; Li, B.; Ji, H.; Guo, F.; Wei, D.; Cao, S.; Zhang, W.; Chen, X. Interfacial adhesion between recycled asphalt binder and aggregates considering aggregate surface anisotropy: A molecular dynamics simulation. Constr. Build. Mater. 2024, 438, 137176. [Google Scholar] [CrossRef]
- Chen, Z.; Yang, Y. Incorporating nuclear quantum effects in molecular dynamics with a constrained minimized energy surface. J. Phys. Chem. Lett. 2023, 14, 279–286. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, L.; Shi, Y. Molecular dynamics study on the viscosity of glass-forming systems near and below the glass transition temperature. J. Am. Ceram. Soc. 2021, 104, 6227–6241. [Google Scholar] [CrossRef]
- Nguele, R.; Poupi, A.B.M.; Anombogo, G.A.M.; Alade, O.S.; Saibi, H. Influence of asphaltene structural parameters on solubility. Fuel 2022, 311, 122559. [Google Scholar] [CrossRef]
- Khrapak, S.A. System size dependence of the diffusion coefficients in MD simulations: A simple correction formula for pure dense fluids. J. Phys. Chem. B 2024, 128, 287–290. [Google Scholar] [CrossRef]
- Andrea, C.; David, C.; Piero, B. Advanced methodologies for the cleaning of works of art. Sci. China Technol. Sci. 2023, 66, 2162–2182. [Google Scholar]
- Wang, P.; Wei, H.; Liu, X.; Ren, R.; Wang, L. Identifying the long-term thermal storage stability of SBS-Polymer-Modified asphalt, including physical indexes, rheological properties, and micro-structures characteristics. Sustainability 2021, 13, 10582. [Google Scholar] [CrossRef]
- Chen, R. Application of solubility parameters in disperse dye blending dyeing (continued). Dye. Finish. Technol. 2019, 41, 1–5+10. [Google Scholar]
- Yan, Y.; Hernando, D.; Roque, R. A solvent free method to characterize the effect of recycled asphalt shingles on virgin asphalt binder. J. Clean. Prod. 2019, 208, 795–805. [Google Scholar] [CrossRef]
- Niu, D.; Xie, H.; Yang, Z.; Zhang, W.; Sheng, Y.; Xiong, R. Molecular structure changes and anti-aging properties of WFO-CR modified asphalt. J. Shenzhen Univ. (Sci. Technol. Ed.) 2020, 37, 589–596. [Google Scholar]
- Chen, H.; Huang, S.; Niu, D.; Gao, Y.; Zhang, Z. Fatigue characterization and assessment methods for the terminal blend crumb rubber/SBS composite modified asphalt binders. Constr. Build. Mater. 2024, 430, 136357. [Google Scholar] [CrossRef]
- Mikhailenko, P.; Baaj, H. Comparison of chemical and microstructural properties of virgin and reclaimed asphalt pavement binders and their saturate, aromatic, resin, and asphaltene fractions. Energy Fuels 2019, 33, 2633–2640. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SARA Component | Molecule Name | Number of Molecules | Molecular Formula | Molecular Weight (g/mol) | Mass Fraction (%) |
|---|---|---|---|---|---|
| Saturated fraction | Squalane | 4 | C30H62 | 422.9 | 5.2 |
| Hopane | 4 | C35H62 | 482.8 | 5.8 | |
| Aroma | PHPN | 11 | C35H44 | 464.8 | 15.7 |
| DOCHN | 13 | C30H46 | 406.8 | 16.2 | |
| Resin | Pyridinohopane | 4 | C36H57N | 530.9 | 6.2 |
| Thio-isorenieratane | 4 | C40H60S | 572.9 | 7.0 | |
| Trimethylbenzene-oxane | 5 | C29H50O | 414.7 | 6.4 | |
| Quinolinohopane | 4 | C40H59N | 554.0 | 6.8 | |
| Benzobisbenzothiophene | 15 | C18H10S2 | 290.4 | 13.4 | |
| Asphaltene | Phenol | 3 | C42H54O | 575 | 5.3 |
| Pyrrole | 2 | C66H81N | 888.5 | 5.5 | |
| Thiophene | 3 | C51H62S | 707.2 | 6.5 |
| Item | Unit | Test Value | |
|---|---|---|---|
| Color | - | dark colored | |
| Density at 15 °C | g/cm3 | 0.874 | |
| Flash point | °C | 212.1 | |
| Chemical composition | Asphaltene | % | 0.73 |
| Gum | % | 9.53 | |
| Aromatic | % | 28.43 | |
| Saturated | % | 61.31 | |
| SBS | Base Asphalt Binder | SBSMA System | |
|---|---|---|---|
| Solubility parameters ((J/cm3)1/2) | 18.088 | 19.024 | 17.872 |
| SPD | - | 0.936 | −0.216 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, N.; Liu, Z.; Yin, J.; Zhang, H.; Dou, H.; Li, B. Investigation of Compatibility Mechanisms and Diffusion Behavior of Polymer SBS-Modified Asphalt Compatibilizer Using Molecular Dynamics Simulation. Materials 2025, 18, 2238. https://doi.org/10.3390/ma18102238
Li N, Liu Z, Yin J, Zhang H, Dou H, Li B. Investigation of Compatibility Mechanisms and Diffusion Behavior of Polymer SBS-Modified Asphalt Compatibilizer Using Molecular Dynamics Simulation. Materials. 2025; 18(10):2238. https://doi.org/10.3390/ma18102238
Chicago/Turabian StyleLi, Ning, Zhenzheng Liu, Jiaqi Yin, Hai Zhang, Hui Dou, and Bo Li. 2025. "Investigation of Compatibility Mechanisms and Diffusion Behavior of Polymer SBS-Modified Asphalt Compatibilizer Using Molecular Dynamics Simulation" Materials 18, no. 10: 2238. https://doi.org/10.3390/ma18102238
APA StyleLi, N., Liu, Z., Yin, J., Zhang, H., Dou, H., & Li, B. (2025). Investigation of Compatibility Mechanisms and Diffusion Behavior of Polymer SBS-Modified Asphalt Compatibilizer Using Molecular Dynamics Simulation. Materials, 18(10), 2238. https://doi.org/10.3390/ma18102238