
Composite Cryogels Based on Hydroxyapatite and Polyvinyl Alcohol and the Study of Physicochemical and Mechanical Properties


Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
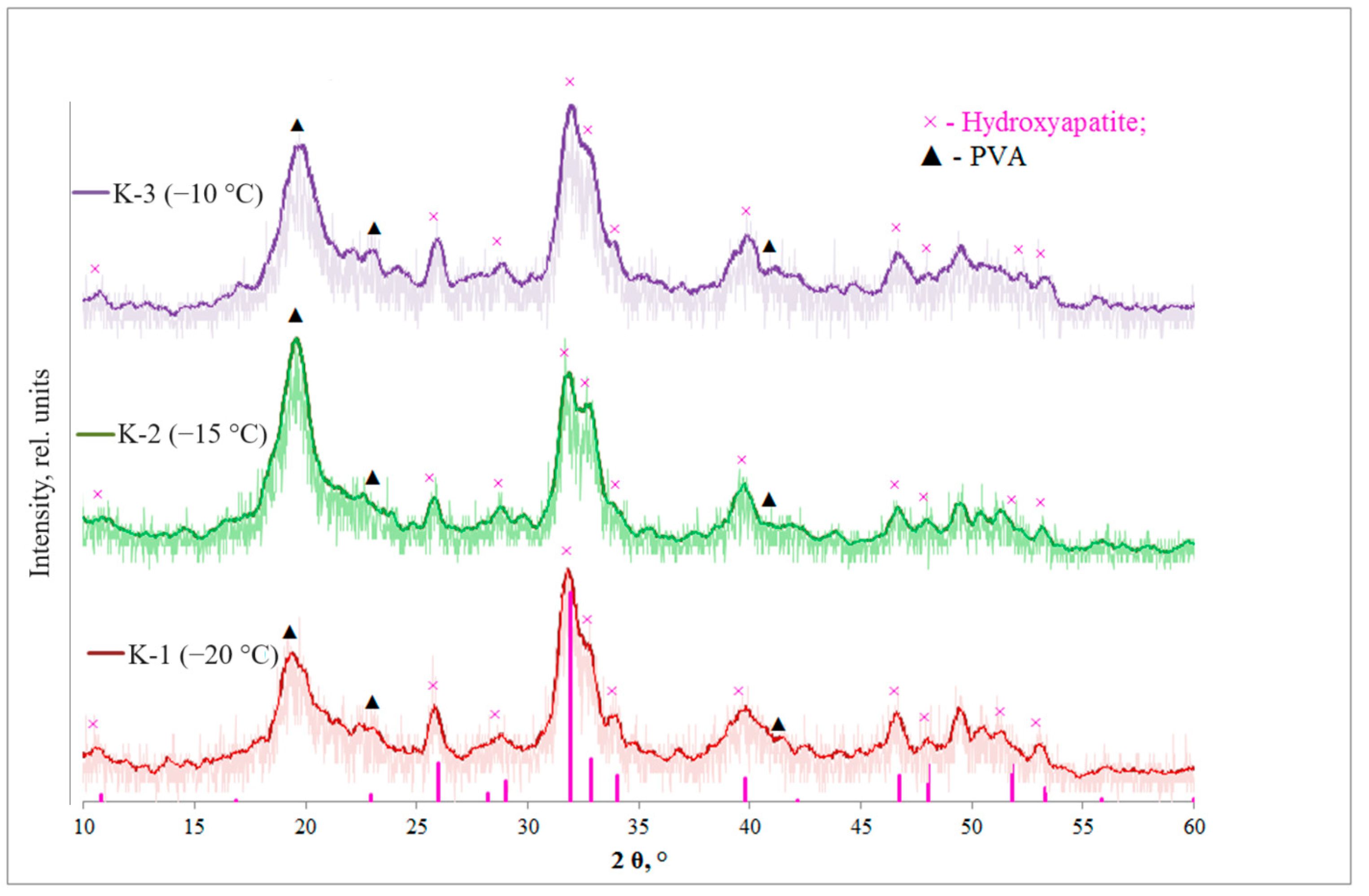
3.1. Determination of the Phase Composition
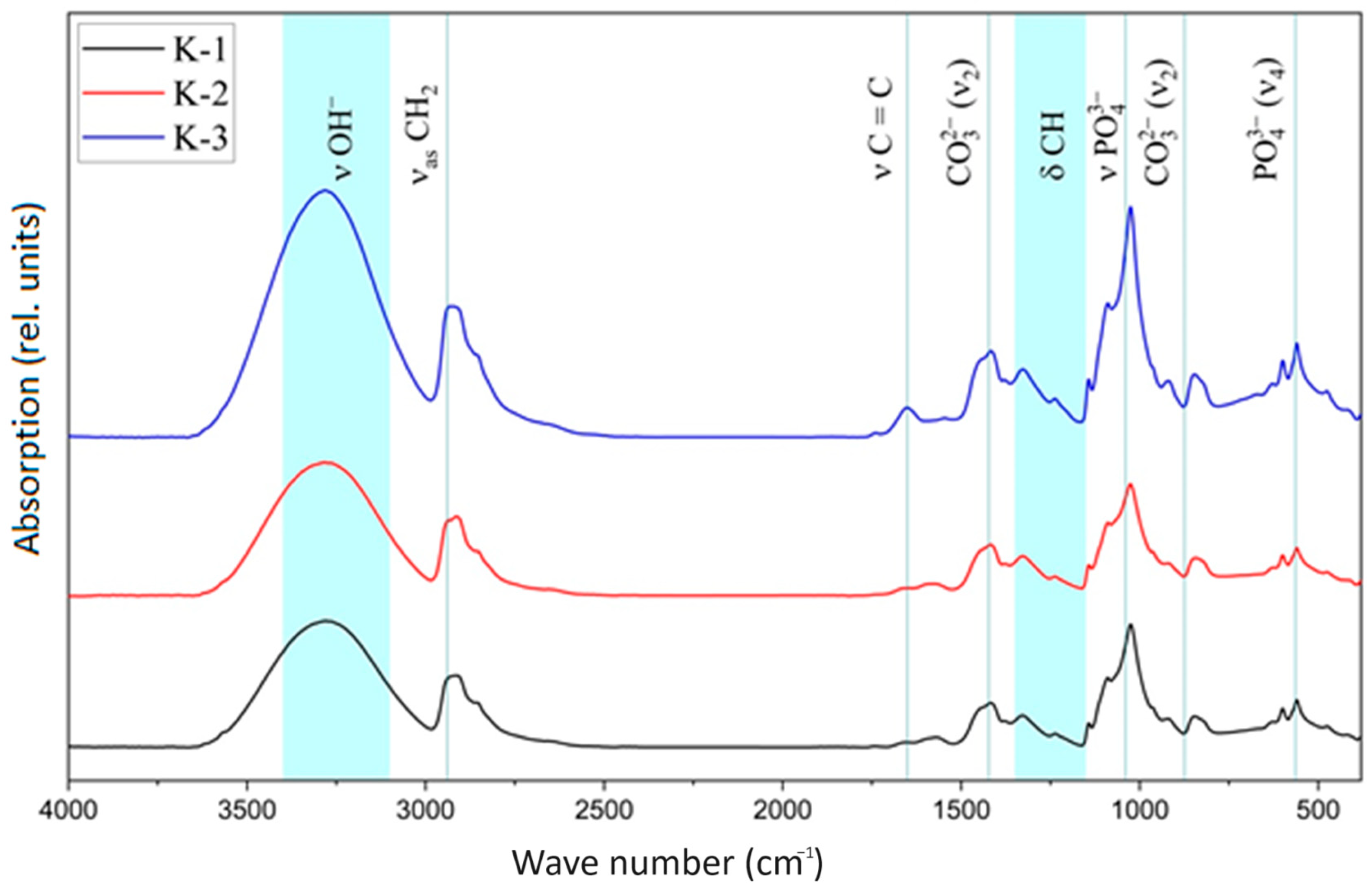
3.2. IR Spectroscopy
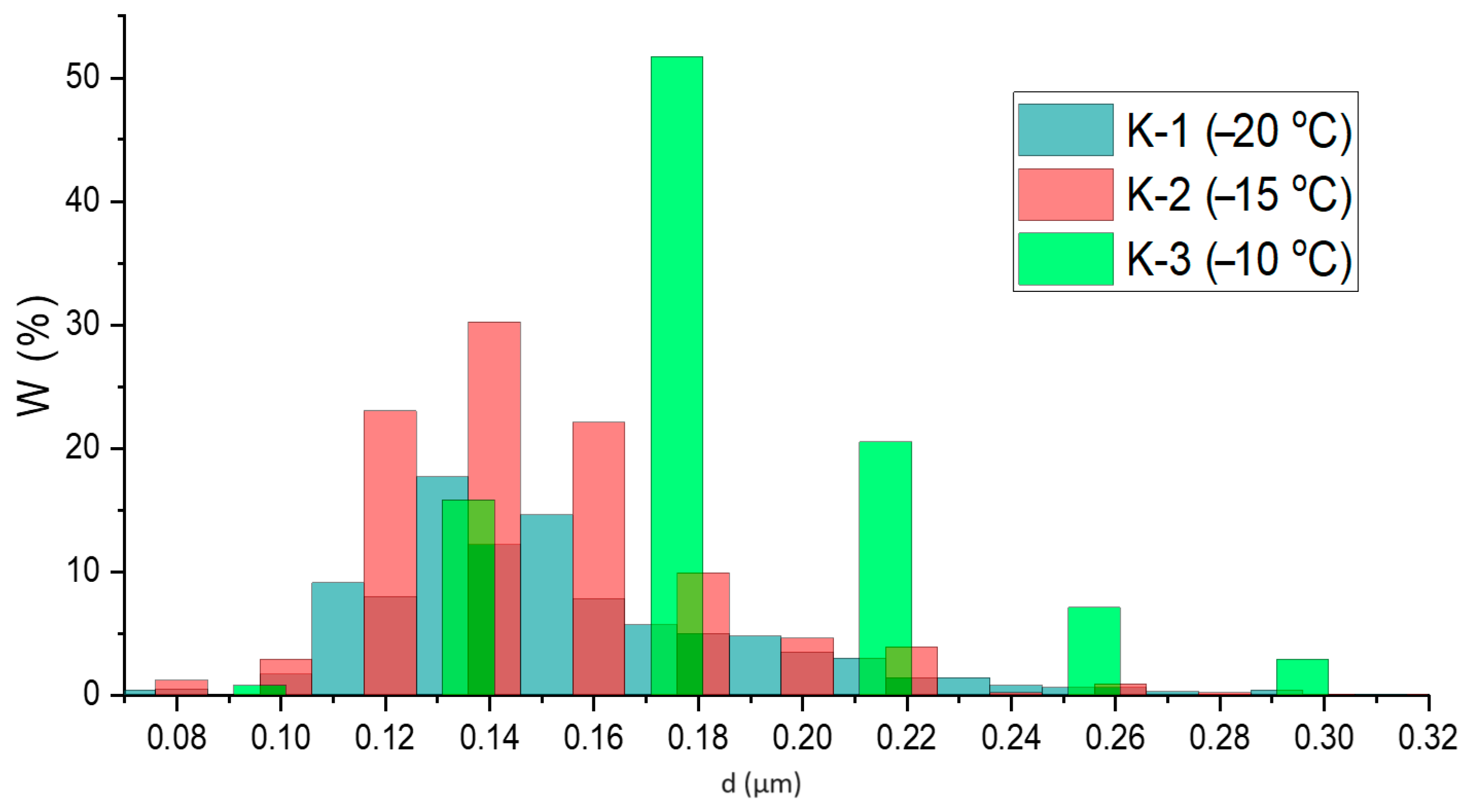
3.3. Morphology and Elemental Composition (EDX) of Materials Surface
3.4. Contact Angle and Surface Energy
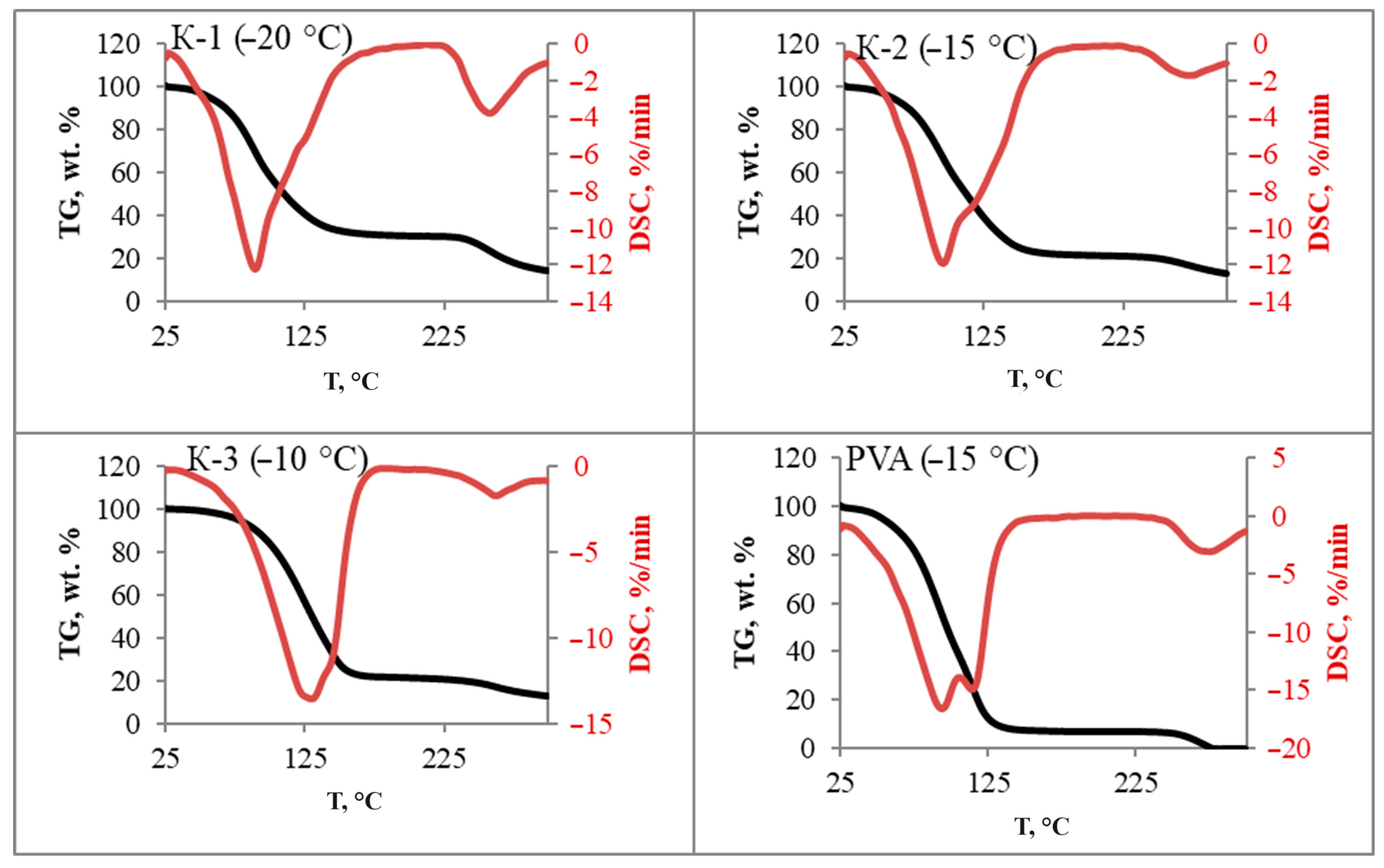
3.5. Thermal Analysis
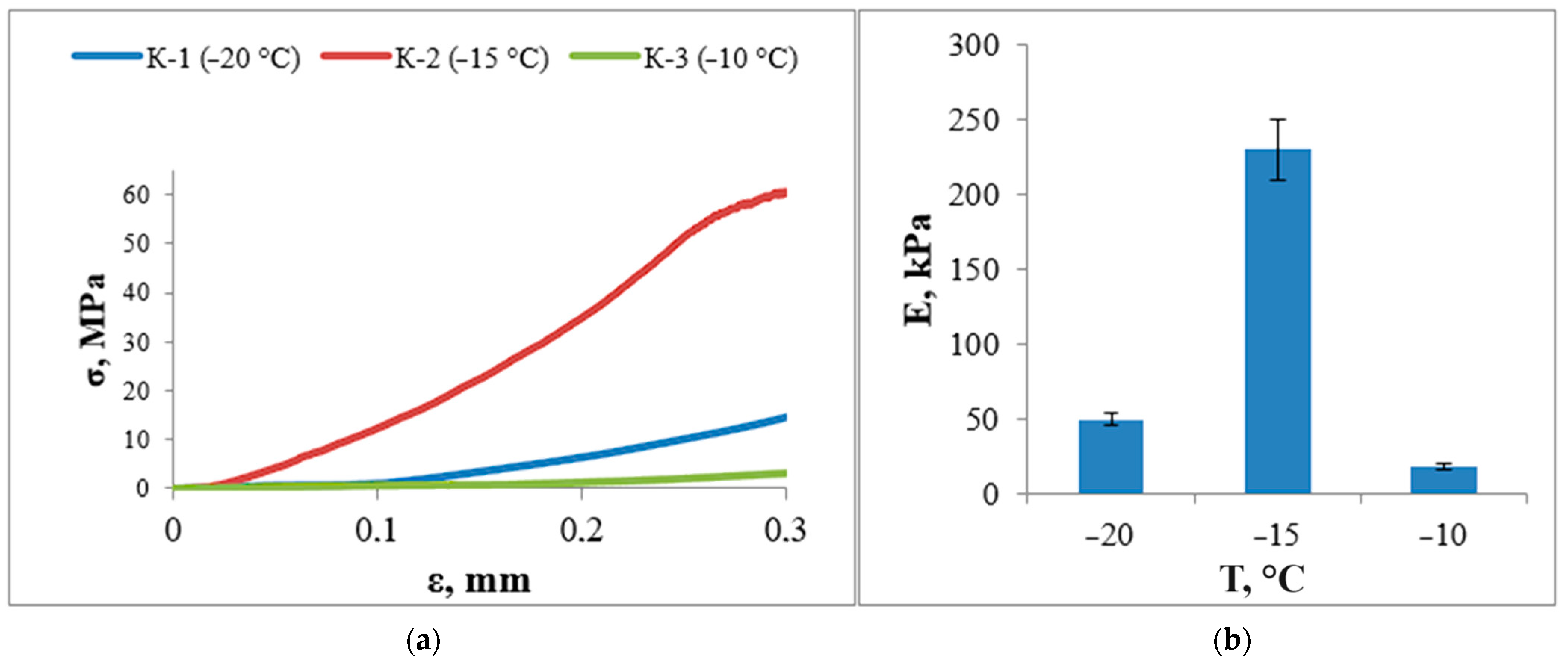
3.6. Study of Mechanical Properties
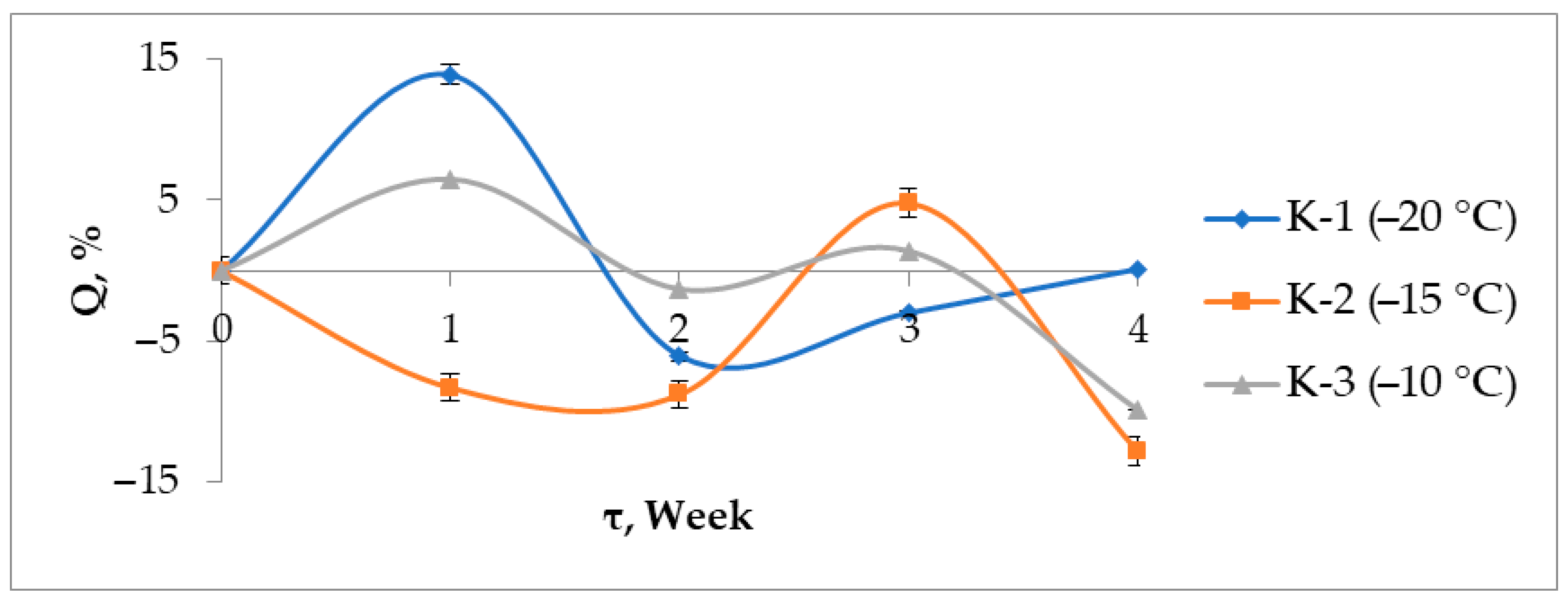
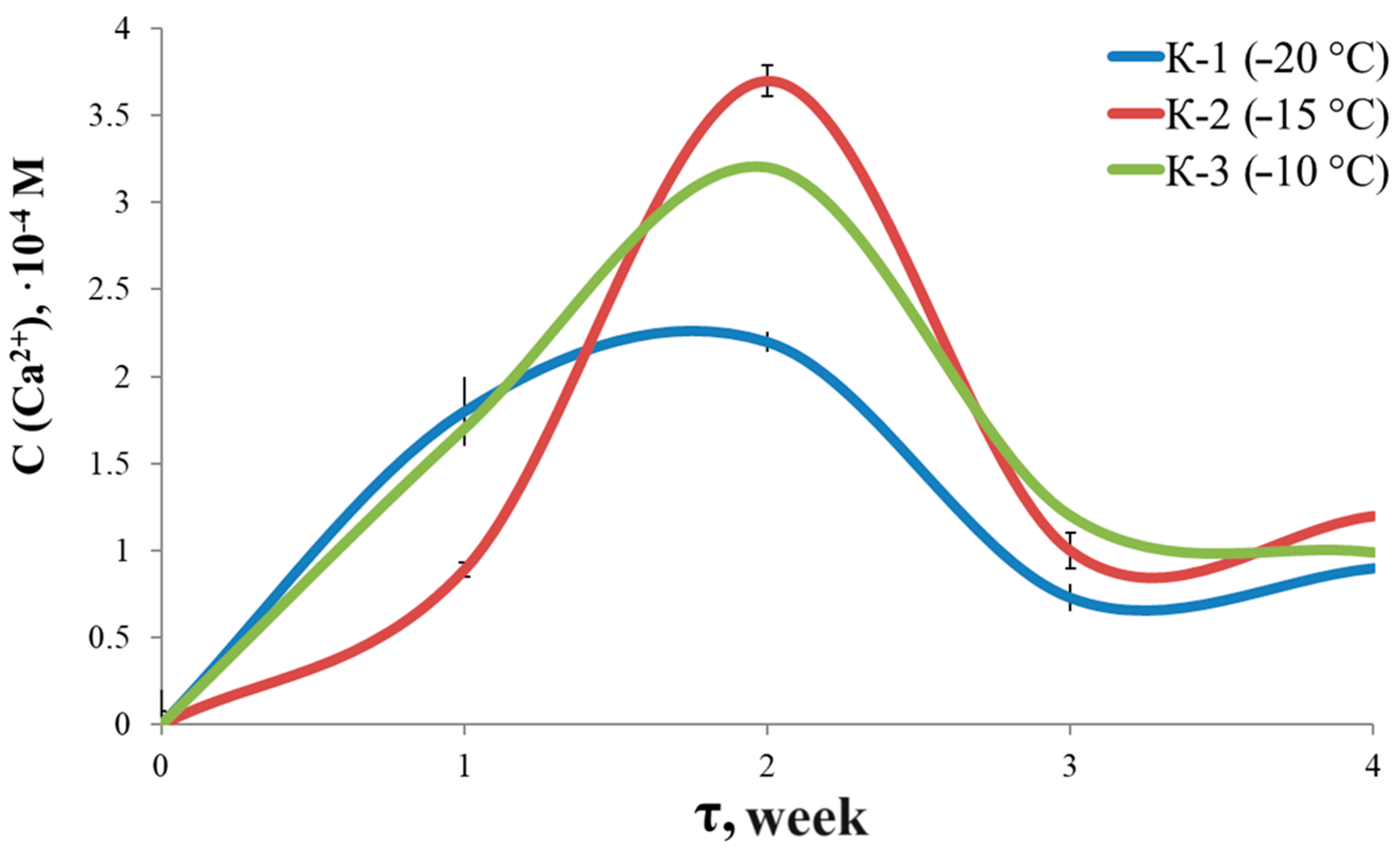
3.7. Solubility and Swelling
3.8. Cell Viability
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization: Official. Website. Updated During the Day. Available online: https://www.who.int/ru/news-room/fact-sheets/detail/musculoskeletal-conditions (accessed on 2 November 2023).
- Health Care in Russia. 2021: Stat./Rosstat. Available online: https://rosstat.gov.ru/storage/mediabank/Zdravoohran-2021.pdf (accessed on 5 April 2023).
- Bykov, L.V. Cytology and General Histology: Textbook for Medical Students. Available online: https://jennyk.ucoz.ru/Bykov_V_L_Tsitologia_i_obschaya_gistologia.pdf (accessed on 5 April 2023).
- Korel, A.V. Tissue engineering strategies for the restoration of bone defects. Current state. Int. J. Appl. Fundam. Res. 2019, 4, 228–234. [Google Scholar]
- Shumilova, A.A.; Shishatskaya, E.I. Materials for bone tissue restoration. J. Sib. Fed. Univ. Biol. 2014, 2, 209–221. [Google Scholar]
- Okuda, T. The slow resorption with replacement by bone of a hydrothermally syn-thesized pure calcium-deficient hydroxyapatite. Biomaterials 2008, 29, 2719–2728. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, S. Resorption of hydroxyapatite and fluorapatite coatings in man: An experimental study in trabecular bone. J. Bone Joint Surg Br. 1997, 79, 654–659. [Google Scholar] [CrossRef]
- Skrtic, D.; Antonucci, J.M.; Eanes, E.D. Amorphous calcium phosphate-based bioactive polymeric composites for mineralized tissue regeneration. J. Res. Natl. 2003, 108, 167. [Google Scholar] [CrossRef]
- Khashaba, R.M. Polymeric-calcium phosphate cement composites-material prop-erties: In vitro and in vivo investigations. Int. J. Biomater. 2010, 2010, 691452. [Google Scholar] [CrossRef]
- Karalkin, P.A. Biocompatibility and osteoplastic properties of mineral polymer composite materials based on sodium alginate, gelatin, and calcium phosphates in-tended for 3D-printing of the constructions for bone replacement. Genes Cells 2016, 11, 94–101. [Google Scholar]
- Fumio, U. Swelling and mechanical properties of poly (vinyl alcohol) hydrogels. Int. J. Pharm. 1990, 58, 135–142. [Google Scholar] [CrossRef]
- Suzuki, A.; Sasaki, S. Swelling and mechanical properties of physically crosslinked poly (vinyl alcohol) hydrogels. Proc. Inst. Mech. Eng. 2015, 229, 828–844. [Google Scholar] [CrossRef]
- Oliveira, R.N. Mechanical properties and in vitro characterization of polyvinyl alcohol-nano-silver hydrogel wound dressings. Interface Focus 2014, 4, 20130049. [Google Scholar] [CrossRef]
- Inoue, K.; Fujisato, T.; Gu, Y.J.; Burczak, K.; Sumi, S.; Kogire, M.; Ikada, Y. Experimental hybrid islet transplantation: Application of polyvinyl alcohol membrane for entrapment of islets. Pancreas 1992, 7, 562–568. [Google Scholar] [CrossRef]
- Baker, M.I.; Walsh, S.P.; Schwartz, Z.; Boyan, B.D. A review of polyvinyl alcohol and its uses in cartilage and orthopedic applications. J. Biomed. Mater. Res. Part B Appl. Biomater. 2012, 100, 1451–1457. [Google Scholar] [CrossRef]
- Nair, B. Final report on the safety assessment of polyvinyl alcohol. Int. J. Toxicol. 1998, 17, 67–92. [Google Scholar] [CrossRef]
- Wan, W. Poly (vinyl alcohol) cryogels for biomedical applications. In Polymeric Cryogels: Macroporous Gels with Remarkable Properties; Springer: Berlin/Heidelberg, Germany, 2014; pp. 283–321. [Google Scholar]
- Plieva, F.M.; Karlsson, M.; Aguilar, M.R.; Gomez, D.; Mikhalovsky, S.; Galaev, I.Y.; Mattiasson, B. Pore structure of macroporous monolithic cryogels prepared from poly (vinyl alcohol). J. Appl. Polym. Sci. 2006, 100, 1057–1066. [Google Scholar] [CrossRef]
- Lytkina, D.N.; Fedorishin, D.A.; Kalachikova, P.M.; Plyaskina, A.A.; Babeshin, A.R.; Kurzina, I.A. Cryo-structured materials based on polyvinyl alcohol and hydroxyapatite for osteogenesis. J. Funct. Biomater. 2021, 12, 18. [Google Scholar] [CrossRef]
- Mollazadeh, S. In situ synthesis and characterization of nano-size hydroxyapatite in poly(vinyl alcohol) matrix. Ceram. Int. 2007, 33, 1579–1583. [Google Scholar] [CrossRef]
- Ali Poursamar, S. Controllable synthesis and characterization of porous polyvinyl alcohol/hydroxyapatite nanocomposite scaffolds via an in situ colloidal technique. Colloids Surf. B Biointerfaces 2011, 84, 310–316. [Google Scholar] [CrossRef]
- Sheikh, F.A. Synthesis of poly(vinyl alcohol) (PVA) nanofibers incorporating hydroxyapatite nanoparticles as future implant materials. Macromol. Res. 2010, 18, 59–66. [Google Scholar] [CrossRef]
- Zeng, S. Preparation and characterization of nano-hydroxyapatite/poly(vinyl alcohol) composite membranes for guided bone regeneration. Biomed. Nanotechnol. 2011, 7, 549. [Google Scholar] [CrossRef]
- Maximum Allowable Concentrations (MPC) of Chemicals in the Water of Water Bodies for Drinking and Domestic Water Use: GN 2.1.5.1315-03. Available online: https://docs.cntd.ru/document/901862249 (accessed on 11 January 2023).
- Zhuravlev, V.F. Toxicity of nitrates and nitrites. Hyg. Sanit. 1983, 1, 62–66. [Google Scholar]
- Lozinsky, V.I. Cryotropic gelation of poly (vinyl alcohol) solutions. Russ. Chem. Rev. 1998, 67, 573–586. [Google Scholar] [CrossRef]
- Lozinsky, V.I.; Okay, O. Basic principles of cryotropic gelation. Polym. Cryogels Macroporous Gels Remarkable Prop. 2014, 263, 49–101. [Google Scholar] [CrossRef]
- Using the method of undisturbed total internal reflection in the analysis of streptocide and its dosage forms. Bull. VSU Ser. Chem. Biol. Pharm. 2008, 2, 150–152.
- Osiris, W.; Guirguis, M.; Moselhey, T.H. Thermal and structural studies of poly(vinyl alcohol) and hydroxypropyl cellulose blends. Nat. Sci. 2012, 4, 57–67. [Google Scholar]
- Gratchev, A.; Kzhyshkowska, J.; Köthe, K.; Muller-Molinet, I.; Kannookadan, S.; Utikal, J.; Goerdt, S. Mϕ1 and Mϕ2 Can be Re-polarized by Th2 or Th1 Cytokines Respectively, and Respond to Exogenous Danger Signals. Immunobiology 2006, 211, 473–486. [Google Scholar] [CrossRef]
- Shubnikov, A.V. How Crystals Grow; Publishing House of the Academy of Sciences of the USSR: Moscow, Russia, 1935; p. 175. [Google Scholar]
- De Rosa, C.; Auriemma, F.; Di Girolamo, R. Kinetic analysis of cryotropic gelation of poly (vinyl alcohol)/water solutions by small-angle neutron scattering. In Polymeric Cryogels: Macroporous Gels with Remarkable Properties; Springer: Berlin/Heidelberg, Germany, 2014; pp. 159–197. [Google Scholar]
- Lozinsky, V.I.; Damshkaln, L.G.; Kurochkin, I.N.; Kurochkin, I.I. Study of cryostructuring of polymer systems. Effect of rate of chilling aqueous poly (vinyl alcohol) solutions during their freezing on physicochemical properties and porous structure of resulting cryogels. Colloid J. 2012, 74, 319–327. [Google Scholar] [CrossRef]
- Hossain, M.S.; Ahmed, S. FTIR spectrum analysis to predict the crystalline and amorphous phases of hydroxyapatite: A comparison of vibrational motion to reflection. RSC Adv. 2023, 13, 14625–14630. [Google Scholar] [CrossRef]
- Bakan, F.; Lacin, O.; Sarac, H. A novel low temperature sol–gel synthesis process for thermally stable nano crystalline hydroxyapatite. Powder Technol. 2013, 233, 295–302. [Google Scholar] [CrossRef]
- Chaikina, M.V.; Bulina, N.V.; Ishchenko, A.V.; Prosanov, I.Y. Study of the process of substitution of phosphate for aluminate in the structure of hydroxyapatite during mechanochemical synthesis and annealing. Chem. Interests Sustain. Dev. 2016, 24, 669–678. [Google Scholar]
- Hui, P.; Meena, S.L.; Singh, G.; Agarawal, R.D.; Prakash, S. Synthesis of hydroxyapatite bio-ceramic powder by hydrothermal method. J. Miner. Mater. Charact. Eng. 2010, 9, 683–692. [Google Scholar] [CrossRef]
- Berzina-Cimdina, L.; Borodajenko, N. Research of calcium phosphates using Fourier transform infrared spectroscopy. Infrared Spectrosc. Mater. Sci. Eng. Technol. 2012, 12, 251–263. [Google Scholar]
- Redkol, O.A.; Laput, I.V.; Vasenina, K.P.; Savkin, D.A.; Kurzina, I.A. Modification of the Surface Physical and Chemical Properties of Polyvinyl Alcohol. Influence of Silver Ion Implantation, Electronic and Plasma Treatment; Ulan-Ude Publishing House of the Buryat Scientific Center SB RAS: Ulan-Ude, Russia, 2018; pp. 178–184. [Google Scholar]
- Orilisi, G.; Monterubbianesi, R.; Tosco, V.; Conti, C.; Procaccini, M.; Putignano, A.; Orsini, G. Effect of a sodium fluoride-releasing rubber cup on hydroxyapatite crystallinity of human enamel: FTIR spectroscopy analysis. Dent. Res. Oral Health 2020, 3, 24–34. [Google Scholar] [CrossRef]
- Mansur, H.S.; Sadahira, C.M.; Souza, A.N.; Mansur, A.A. FTIR spectroscopy characterization of poly (vinyl alcohol) hydrogel with different hydrolysis degree and chemically crosslinked with glutaraldehyde. Mater. Sci. Eng. C 2008, 28, 539–548. [Google Scholar] [CrossRef]
- Linnikov, O.D. The mechanism of sediment formation during spontaneous crystallization of salts from supersaturated aqueous solutions. Uspekhi Khimi 2014, 83, 343–364. [Google Scholar] [CrossRef]
- Egorova, E.N. Biochemistry of the Oral Cavity: Proc. Allowance for Students of Dentistry. 2018, 155. Available online: https://main.isuct.ru/files/publ/PUBL_ALL/fkh/fkh18052009.pdf (accessed on 5 April 2023).
- Anselme, K. Osteoblast adhesion on biomaterials. Biomaterials 2000, 21, 667–681. [Google Scholar] [CrossRef]
- Kocijan, A.; Conradi, M.; Hočevar, M. The influence of surface wettability and topography on the bioactivity of TiO2/epoxy coatings on AISI 316L stainless steel. Materials 2019, 12, 1877. [Google Scholar] [CrossRef]
- Han, D.K.; Park, K.D.; Ryu, G.H.; Kim, U.Y.; Min, B.G.; Kim, Y.H. Plasma protein adsorption to sulfonated poly (ethylene oxide)-grafted polyurethane surface. J. Biomed. Mater. Res. 1996, 30, 23–30. [Google Scholar] [CrossRef]
- Desai, N.P.; Hubbell, J.A. Biological responses to polyethylene oxide modified polyethylene terephthalate surfaces. J. Biomed. Mater. Res. 1991, 25, 829–843. [Google Scholar] [CrossRef]
- Hassan, C.M.; Peppas, N.A. Structure and applications of poly (vinyl alcohol) hydrogels produced by conventional crosslinking or by freezing/thawing methods. In Biopolymers PVA Hydrogels, Anionic Polymerisation Nanocomposites; Springer: Berlin/Heidelberg, Germany, 2000; pp. 37–65. [Google Scholar]
- Nugent, M.J.; Hanley, A.; Tomkins, P.T.; Higginbotham, C.L. Investigation of a novel freeze-thaw process for the production of drug delivery hydrogels. J. Mater. Sci. Mater. Med. 2005, 16, 1149–1158. [Google Scholar] [CrossRef]
- Lytkina, D.; Heinrich, L.; Churina, E.; Kurzina, I. Biocompatible composite materials based on porous hydroxyapatite ceramics and copolymer of lactide and glycolide. Materials 2021, 14, 2168. [Google Scholar] [CrossRef]
- Sadykov, R.; Lytkina, D.; Stepanova, K.; Kurzina, I. Synthesis of biocompatible composite material based on cryogels of polyvinyl alcohol and calcium phosphates. Polymers 2022, 14, 3420. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | K-1 | K-2 | K-3 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T, °C | −20 | −7 | +7 | +20 | −15 | −7 | +7 | +20 | −10 | −7 | +7 | +20 |
| τ, min | 240 | 15 | 700 | 35 | 240 | 15 | 700 | 35 | 240 | 15 | 700 | 35 |
| Cooling rate, °C/min | 0.167 | 0.146 | 0.125 | |||||||||
| Characteristics | Samples | |||
|---|---|---|---|---|
| K-1 | K-2 | K-3 | ||
| Xs HA (211), % | 22 | 16 | 9 | |
| Xs PVA (101), % | 18 | 42 | 23 | |
| D (PVA) (101), nm | 6 | 6 | 6 | |
| D (HA) (211), nm | 10 | 10 | 12 | |
| HA crystal lattice parameters | a = b, Å | 9.43 | 9.51 | 9.45 |
| c, Å | 6.89 | 6.98 | 6.90 | |
| V hex, Å3 | 77.01 | 78.32 | 77.34 | |
| Vibrations | Wavenumber (WN), cm−1 | K-1 WN, cm−1 | K-2 WN, cm−1 | K-3 WN, cm−1 |
|---|---|---|---|---|
| ν, stretching vibrations, OH- (strong) | 3400–3100 | 3271 | 3285 | 3281 |
| asymmetric stretching vibrations, CH2 | 2940 | 2908 | 2914 | 2920 |
| symmetrical stretching vibrations, CH2 | 2900 | 2854 | 2854 | 2856 |
| stretching vibrations, C=C | 1651 | 1655 | 1647 | 1653 |
| Stretching vibrations, -C-O-H | 1560 | 1572 | 1582 | 1545 |
| scissor oscillations, CH2 | 1432 | 1416 | 1418 | 1416 |
| stretching vibrations, C-O-C | 1150–1060 | 1142 | 1142 | 1142 |
| torsional vibrations, δCH | 1150−1350 | 1329 | 1327 | 1327 |
| stretching vibrations C-C bonds between the carbon of CH2 groups and carbon atoms related to unsaturated bonds | 914 | 920 | 920 | 920 |
| ν1 symmetrical stretching vibrations, PO43− (weak) | 962 | 964 | 964 | - |
| ν3 asymmetric stretching vibrations, PO43− | 1094 | 1090 | 1088 | 1090 |
| ν3 asymmetric stretching vibrations, PO43− | 1039 | 1024 | 1026 | 1026 |
| ν4 stretching vibrations, PO43− (strong) | 564, 604 | 557, 598 | 559, 600 | 559 |
| librational vibrations, OH- (middle) | 633 | 627 | 627 | 629 |
| stretching vibrations, OH- (weak) | 3546 | 3271 | 3285 | 3281 |
| ν2 deformation vibrations, OH- (weak) | 1635 | 1655 | 1647 | 1653 |
| ν2 deformation vibrations, CO32− | 875 | 847 | 843 | 847 |
| ν3 stretching vibrations CO32− | 1422 | 1416 | 1418 | 1416 |
| Sample | D, µm | σ2 (D, μm) |
|---|---|---|
| K-1 (−20 °C) | 0.15 | 0.04 |
| K-2 (−15 °C) | 0.14 | 0.04 |
| K-3 (−10 °C) | 0.17 | 0.07 |
| Sample | Ca, At % | P, At % | Ca/P |
|---|---|---|---|
| K-1 | 5.32 | 2.73 | 1.95 |
| K-2 | 5.60 | 3.00 | 1.87 |
| K-3 | 4.63 | 2.29 | 2.02 |
| Sample | HA | PVA | K-1 | K-2 | K-3 |
|---|---|---|---|---|---|
| θ water, ° | 9.6 | 10.2 | 9 | 10.2 | 9.3 |
| θ glycerol, ° | 25.5 | 15.3 | 18.9 | 23.4 | 10.9 |
| Sample | σD, mJ/m2 | σP, mJ/m2 | σ, mJ/m2 |
|---|---|---|---|
| HA | 9.28 | 66.10 | 75.38 |
| PVA | 14.34 | 58.33 | 72.67 |
| K-1 | 12.52 | 61.13 | 73.65 |
| K-2 | 10.53 | 63.88 | 74.41 |
| K-3 | 15.71 | 56.71 | 72.49 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shalygina, K.; Lytkina, D.; Sadykov, R.; Kurzina, I. Composite Cryogels Based on Hydroxyapatite and Polyvinyl Alcohol and the Study of Physicochemical and Mechanical Properties. Materials 2024, 17, 403. https://doi.org/10.3390/ma17020403
Shalygina K, Lytkina D, Sadykov R, Kurzina I. Composite Cryogels Based on Hydroxyapatite and Polyvinyl Alcohol and the Study of Physicochemical and Mechanical Properties. Materials. 2024; 17(2):403. https://doi.org/10.3390/ma17020403
Chicago/Turabian StyleShalygina, Kseniia, Daria Lytkina, Rustam Sadykov, and Irina Kurzina. 2024. "Composite Cryogels Based on Hydroxyapatite and Polyvinyl Alcohol and the Study of Physicochemical and Mechanical Properties" Materials 17, no. 2: 403. https://doi.org/10.3390/ma17020403
APA StyleShalygina, K., Lytkina, D., Sadykov, R., & Kurzina, I. (2024). Composite Cryogels Based on Hydroxyapatite and Polyvinyl Alcohol and the Study of Physicochemical and Mechanical Properties. Materials, 17(2), 403. https://doi.org/10.3390/ma17020403