
Molecular Dynamics Simulations Guide the Gasification Process of Carbon-Supported Nickel Catalysts in Biomass Supercritical Water


Abstract
1. Introduction
2. The Simulation Method
2.1. Simulation Software and Modules
2.2. Simulation Methodology
3. Model Construction and Optimization
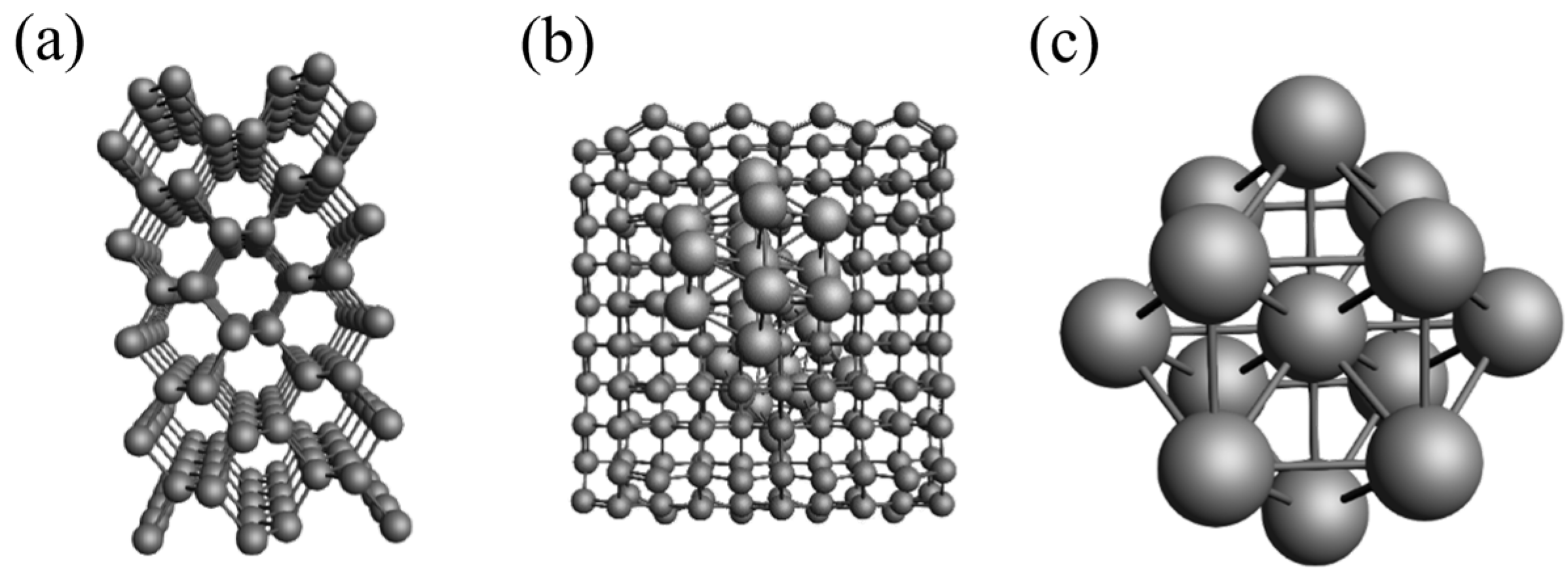
3.1. Carbon Support Model and Nickel Metal Cluster Model
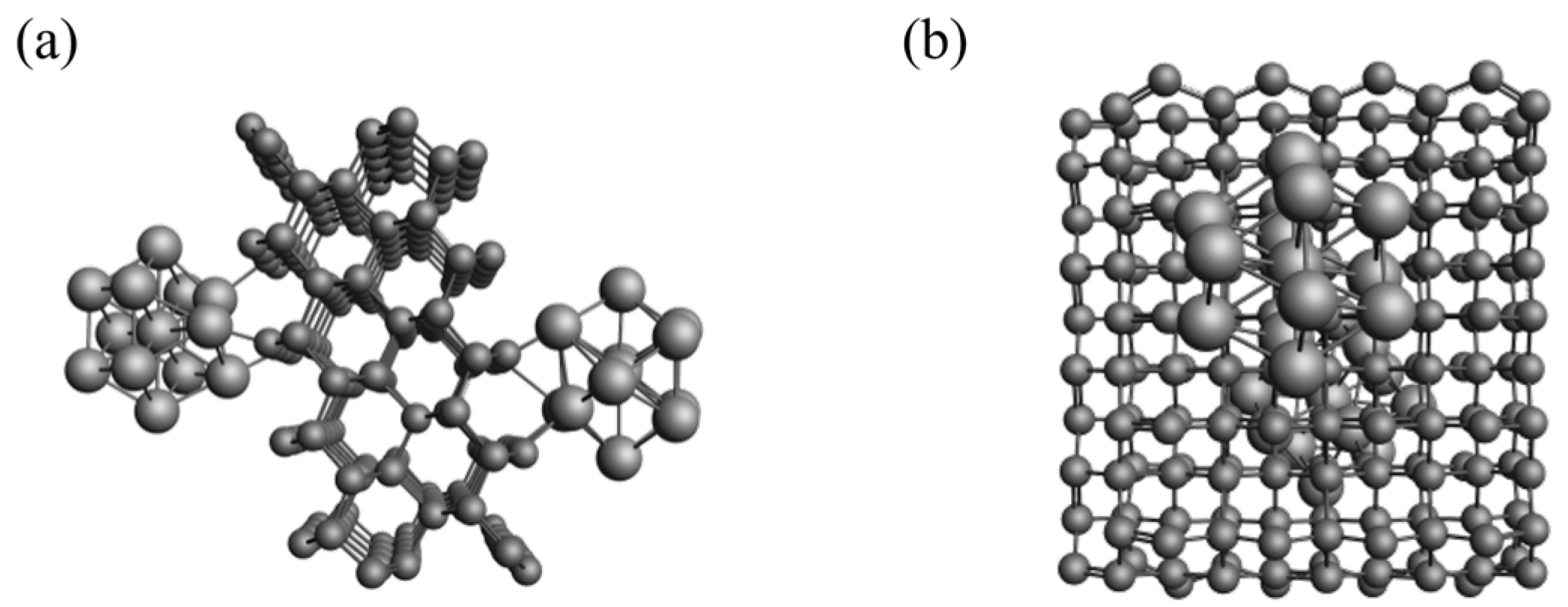
3.2. Model of Carbon-Supported Nickel Metal Catalyst
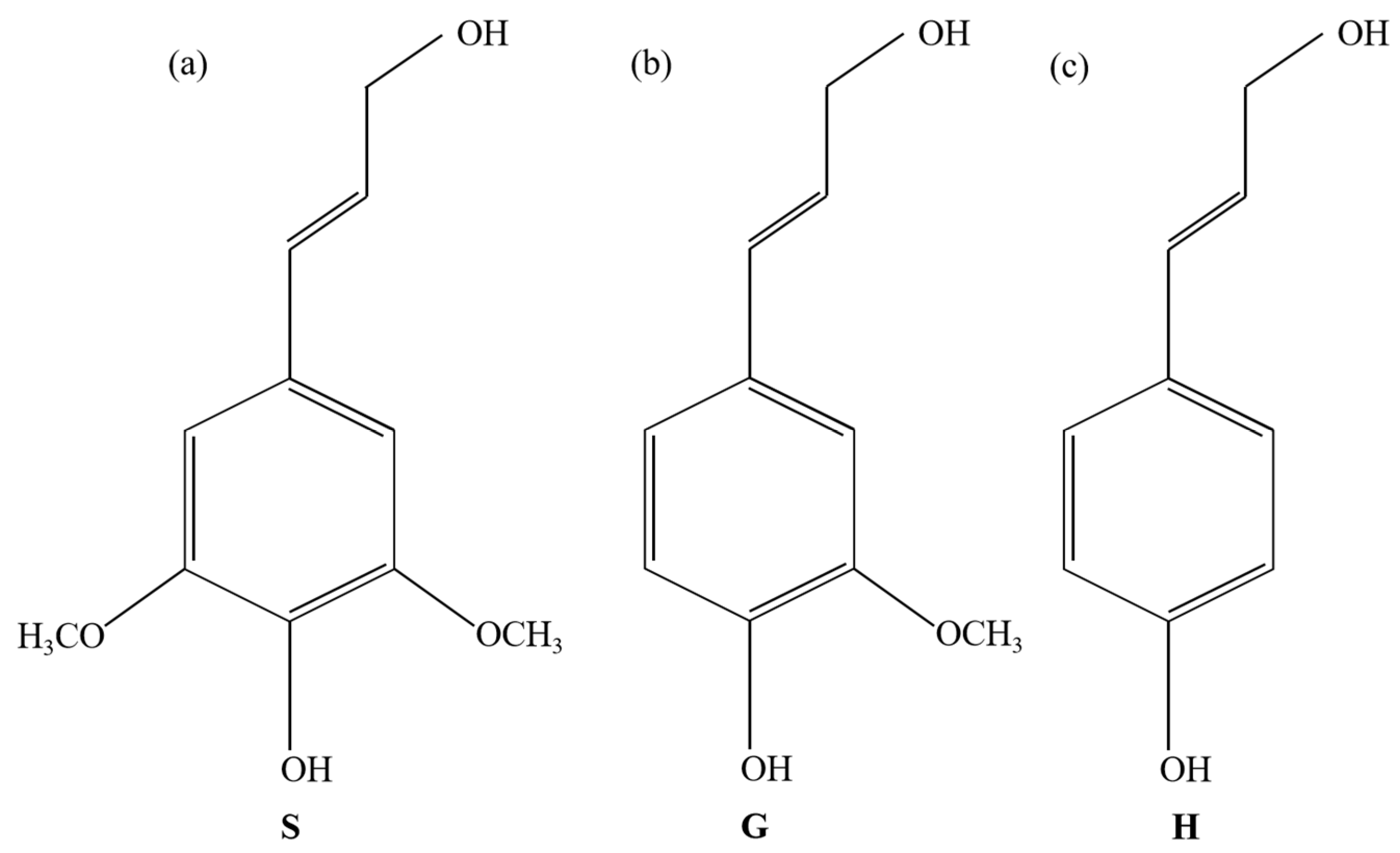
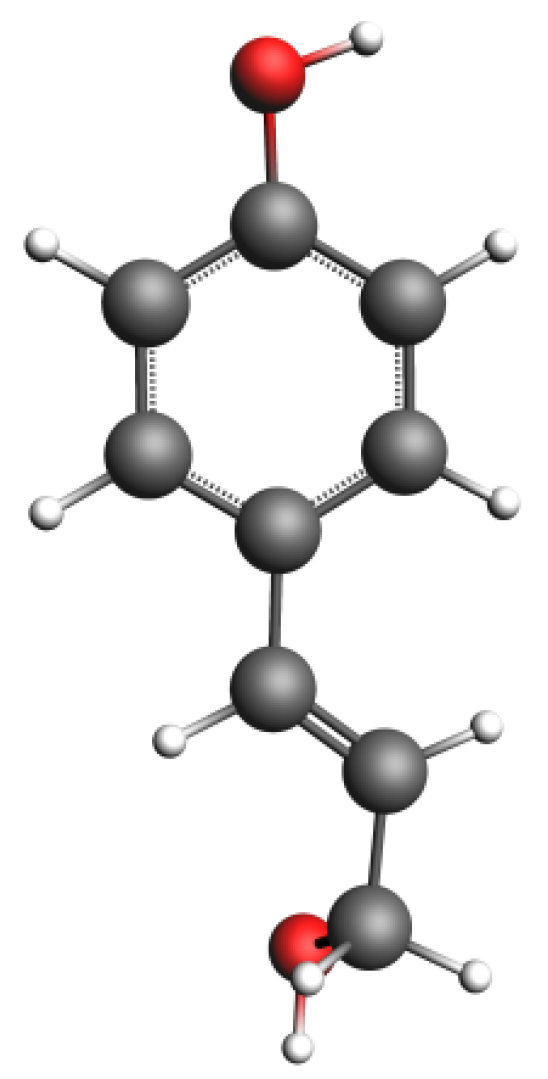
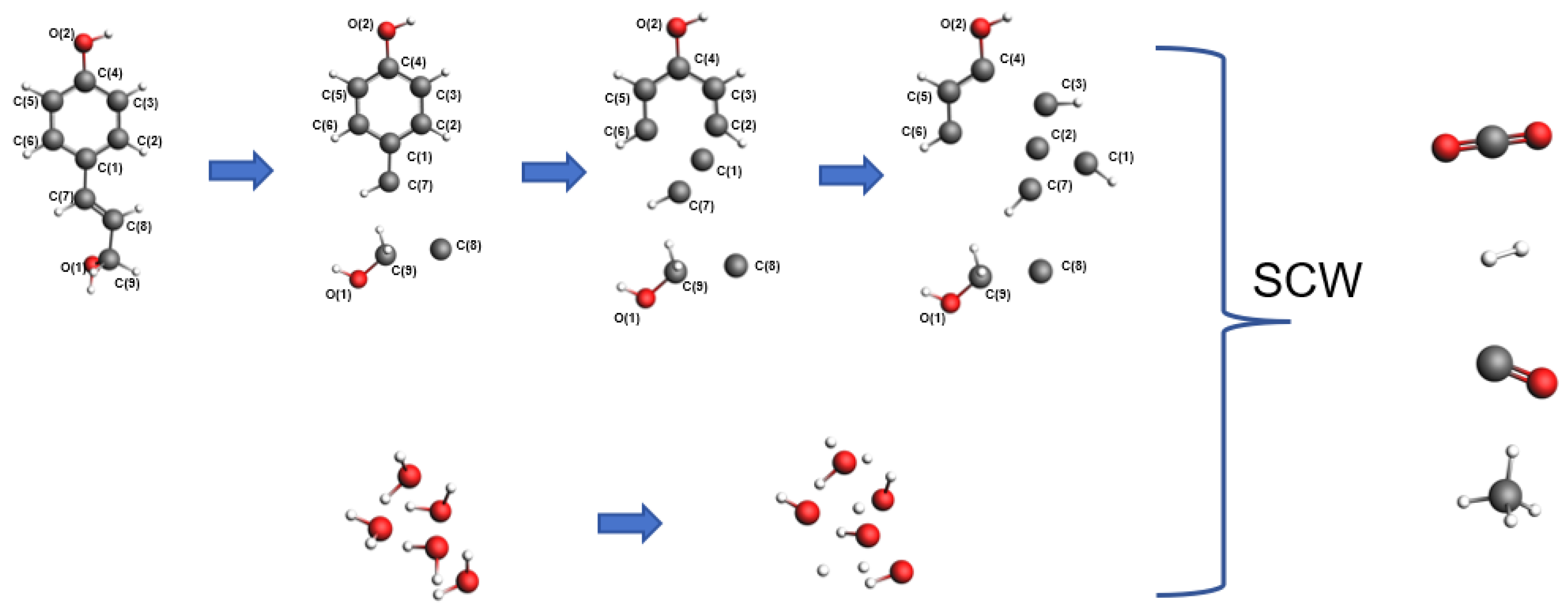
3.3. Biomass Model
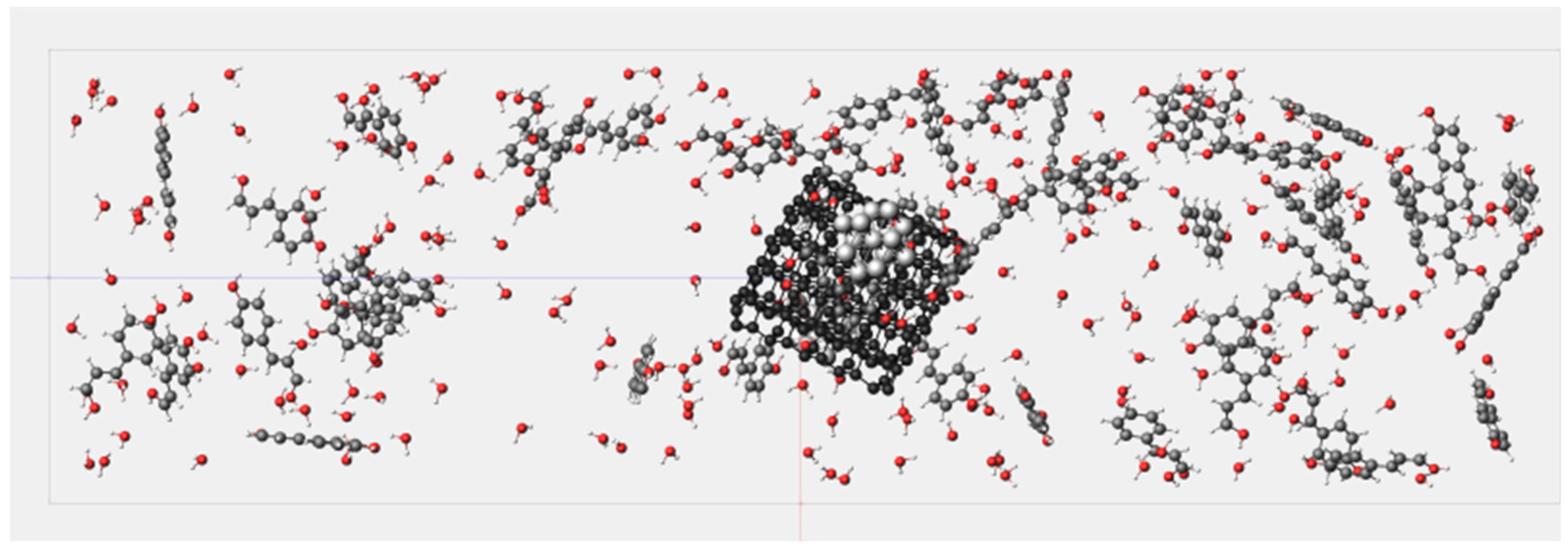
3.4. Molecular Dynamics Simulation System
4. Simulation Results and Discussion
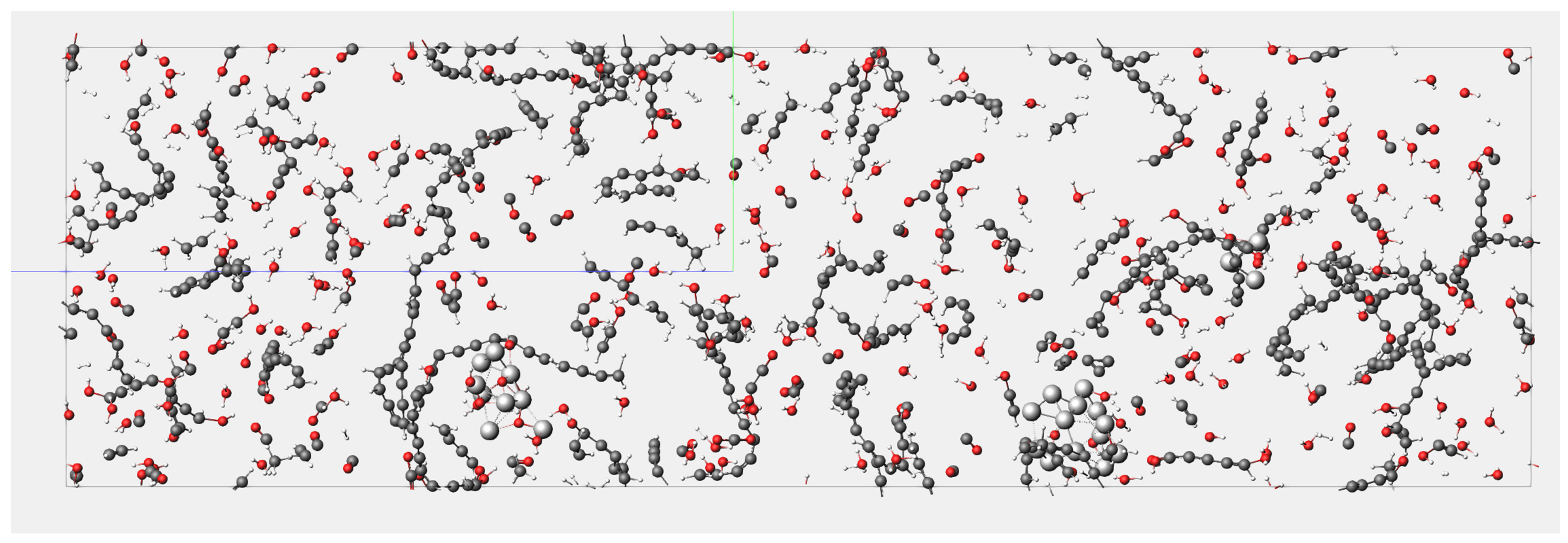
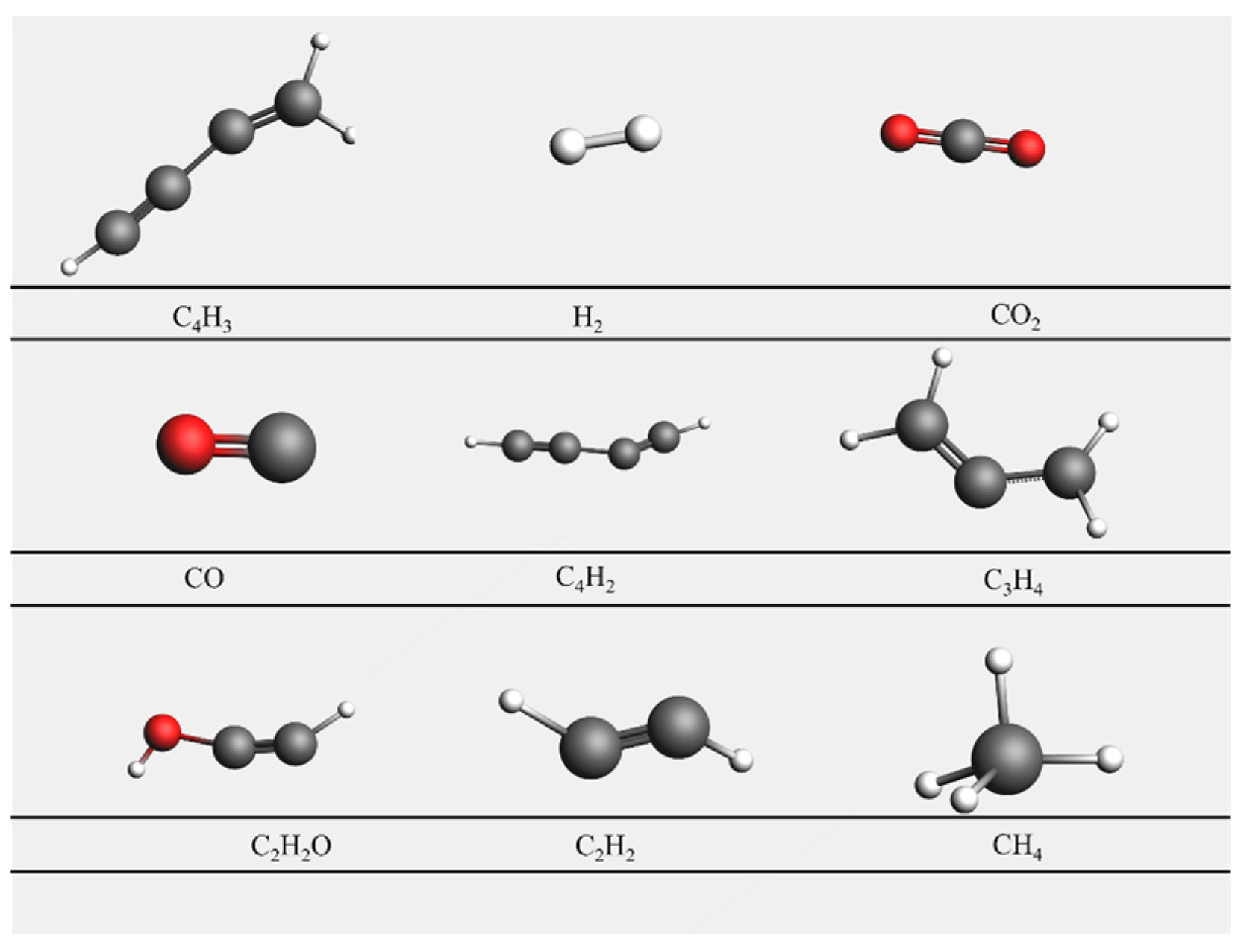
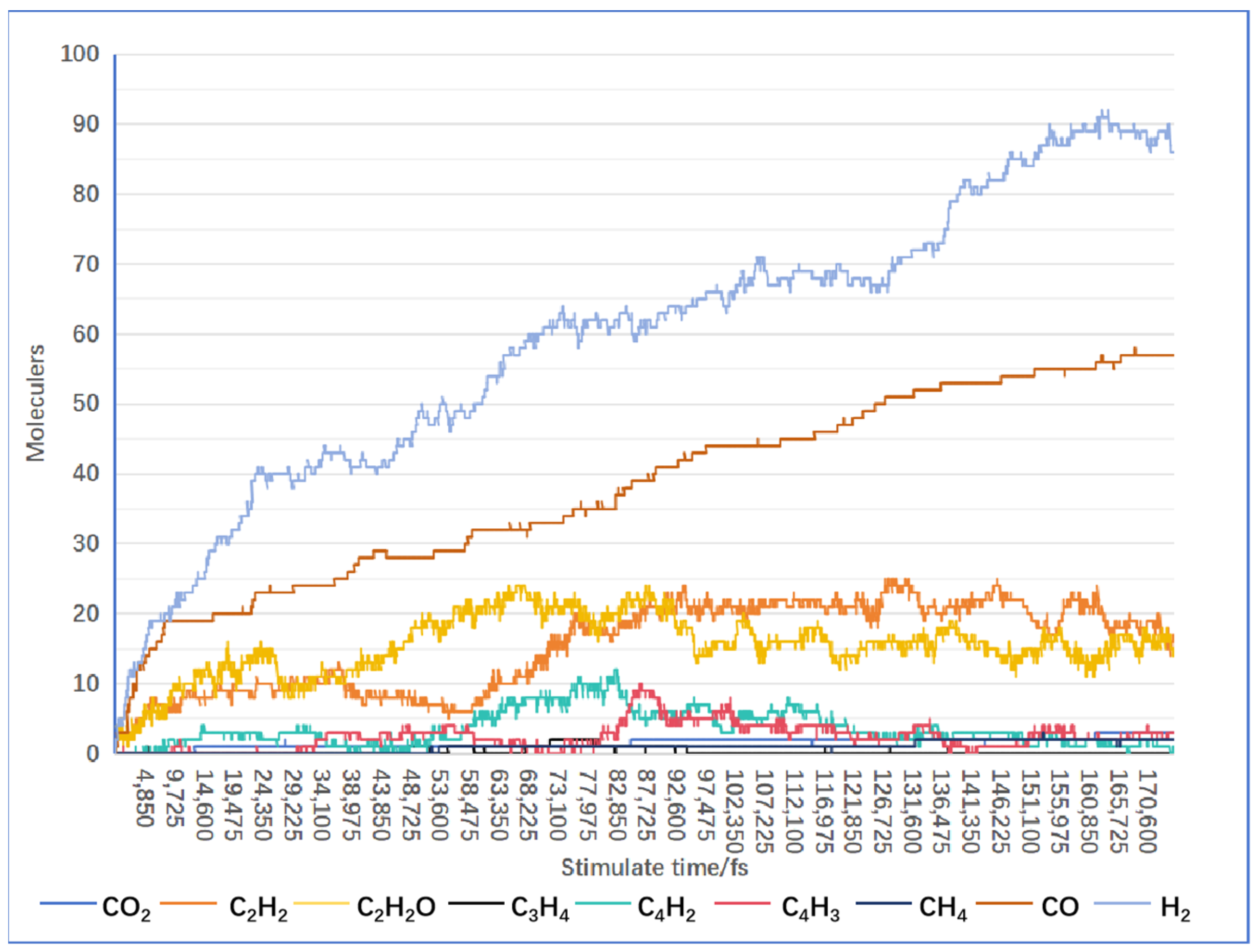
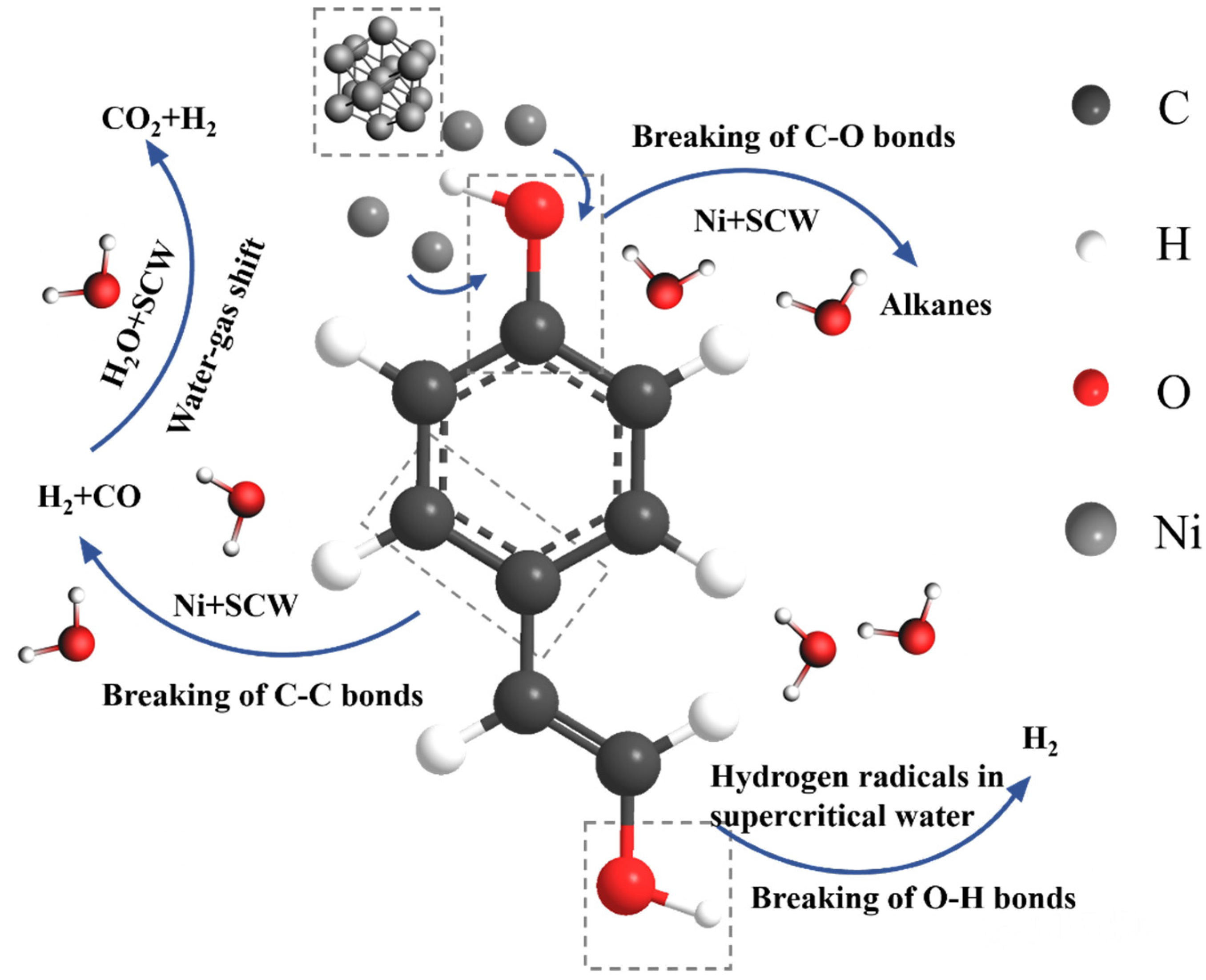
4.1. Results of Lignin Gasification
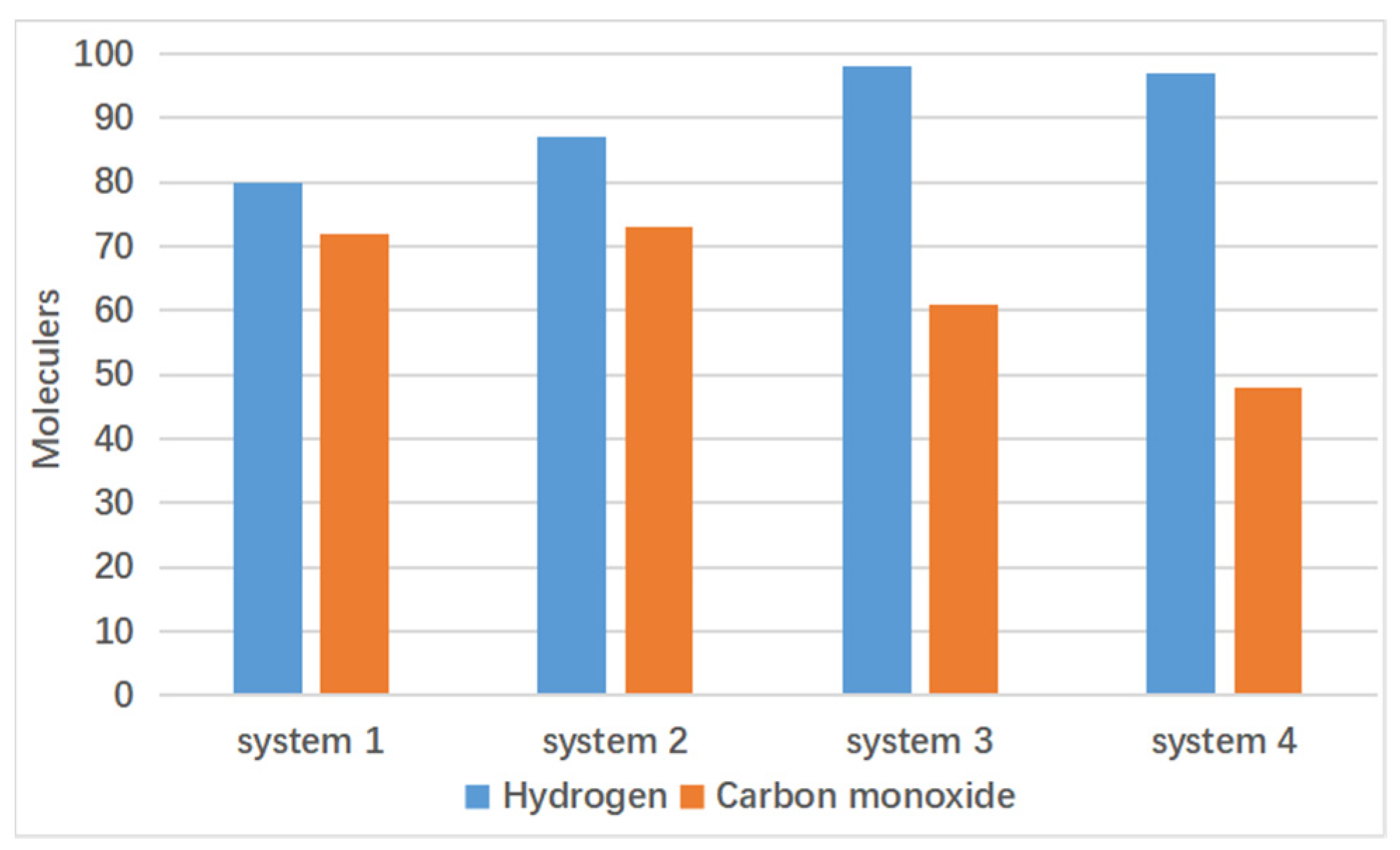
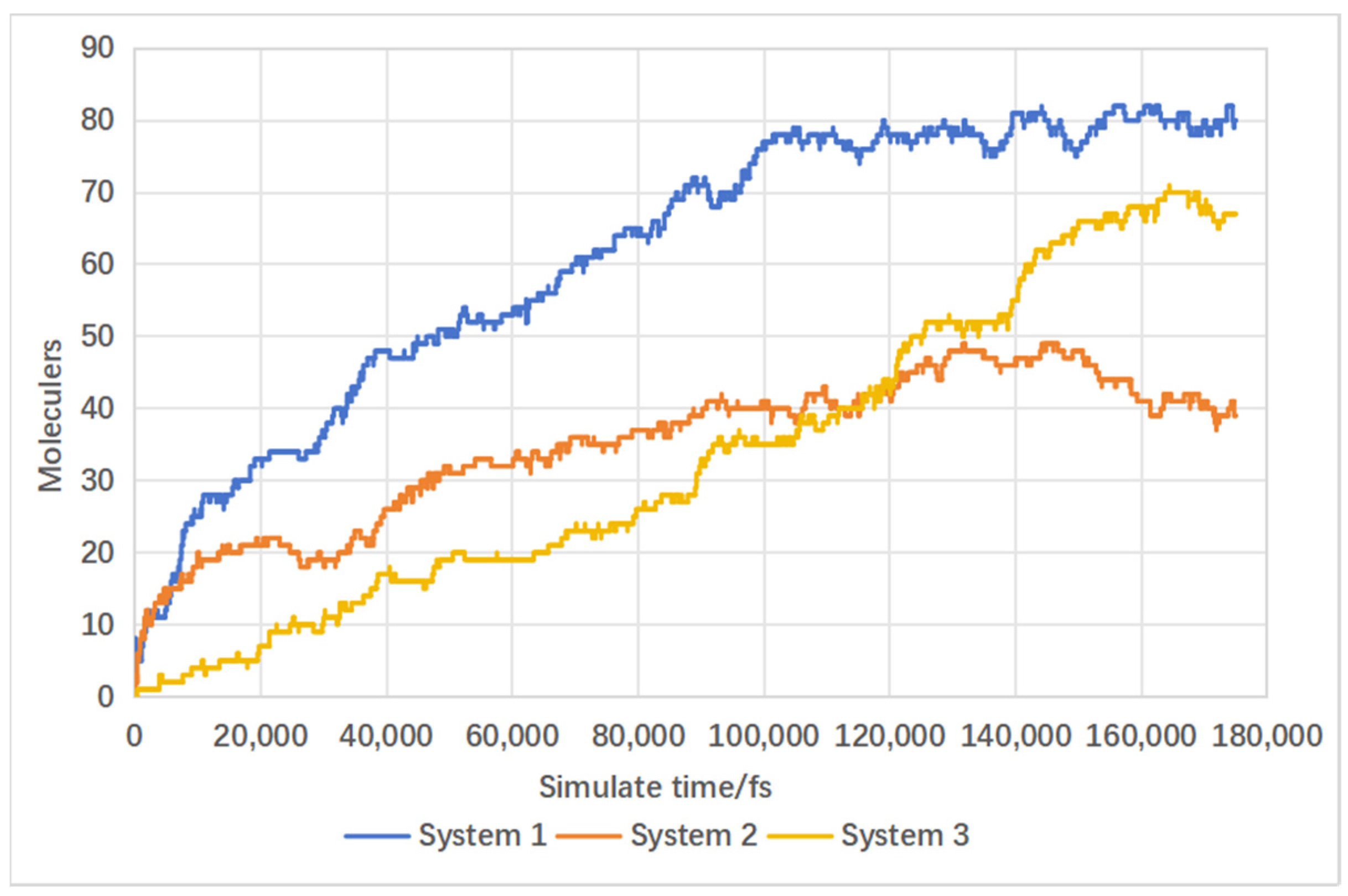
4.2. Effect of Supercritical Water on Gas Product Formation
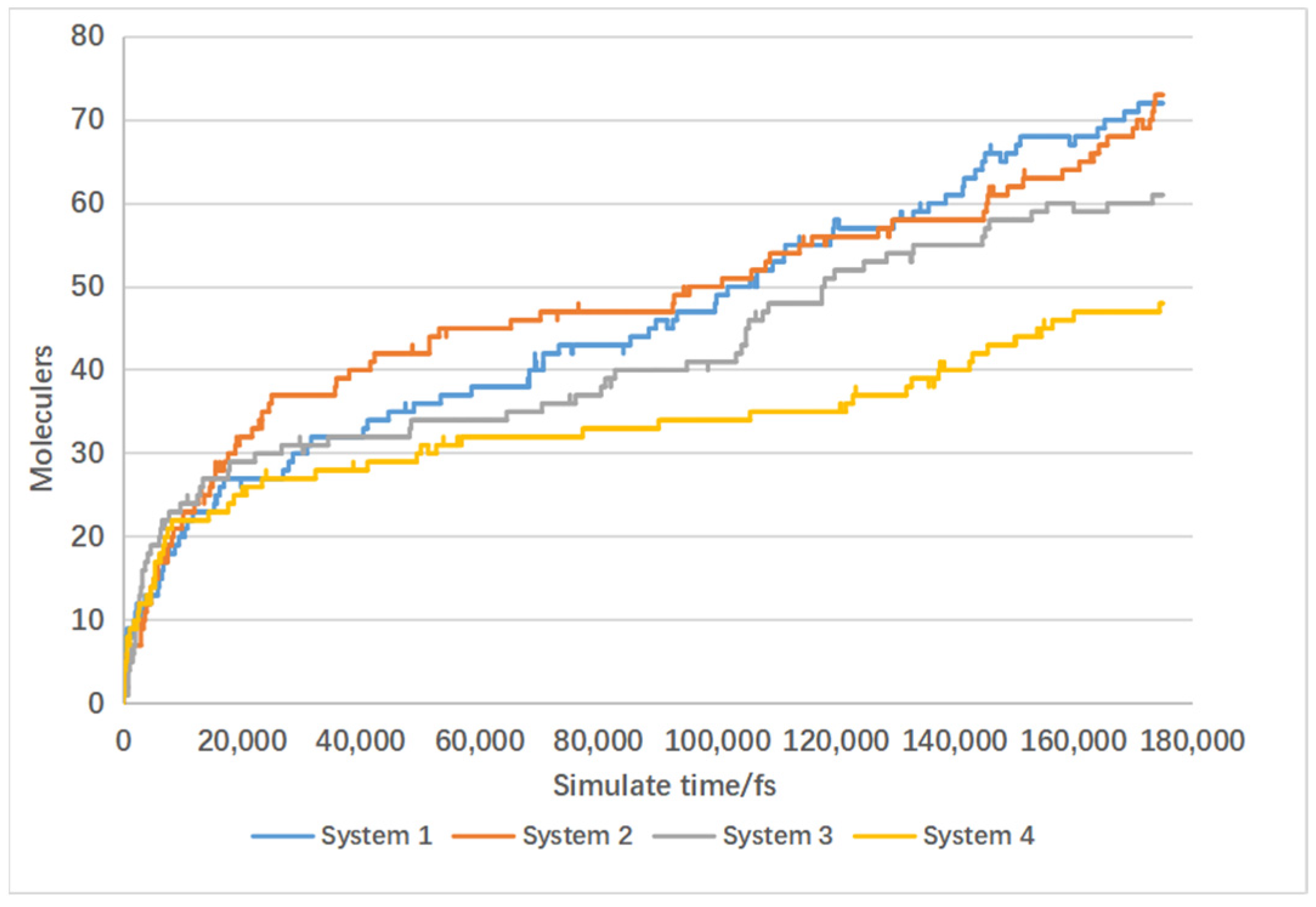
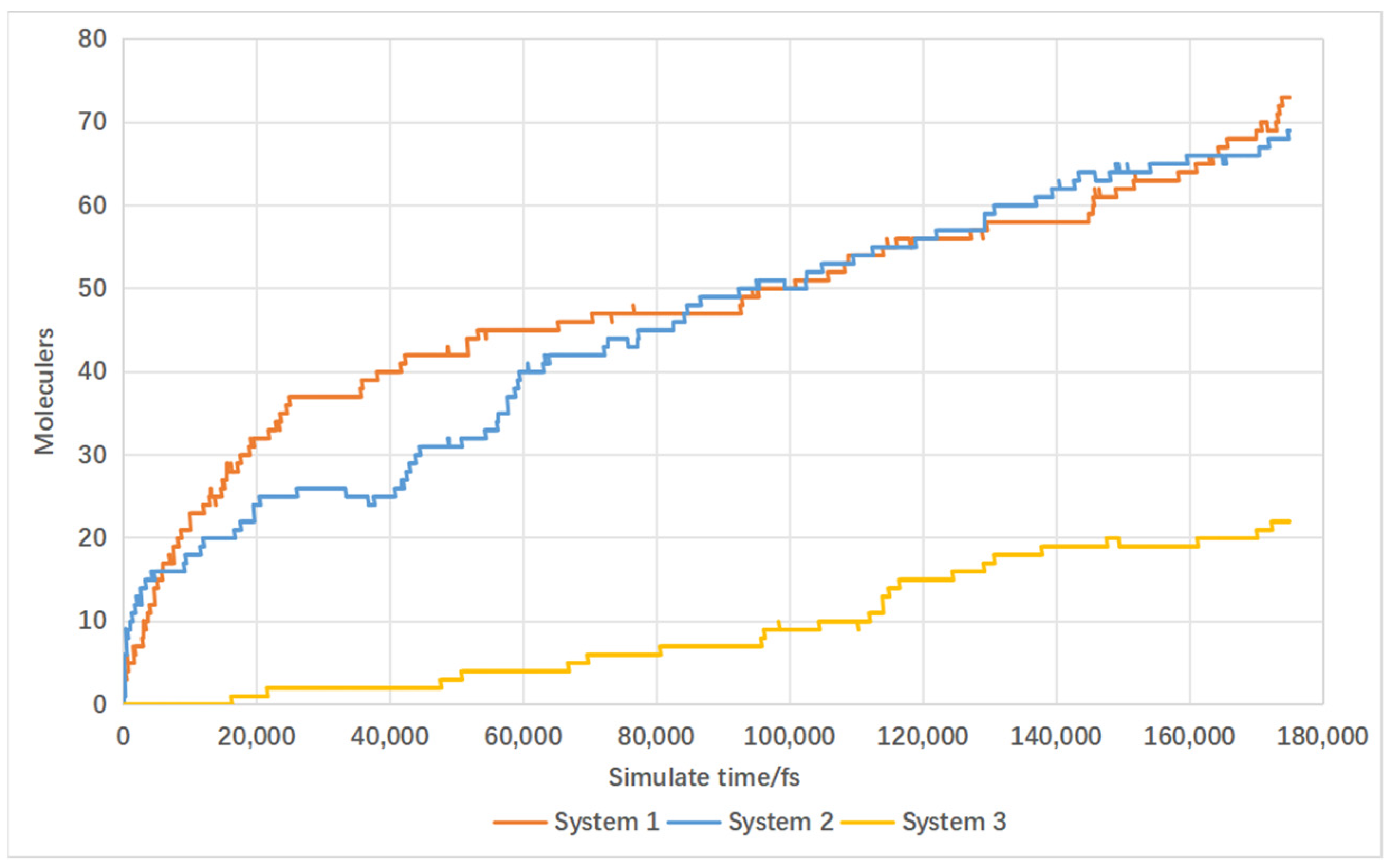
4.3. Effect of Catalyst on Gas Product Formation
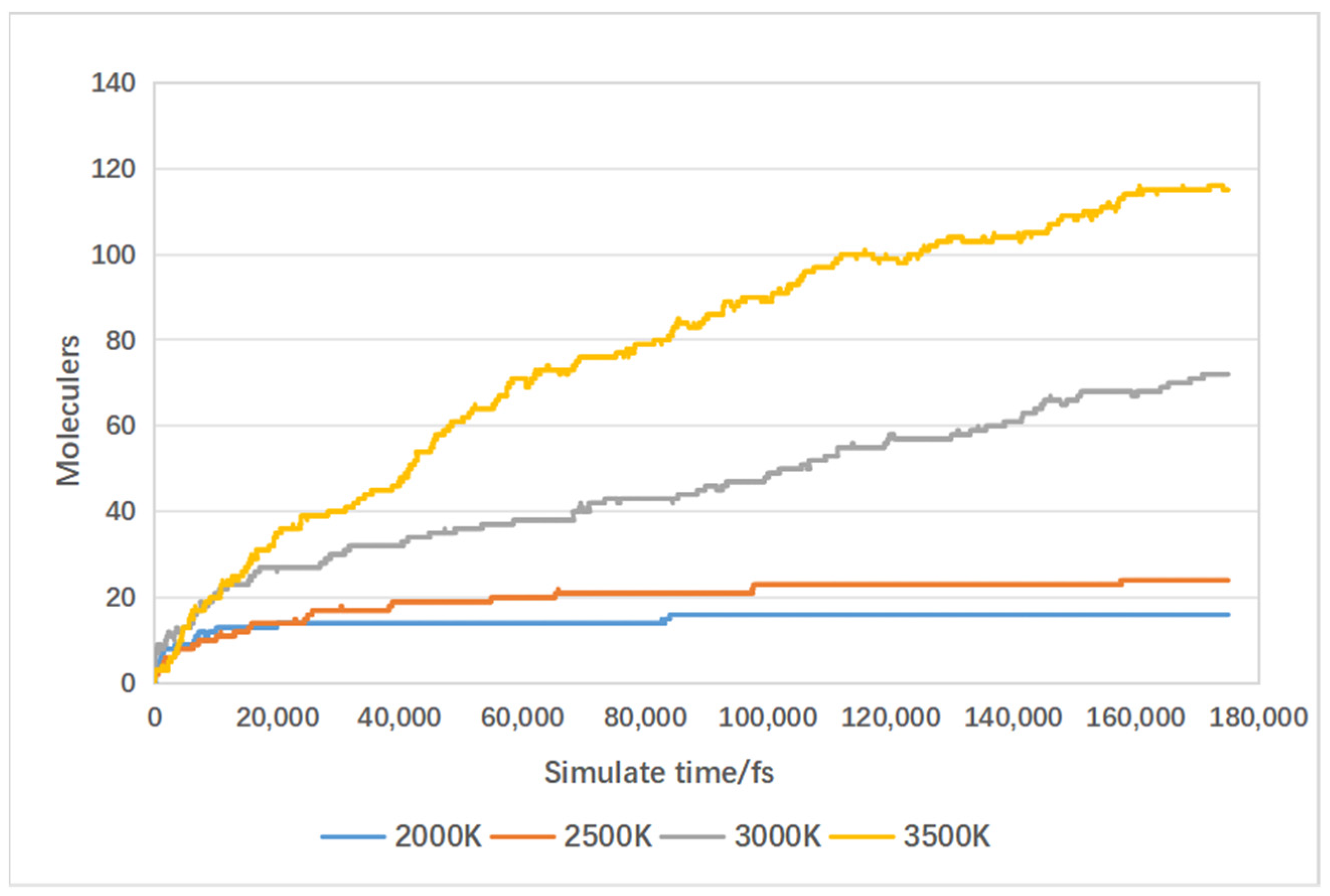
4.4. Effect of Temperature on Gas Production
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, S. Review on the current development of catalysts in China. Sci. Technol. Wind 2018, 33, 240. [Google Scholar]
- Xiong, H.; Han, X.; Zhang, X. Development and prospects of zeolite catalysts. Mater. Rev 2021, 35, 137–142. [Google Scholar]
- He, L.; Zhao, Y.; Zeng, H. Types and current status of catalysts. Salt Sci. Chem. Eng. 2023, 52, 7–8+13. [Google Scholar]
- Sun, S.; Li, R. Current situation and development trends of catalyst use in China. Chem. Eng. Manag. 2016, 15, 7. [Google Scholar]
- Luo, X.; Hou, Z.; Ming, P.; Shao, Z.; Yi, B. Influence of graphitized carbon support on the stability of Pt/C catalysts for proton exchange membrane fuel cells. Acta Catal. Sin. 2008, 4, 330–334. [Google Scholar]
- Yu, L.; You, W. Brief discussion on the development of noble metal catalysts. Shandong Chem. Ind. 2018, 47, 116–117. [Google Scholar]
- Wu, L.; Yu, H.; Wang, Y. Research progress on non-noble metal oxygen reduction electrocatalysts based on porous carbon. Inorg. Salt Ind. 2023, 55, 13–23. [Google Scholar]
- Yang, D.; Tang, K. Research and development of nano-nickel hydrogenation catalysts. Guangdong Chem. Ind. 2017, 44, 95–96+94. [Google Scholar]
- Li, M.; Yuan, M.; Huang, J.; Wu, H.; Li, J.; You, Z. Applications of graphene and its derivatives in catalysis. Mol. Catal. 2019, 33, 190–200. [Google Scholar]
- Wang, D.; Yao, D.; Yang, H.; Wang, X.; Chen, H. Influence of Ni/C catalysts on hydrogen production from biomass gasification. Chin. J. Electr. Eng. 2017, 37, 5682–5687+5845. [Google Scholar]
- Azadi, P.; Khan, S.; Strobel, F.; Azadi, F.; Farnood, R. Hydrogen production from cellulose, lignin, bark and model carbohydrates in supercritical water using nickel and ruthenium catalysts. Appl. Catal. B Environ. 2012, 117–118, 330–338. [Google Scholar] [CrossRef]
- Yu, L.; Zhang, R.; Cao, C.; Liu, L.; Fang, J.; Jin, H. Hydrogen production from supercritical water gasification of lignin catalyzed by Ni supported on various zeolites. Fuel 2022, 319, 123744. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, H.; Wu, X.; Li, T.; Yuan, P.; Bao, X. Current status and development trends of green clean energy technologies under the “dual carbon” goal. Pet. Sci. Bull. 2023, 8, 555–576. [Google Scholar]
- Yin, Z.; Fu, C.; Han, K.; Li, Y.; Gao, M.; He, S.; Qi, J. Review on biomass hydrogen production technology. Therm. Power Gener. 2022, 51, 37–48. [Google Scholar]
- Yu, J.; Chen, Q.; Sun, S.; Guan, Q.; Ning, P.; Gu, J. Research progress on transition metal catalysts for biomass supercritical water gasification hydrogen production. Polym. Bull. 2015, 1, 19–24. [Google Scholar]
- Zhang, H.; Liu, X.; Fu, S. Review and research progress of biomass hydrogen production technology. Chin. J. Pap. 2019, 38, 68–74. [Google Scholar]
- Luo, W.; Liao, C.; Chen, H.; Zhu, Y. Research progress on biomass supercritical water gasification hydrogen production technology. Nat. Gas Chem. Eng. (Chem. Ind. Chem. Eng.) 2016, 41, 84–90. [Google Scholar]
- Dai, Y.; Zhou, M.; Shen, J.; Jiang, H.; Li, C. Molecular Dynamics Simulation of sintering mechanism of TiO2 nanoparticles[J/OL]. Chem. Ind. Eng. Prog. 2024, 1–16. [Google Scholar] [CrossRef]
- Yan, Q.; Guo, L.; Lv, Y. Major influencing factors of biomass/coal supercritical water gasification hydrogen production. J. Xi’an Jiaotong Univ. 2008, 42, 368–371. [Google Scholar]
- Yu, T.; Li, Q.; Tan, Y.; Chen, B.; Hu, H. Research progress on CO2 flooding in tight sandstone reservoirs based on molecular dynamics. Nat. Gas Ind. 2024, 44, 146–159. [Google Scholar]
- Abolfath, R.M.; Van Duin, A.C.T.; Brabec, T. Extension of the reaxff combustion force field toward syngas combustion and initial oxidation kinetics. J. Phys. Chem. A 2017, 121, 1051–1068. [Google Scholar]
- Abolfath, R.M.; Van Duin, A.C.T.; Brabec, T. Reactive Molecular Dynamics study on the first steps of DNA damage by free hydroxyl radicals. J. Phys. Chem. A 2011, 115, 11045–11049. [Google Scholar] [CrossRef]
- Ye, W.; Ren, Q.; Wang, C.; Qu, Y. Effect of S-doped carbon carrier on Pt catalyst for oxygen reduction reaction based on density functional theory. Acta Pet. Sin. (Pet. Process.) 2024, 40, 931–941. [Google Scholar]
- Fu, W.; Liao, X.; Wang, J.; Du, J. Lignin in plant: A review. Bull. Biol. 2004, 39, 12–14. [Google Scholar]
- Wang, F.; Han, S.; Ye, S.; Yin, S.; Li, S.; Wang, X. DFT study on the structure and Raman spectra of lignin monomer and dimer. Spectrosc. Spectr. Anal. 2024, 44, 76–81. [Google Scholar]
- Zhang, Y.; Chen, C.; Zhan, Y.; Chen, Y.; Lou, B.; Zheng, G.; Lin, Q. Hydrogen production via water-gas shift reaction over CuO/ZrO2 catalyst: Effect of ZrO2 support calcination temperature. Fuel Chem. J. 2019, 47, 464–473. [Google Scholar]
- Hao, J.; Cao, X.; Hu, Z. Study on the band gap of bilayer graphene with different stacking patterns on highly polarized substrates. J. Nankai Univ. (Nat. Sci. Ed.) 2020, 53, 56–61. [Google Scholar]
- Feng, P.; Yu, X.; Li, Y.; Yu, S.; Li, R.; Xie, G. Preparation and application of graphene. New Chem. Mater. 2018, 46, 14–17+21. [Google Scholar] [CrossRef]
- Tan, Z.; Wang, X.; Zeng, M.; Wang, J. Preparation of carbon nanocages and graphene sheets by spray pyrolysis and their application as supports for platinum catalysts. Mater. Rev. 2014, 28, 12–17. [Google Scholar]
- Huang, M.; Hu, M.; Yao, Z. Research progress on heteroatom-doped carbon-based oxygen reduction catalysts. Mod. Chem. Ind. 2024, 44, 25–29. [Google Scholar]
- Guo, S.; Guo, L.; Lv, Y. Reaction modeling and numerical simulation of biomass supercritical water gasification for hydrogen production. Sol. Energy 2011, 32, 929–935. [Google Scholar]
- Zhan, X.; Wu, W.; Cui, G. Formation and homogeneous conversion mechanism of pyrolysis tar during biomass gasification process. Energy Res. Inf. 2019, 35, 125–133. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Task | xc Function | Relativity | Basis Set | Frozen Core | Numerical Quality |
|---|---|---|---|---|---|
| Geometry Optimization | GGA: PBE | Scalar | DZP | Large | Normal |
| Task | Time Step (fs) | Torsions | Periodicity | Number of Steps | Thermostat |
|---|---|---|---|---|---|
| Molecular Dynamics | 0.25 | Original | Bulk | 700,000 | NHC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Wu, L.; Liu, F.; Qiu, Y.; Dong, R.; Chen, J.; Liu, D.; Wang, L.; Yi, L. Molecular Dynamics Simulations Guide the Gasification Process of Carbon-Supported Nickel Catalysts in Biomass Supercritical Water. Materials 2024, 17, 4192. https://doi.org/10.3390/ma17174192
Wu Y, Wu L, Liu F, Qiu Y, Dong R, Chen J, Liu D, Wang L, Yi L. Molecular Dynamics Simulations Guide the Gasification Process of Carbon-Supported Nickel Catalysts in Biomass Supercritical Water. Materials. 2024; 17(17):4192. https://doi.org/10.3390/ma17174192
Chicago/Turabian StyleWu, Yuhui, Liang Wu, Fan Liu, Yue Qiu, Runqiu Dong, Jingwei Chen, Daoxiu Liu, Le Wang, and Lei Yi. 2024. "Molecular Dynamics Simulations Guide the Gasification Process of Carbon-Supported Nickel Catalysts in Biomass Supercritical Water" Materials 17, no. 17: 4192. https://doi.org/10.3390/ma17174192
APA StyleWu, Y., Wu, L., Liu, F., Qiu, Y., Dong, R., Chen, J., Liu, D., Wang, L., & Yi, L. (2024). Molecular Dynamics Simulations Guide the Gasification Process of Carbon-Supported Nickel Catalysts in Biomass Supercritical Water. Materials, 17(17), 4192. https://doi.org/10.3390/ma17174192