
Effect of a Hybrid Pumice–Portland Cement Extract on Corrosion Activity of Stainless Steel SS304 and Carbon Mild Steel A36



Abstract
1. Introduction
2. Materials and Methods
2.1. Steel Samples
2.2. Hybrid Cement “HB1” and Its Cement Extract Solution
2.3. Immersion Test
2.4. Electrochemical Measurements
3. Results and Discussion
3.1. Change in Time of pH of the “HB1” CE Solution and OCP of the Steels
3.2. Stainless Steel 304 Surface Characterization after Immersion in the “HB1” CE Solution
3.3. Carbon Steel A36 Surface Characterization after the Immersion in the “HB1” CE Solution
3.4. Steel Surface Deterioration after the Immersion Tests in “HB1” CE Solution
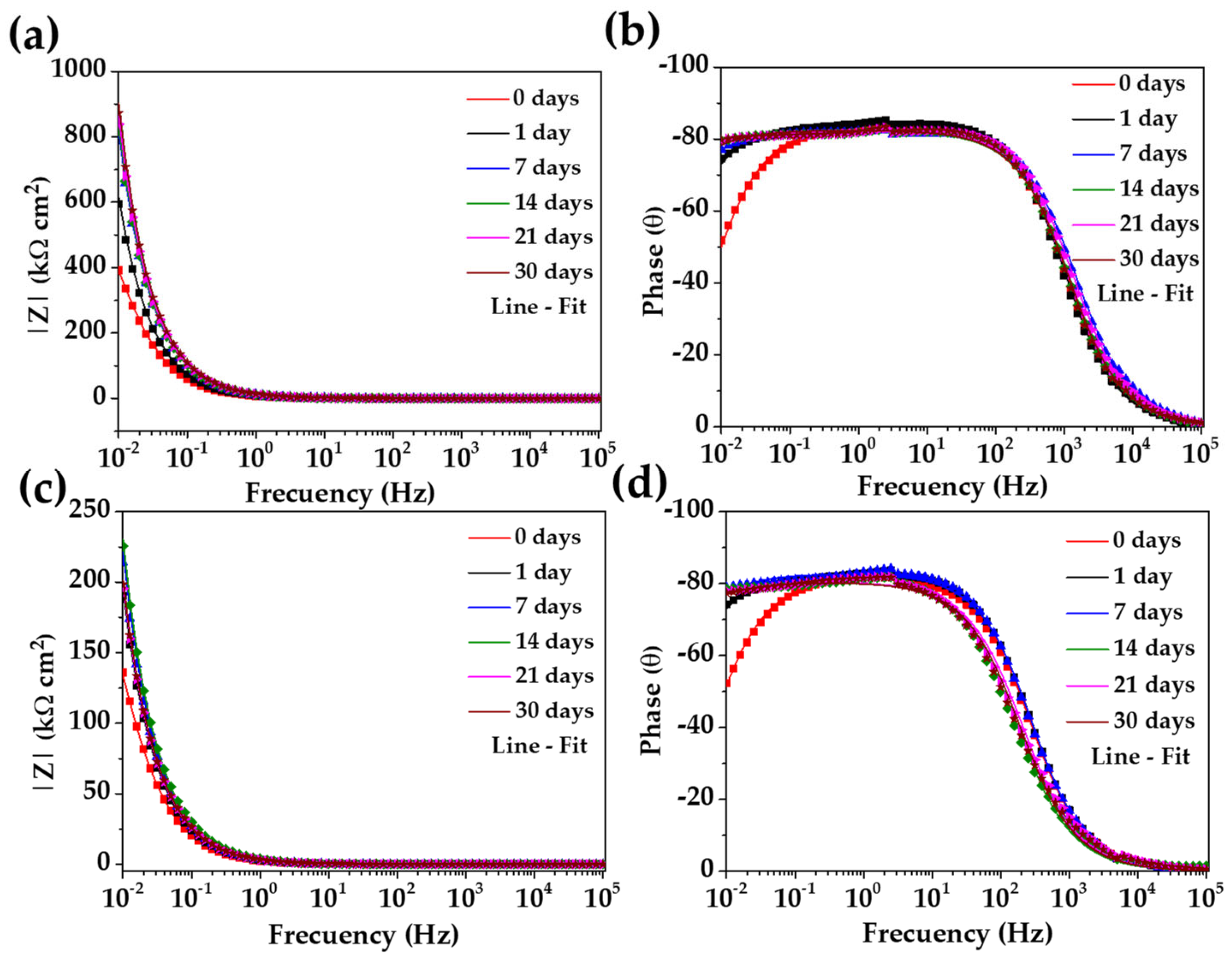
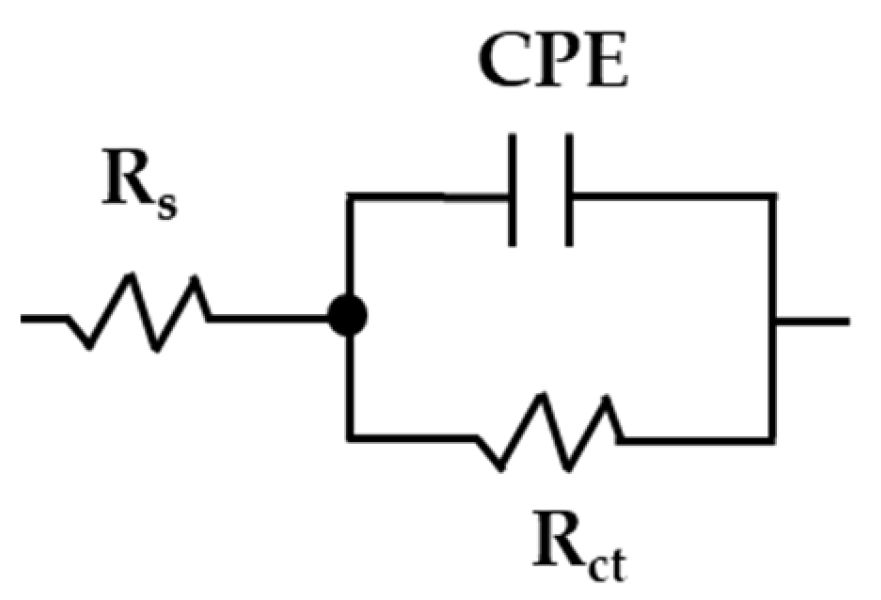
3.5. EIS Diagrams (Nyquist and Bode)
4. Conclusions
- The change in the corrosion activities of SS304 and carbon steel A36 were studied during their exposure for 30 days to an aqueous extract solution of a hybrid cement, labeled as “HB1”, composed of CPC30R, partially replaced by volcanic pumice, in the presence of , as alkaline activators.
- The ionic composition of the “HB1” CE is influenced by the reduction of PC content, the composition of the volcanic pumice and the presence of alkaline activators.
- The initial pH of the “HB1” CE solution was 12.99 (similar to that of the “PC–CE” solution) due to the contribution of the greater contents of and (volcanic pumice) and the reaction of the activators. The pH diminished and maintained an almost constant value ≈ 9.10 for the 30 days, indicating an established dynamic ionic equilibrium because of the air dissolution.
- In the meantime, the OCP of the SS304 turned towards a positive value (≈265 mV) and that of the carbon steel A36 also reached a positive value (≈88 mV), as an indication that the “HB1” promoted the passivation of both steels. However, for the carbon steel, the OCP value may indicate an intermediate risk of corrosion.
- After exposure to the “HB1” CE solution, SEM–EDS and XPS analysis suggested that the passive layer of SS304 was composed of , and , while , and were characteristic of the carbon steel A36 layer.
- Probably due to the presence of ions (originated from the pumice), isolated initial pits were observed in the vicinity of the carbides (local cathodes), causing a local depassivation. However, the preferential accumulation of ions, with a higher charge than that of the ions, may cause a retarding effect of the localized chloride attack over a longer time of exposure, which in fact agrees with the positive shift in the OCP.
- The quantitative analysis of the EIS diagrams, based on the equivalent electrical circuit proposed, allowed us to characterize the corrosion activity of the studied steels at the metal–CE solution interface. Due to the passive state reached during the exposure to the “HB1” CE solution, both steels presented an increased value, being ≈4 higher for the SS304 than that of the carbon steel A36. The thickness of the passive layer formed on the SS304 surface was ≈1.8 nm, while that on the A36 was ≈0.3 nm. These differences could be attributed to the nature of the steels.
- The reported results of this study, using the “HB1” CE solution to simulate the concrete–pore environment, suggest that the hybrid cement “HB1” may be considered as a “green” alternative for the partial replacement of PC with volcanic pumice. The corrosion behavior of the reinforcement steels will depend mainly on their nature and the composition of the concrete–pore environment.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poudyal, L.; Adhikari, K. Environmental Sustainability in Cement Industry: An Integrated Approach for Green and Economical Cement Production. Resour. Environ. Sustain. 2021, 4, 100024. [Google Scholar] [CrossRef]
- Andrew, R.M. Global CO2 Emissions from Cement Production. Earth Syst. Sci. 2018, 10, 195–217. [Google Scholar] [CrossRef]
- Damtoft, J.S.; Lukasik, J.; Herfort, D.; Sorrentino, D.; Gartner, E.M. Sustainable development and climate change initiatives. Cem. Concr. Res. 2008, 38, 115–127. [Google Scholar] [CrossRef]
- Neville, A.M. Properties of Concrete, 4th ed.; Prentice/Pearson Hill: Harlow, UK, 2009; pp. 1–103. [Google Scholar]
- Ramezanianpour, A.A. Cement Replacement Materials: Properties, Durability, Sustainability, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 1–43. [Google Scholar] [CrossRef]
- Juenger, M.C.G.; Winnefeld, F.; Provis, J.L.; Ideker, J.H. Advances in alternative cementitious binders. Cem. Concr. Res. 2011, 41, 1232–1243. [Google Scholar] [CrossRef]
- Juenger, M.C.G.; Snellings, R.; Bernal, S. Supplementary cementitious materials: New sources, characterization and performance insights. Cem. Concr. Res. 2019, 122, 257–273. [Google Scholar] [CrossRef]
- Hossain, K.M.A. Blended cement using volcanic ash and pumice. Cem. Concr. Res. 2003, 33, 1601–1605. [Google Scholar] [CrossRef]
- Játiva, A.; Etxeberria, M. Exploring the Utilization of Activated Volcanic Ash as a Substitute for Portland Cement in Mortar Formulation: A Thorough Experimental Investigation. Materials 2024, 17, 1123. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Qu, S.; Gao, K.; Hailu Tekle, B.; Bao, J.; Zhan, P. Effect of corrosion on the bond behavior of steel-reinforced Alkali-Activated Slag Concrete. Materials 2023, 16, 2262. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, M.N.; Mustapha, F.; Yusof, N.‘I.; Khan, T.; Sebaey, T.A. Thermal Properties and Drying Shrinkage Performance of Palm Kernel Shell Ash and Rice Husk Ash-Based Geopolymer Concrete. Materials 2024, 17, 1298. [Google Scholar] [CrossRef]
- Cui, Y.; Qu, S.; Bao, J.; Zhang, P. Bond Performance of Steel Bar and Fly Ash-Based Geopolymer Concrete in Beam End Tests. Polymers 2022, 14, 2012. [Google Scholar] [CrossRef]
- Cabrera-Luna, K.; Maldonado-Bandala, E.E.; Nieves-Mendoza, D.; Castro-Borges, P.; Escalante García, J.I. Supersulfated cements based on pumice with quicklime, anhydrite, and hemihydrate; characterization and environmental impact. Cem. Concr. Compos. 2021, 124, 104236. [Google Scholar] [CrossRef]
- Lopez-Salas, J.; Escalante-Garcia, J.I. Hybrid binders based on volcanic pumice: Effect of the chemical composition on strength and microstructures. Cem. Concr. Res. 2024, 176, 107393. [Google Scholar] [CrossRef]
- Glasser, F.P.; Luke, K.; Angus, M.J. Modification of cement pore fluid compositions by pozzolanic additives. Cem. Concr. Res. 1988, 18, 165–178. [Google Scholar] [CrossRef]
- Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Solutions, 2nd ed.; NACE: Houston, TX, USA, 1974; p. 561. [Google Scholar]
- Freire, L.; Carmezim, M.J.; Ferreira, M.G.S.; Montemor, M.F. The passive behaviour of AISI 316 in alkaline media and the effect of pH: A combined electrochemical and analytical study. Electrochim. Acta 2010, 55, 6174–6181. [Google Scholar] [CrossRef]
- Luo, H.; Su, H.; Dong, C.; Li, X. Passivation and electrochemical behavior of 316 stainless steel in chlorinated simulated concrete pore solution. Appl. Surf. Sci. 2017, 400, 38–48. [Google Scholar] [CrossRef]
- Ghods, P.; Isgor, O.B.; McRae, G.; Miller, T. The effect of concrete pore solution composition on the quality of passive oxide films on black steel reinforcement. Cem. Concr. Compos. 2009, 31, 2–11. [Google Scholar] [CrossRef]
- Scott, A.; Alexander, M.G. Effect of suplementary cementitous materials (binder type) on the pore solution chemistry and the corrosion of steen in alkaline environments. Cem. Concr. Res. 2016, 89, 45–55. [Google Scholar] [CrossRef]
- Page, C.L.; Vennesland, O. Pore solution compositions and chloride binding capacity of silica fume cement paste. Mat. Struct. 1983, 16, 19–25. [Google Scholar] [CrossRef]
- Duchesne, J.; Berube, M. Evaluation of the validity of the pore solution expression method from hardened cement pastes and mortars. Cem. Concr. Res. 1994, 24, 456–462. [Google Scholar] [CrossRef]
- Puertas, F.; Fernández-Jiménez, A.; Blanco-Varela, M.T. Pore solution in alkali-activated slag cement pastes. Relation to the composition and structure of calcium silicate hydrate. Cem. Concr. Res. 2004, 34, 139–148. [Google Scholar] [CrossRef]
- Andersson, K.; Allard, B.; Begtsson, M.; Magnusson, B. Chemical composition of cement pore solutions. Cem. Concr. Res. 1989, 19, 327–332. [Google Scholar] [CrossRef]
- Goñi, S.; Andrade, C. Sinthetic concrete pore solution chemistry and rebar corrosion rate in the presence of chlorides. Cem. Concr. Res. 1990, 20, 525–539. [Google Scholar] [CrossRef]
- Moragues, A.; Macias, A.; Andrade, C. Equilibria of the chemical composition of the concrete pore solution. Part I: Comparative study of synthetic and extracted solutions. Cem. Concr. Res. 1987, 17, 173–182. [Google Scholar] [CrossRef]
- Poursaee, A. Corrosion of steel bars in saturated Ca(OH)2 and concrete pore solution. Concr. Res. Lett. 2010, 1, 90–97. [Google Scholar]
- Jiang, H.; Jin, Z.; Zhang, X.; Qian, L.; Zhou, Z. The Effect of Temperatures on the Passivation Behavior of Q235 Steel in the Simulated Concrete Pore Solution. Materials 2023, 16, 588. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Sagüés, A.A. Chloride corrosion threshold of reinforcing steel in alkaline solutions-open circuit immersion tests. Corrosion 2001, 57, 19–28. [Google Scholar] [CrossRef]
- Zhang, F.; Jinshan, P.; Changjian, L. Localized corrosion behaviour of reinforcement steel in simulated concrete pore solution. Corros. Sci. 2009, 51, 2130–2138. [Google Scholar] [CrossRef]
- Behera, P.K.; Misra, S.; Mondal, K. Corrosion Behavior of Strained Rebar in Simulated Concrete Pore Solution. J. Mater. Eng. Perform. 2020, 29, 1939–1954. [Google Scholar] [CrossRef]
- Pokorný, P.; Vacek, V.; Prodanovic, N.; Zabloudil, A.; Fojt, J.; Johánekm, V. The Influence of Graded Amount of Potassium Permanganate on Corrosion of Hot-Dip Galvanized Steel in Simulated Concrete Pore Solutions. Materials 2022, 15, 7864. [Google Scholar] [CrossRef]
- Zakroczymski, T.; Fan, C.J.; Szklarska-Smialowska, Z. Kinetics of passive film formation on iron in 0.05M NaOH. J. Electrochem. Soc. 1985, 132, 2282–2287. [Google Scholar] [CrossRef]
- Montemor, M.F.; Simoes, A.M.; Ferreira, M.G. Analytical characterization of the passive film formed on steel in solutions simulating the concrete interstitial electrolyte. Corrosion 1998, 54, 347–353. [Google Scholar] [CrossRef]
- Veleva, L.; Alpuche-Aviles, M.A.; Graves-Brook, M.K.; Wipf, D.O. Comparative cyclic voltammetry and surface analysis of passive films grown on stainless steel 316 in concrete pore model solutions. J. Electroanal. Chem. 2002, 537, 85–93. [Google Scholar] [CrossRef]
- Veleva, L.; Alpuche-Aviles, M.A.; Graves-Brook, M.K.; Wipf, D.O. Voltammetry and surface analysis of AISI 316 stainless steel in chloride-containing simulated concrete pore environment. J. Electroanal. Chem. 2005, 578, 45–53. [Google Scholar] [CrossRef]
- Miserque, F.; Huet, B.; Azou, G.; Bendjaballah, D.; L’Hostis, V. X-ray photoelectron spectroscopy and electrochemical studies of mild steel FeE500 passivation in concrete simulated water. J. Phys. IV 2006, 136, 89. [Google Scholar] [CrossRef]
- Fan, L.F.; Zhong, W.L.; Zhang, Y.H. Effect of the composition and concentration of geopolymer pore solution on the passivation characteristics of reinforcement. Constr. Build. Mater. 2022, 319, 126128. [Google Scholar] [CrossRef]
- Albani, O.A.; Zerbino, J.O.; Vilche, J.R.; Arvia, A.J. A comparative electrochemical and ellipsometric study of iron electrodes in different alkaline electrolytes. Electrochim. Acta 1986, 31, 1403–1411. [Google Scholar] [CrossRef]
- Oranowska, H.; Szklarska-Smialowska, Z. An electrochemical and ellipsometric investigation of surface films grown on iron in saturated calcium hydroxide solutions with or without chloride ions. Corros. Sci. 1981, 21, 735–747. [Google Scholar] [CrossRef]
- Huet, B.; Hostis, L.; Tricheux, L.; Idrissi, H. Huet Influence of alkali, silicate, and sulfate content of carbonated concrete pore solution on mild steel corrosion behavior. Mater. Corros. 2010, 61, 111–124. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, Y.; Ni, Z.; Huang, R. Corrosion behavior of steel submitted to chloride and sulphate ions in simulated concrete pore solution. Constr. Build. Mater. 2016, 115, 1–5. [Google Scholar] [CrossRef]
- Xu, P.; Jiang, L.; Guo, M.Z.; Zha, J.; Chen, L.; Chen, C.; Xu, N. Influence of sulfate salt type on passive film of steel in simulated concrete pore solution. Constr. Build. Mater. 2019, 223, 352–359. [Google Scholar] [CrossRef]
- Shaheen, F.; Pradhan, B. Influence of sulfate ion and associated cation type on steel reinforcement corrosion in concrete powder aqueous solution in the presence of chloride ions. Cem. Concr. Res. 2017, 91, 73–86. [Google Scholar] [CrossRef]
- Al-Amoudi, O.S.B.; Maslehuddin, M. The effect of chloride and sulfate ions on reinforcement corrosion. Cem. Concr. Res. 1993, 23, 139–146. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, R.; Hu, J.; Zhang, Z.; Huang, H.; Ma, Y.; Wei, J.; Zhang, Z.; Yin, S.; Wang, H.; et al. Surface characteristics and electrochemical behaviors of passive reinforcing steel in alkali-activated slag. Corros. Sci. 2021, 190, 109657. [Google Scholar] [CrossRef]
- Bacelis, A.; Veleva, L.; Feliu, S.; Cabrini, M.; Lorenzi, S. Corrosion Activity of Carbon Steel B450C and Low Chromium Ferritic Stainless Steel 430 in Cement Extract Solution. Buildings 2021, 11, 220. [Google Scholar] [CrossRef]
- Bonfil, D.; Veleva, L.; Feliu, S., Jr.; Escalante-García, J.I. Corrosion Activity of Carbon Steel B450C and Stainless Steel SS430 Exposed to Extract Solution of a Supersulfated Cement. Materials 2022, 15, 8782. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhan, S.; Tang, X.; Xiu, Q.; Qian, K. Pore solution chemistry of calcium sulfoaluminate cement and its effects on steel passivation. Appl. Sci. 2019, 9, 1092. [Google Scholar] [CrossRef]
- ASTM-NACE/ASTM G31-12a; Standard Guide for Laboratory Immersion Corrosion Testing of Metals. ASTM International: West Conshohocken, PA, USA, 2012.
- ASTM G1-03; Standard Practice for Preparing, Cleaning, and Evaluating Corrosion Test Specimens. ASTM International: West Conshohocken, PA, USA, 2017.
- Anstice, D.J.; Page, C.L.; Page, M.M. The pore solution phase of carbonated cement pastes. Cem. Concr. Res. 2005, 35, 377–383. [Google Scholar] [CrossRef]
- Adamczyk, K.; Prémont-Schwarz, M.; Pines, D.; Pines, E.; Nibbering, T.J. Real-time observation of carbonic acid formation in aqueous solutions. Science 2009, 326, 1690–1694. [Google Scholar] [CrossRef] [PubMed]
- Kern, M. The hydration of carbon dioxide. J. Chem. Educ. 1960, 39, 1–14. [Google Scholar] [CrossRef]
- Greve-Dierfeld, S.; Lothenbach, B.; Vollpracht, A.; Wu, B.; Huet, B.; Andrade, C.; Medina, C.; Thiel, C.; Gruyaert, E.; Vanoutrive, H.; et al. Understanding the carbonation of concrete with supplementary cementitous materials. A critical review by RILEM TC 281-CCC. Mater. Struct. 2020, 53, 136. [Google Scholar] [CrossRef]
- ASTM-NACE/ASTM C876-15; Standard Test Method for Corrosion Potentials of Uncoated Reinforcing Steel in Concrete. ASTM International: West Conshohocken, PA, USA, 2015. [CrossRef]
- Galvele, J.R. Transport Processes and the mechanism of pitting of metals. J. Electrochem. 1976, 123, 464. [Google Scholar] [CrossRef]
- Ai, Z.; Sun, W.; Jiang, J.; Song, D.; Ma, H.; Zhang, J.; Wang, D. Passivation Characteristics of Alloy Corrosion-Resistant Steel Cr10Mo1 in Simulating Concrete Pore Solutions: Combination Effects of pH and Chloride. Materials 2016, 9, 749. [Google Scholar] [CrossRef] [PubMed]
- Addari, D.; Elsener, B.; Rossi, A. Electrochemistry and surface chemistry of stainless steel in alkaline media simulating concrete pore solutions. Electrochim. Acta 2008, 53, 8078–8086. [Google Scholar] [CrossRef]
- Luo, H.; Su, H.; Dong, C.; Xiao, K.; Li, X. Electrochemical and passivation behavior investigation of ferritic stainless steel in simulated concrete pore media. Data Brief 2015, 96, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Wahlqvist, M.; Shchukarev, A. XPS spectra and electronic structure of Group IA sulfates. J. Electron. Spectrosc. 2007, 156–158, 310–314. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.F.; Sobol, P.E.; Bomben, K.D. Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data; Physical Electronics: Chanhassen, MN, USA, 1995; p. 81. [Google Scholar]
- Freire, L.; Catarino, M.A.; Godinho, M.I.; Ferreira, M.J.; Simoes, A.M.P.; Montemor, M.F. Electrochemical and analytical investigation of passive films formed on stainless steel in alkaline media. Cem. Concr. Comp. 2012, 34, 1075–1081. [Google Scholar] [CrossRef]
- Carmezim, M.J.; Simões, A.M.P.; Montemor, M.F.; Da Cunha Belo, M. Capacitance behaviour of passive films on ferritic and asutenitic stainless steel. Corros. Sci. 2005, 47, 581–591. [Google Scholar] [CrossRef]
- Hakiki, N.B.; Boudin, S.; Rondot, B.; Da Cunha Belo, M. The electronic structure of passive films formed on stainless steel. Corros. Sci. 1995, 37, 1809–1822. [Google Scholar] [CrossRef]
- Freire, L.; Novoa, X.R.; Montemor, M.F.; Carmezin, M.J. Study of passive films formed on mild steel in alkaline media by the application of anodic potentials. Mater. Chem. Phys. 2009, 114, 962–972. [Google Scholar] [CrossRef]
- Antony, H.; Legrand, L.; Maechal, L.; Perrin, S.; Dillmann, P.; Chaussé, A. Study of lepidocrocite γ-FeOOH electrochemical reduction in neutral and slightly alkaline solutions at 25 °C. Electrochim. Acta 2005, 51, 745–753. [Google Scholar] [CrossRef]
- Burak Gunay, H.; Ghods, P.; Burkan Isgor, O.; Carpenter, G.J.C.; Wu, X. Characterization of atomic structure oxide films on carbon steel in simulated concrete pore solutions using EELS. Appl. Surf. Sci. 2013, 274, 195–202. [Google Scholar] [CrossRef]
- Olsson, C.-O.A.; Landolt, D. Passive film on stainless steel-chemistry, structure and growth. Electrochim. Acta 2003, 48, 1093–1104. [Google Scholar] [CrossRef]
- Tolulope Loto, R. Pitting corrosion evaluation and inhibition of stainless steels: A review. J. Mater. Environ. Sci. 2015, 6, 2750–2762. [Google Scholar]
- Jiang, J.-y.; Liu, Y.; Chu, H.-y.; Wang, D.; Ma, H.; Sun, W. Pitting Corrosion Behaviour of New Corrosion-Resistant Reinforcement Bars in Chloride-Containing Concrete Pore Solution. Materials 2017, 10, 903. [Google Scholar] [CrossRef]
- Jiang, J.-y.; Wang, D.; Chu, H.-y.; Ma, H.; Liu, Y.; Gao, Y.; Shi, J.; Sun, W. The Passive Film Growth Mechanism of New Corrosion-Resistant Steel Rebar in Simulated Concrete Pore Solution: Nanometer Structure and Electrochemical Study. Materials 2017, 10, 412. [Google Scholar] [CrossRef] [PubMed]
- Mundra, S.; Criado, M.; Bernal, S.A.; Provis, J.L. Chloride-induced corrosion of steel rebars in simulated pore solutions of alkali-activated concrete. Cem. Concr. Res. 2017, 100, 385–397. [Google Scholar] [CrossRef]
- Lin, L.F.; Chao, C.Y.; Macdonald, D.D. A point defect model for anodic passive films: II. Chemical breakdown and pit initiation. J. Electrochem. 1981, 128, 1194. [Google Scholar] [CrossRef]
- Lasia, A. Definition of Impedance and Impedance of Electrical Circuits. In Electrochemical Impedance Spectroscopy and Its Applications; Springer: New York, NY, USA, 2014; pp. 7–66. [Google Scholar]
- Shi, J.; Wu, M.; Ming, J. Degradation effect of carbonation on electrochemical behavior of 2304 duplex stainless steel in simulated concrete pore solutions. Corros. Sci. 2020, 177, 109006. [Google Scholar] [CrossRef]
- Wu, M.; Ma, H.; Shi, J. Enhanced corrosion resistance of reinforcing steels in simulated concrete pore solution with low molybdate to chloride ratios. Cem. Concr. Compos. 2020, 110, 103589. [Google Scholar] [CrossRef]
- Brug, G.J.; Van Den Eeden, A.L.G.; Sluyters-Rehbach, M.; Sluyters, J.H. The analysis of electrode impedance complicated by the presence of a constant phase element. J. Electroanal. Chem. 1954, 176, 275–295. [Google Scholar] [CrossRef]
- Rammelt, U.; Reinhard, G. The influence of surface roughness on the impedance data for iron electrodes in acid solutions. Corros. Sci. 1987, 27, 373–382. [Google Scholar] [CrossRef]
- Sarango de Souza, J.; De Oliveira, L.A.; Sayeg, I.J.; Antunes, R.A. Electrochemical study of the AISI 409 ferritic stainless steel: Passive film stability and pitting nucleation and growth. Mater. Res. 2017, 20, 1669. [Google Scholar] [CrossRef]
- Orazem, M.E.; Frateur, I.; Tribollet, B.; Vivier, V.; Marcelin, S.; Pebere, N.; Bunge, A.L.; White, E.A.; Riemer, D.P.; Musiani, M. Dielectric properties of materials showing constant-phase-element (CPE) impedance response. J. Electrochem. Soc. 2013, 160, C215. [Google Scholar] [CrossRef]
- Hirschorn, B.; Orazem, M.E.; Tribollet, B.; Vivier, V.; Frateur, I.; Musiani, M. Determination of effective capacitance and film thickness from constant-phase element parameters. Electrochim. Acta 2010, 55, 6218–6227. [Google Scholar] [CrossRef]
- Ji, H.; Tian, Y.; Zhao, R.; Jin, N.; Tian, Z.; Yan, D.; Ye, H. Passivation and depassivation og HPB335 Carbon steel in simulated concrete pore solution. Int. J. Electrochem. Sci. 2020, 15, 6488–6507. [Google Scholar] [CrossRef]
- Saura, P.; Zornoza, E.; Andrade, C.; Ferrandiz-Mas, V.; Garcés, P. Composition of Corroded Reinforcing Steel Surface in Solutions Simulating the Electrolytic Environments in the Micropores of Concrete in the Propagation Period. Materials 2022, 15, 2216. [Google Scholar] [CrossRef]
- Poursaee, A.; Hansson, C.M. Reinforcing steel passivation in mortar and pore solution. Cem. Concr. Res. 2017, 37, 1127–1133. [Google Scholar] [CrossRef]
- Duffó, G.S.; Farina, S.B. Electrochemical behaviour of steel in mortar and in simulated pore solutions: Analogies and differences. Cem. Concr. Res. 2016, 88, 211–216. [Google Scholar] [CrossRef]
- Ghods, P.; Isgor, O.B.; Carpenter, G.J.C.; Li, J.; McRae, G.A.; Gu, G.P. Nano-scale study of passive films and chloride-induced depassivation of carbon steel rebar in simulated concrete pore solutions using FIB/TEM. Cem. Concr. Res. 2013, 47, 55–68. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element (wt.%) | C | Cr | P | S | Mn | Si | Ni | Fe |
|---|---|---|---|---|---|---|---|---|
| 304 | 0.08 | 18.00 | 0.045 | 0.03 | 2.00 | 1.00 | 8.00 | Balance |
| A36 | 0.0650 | 0.02 | 0.004 | 0.0020 | 0.41 | 0.02 | - | Balance |
| HB1 | CPC30R | Pumice () | Total | ||
|---|---|---|---|---|---|
| g | 300 | 36 | 18.78 | 300 | 654.78 |
| wt.% | 45.82 | 5.49 | 2.87 | 45.82 | 100 |
| SS1 | CPC30 | Hh (CS) () | Pumice () | Total | |
|---|---|---|---|---|---|
| wt.% | 6.90 | 6.90 | 34.48 | 51.72 | 100 |
| Cement | SiO2 | Al2O3 | Fe2O3 | CaO | MgO | SO3 | K2O | Na2O | Cl | LOI |
|---|---|---|---|---|---|---|---|---|---|---|
| HB1 | 43.05 | 8.85 | 2.79 | 29.78 | 0.75 | 4.87 | 2.88 | 4.35 | 0.05 | 2.63 |
| SS1 | 38.22 | 7.82 | 1.63 | 26.07 | 0.49 | 19.46 | 2.98 | 2.00 | 0.06 | 1.27 |
| PC | 22.30 | 4.62 | 2.44 | 58.42 | 1.92 | 2.20 | 0.35 | 0.28 | - | 3.62 |
| Element (mg/L) | Li | Si | Sr | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HB1 | 1.32 | 2480.20 | 17,358.90 | 0.37 | 210.00 | 4.39 | 10,115.00 | 9.74 | 172.00 | 1515.00 |
| SS1 | 0.18 | 496.80 | 310.20 | 0.18 | 396.60 | 5.4 | 563.50 | 9.02 | 45.40 | 407.80 |
| PC [49] | - | 1373.58 | 420.71 | - | 256.51 | - | - | - | - | 958.8 |
| Time (Days) | Initial | 1 | 7 | 14 | 21 | 30 | |
|---|---|---|---|---|---|---|---|
| 304 | pH | 12.99 | 12.92 | 10.48 | 9.61 | 9.18 | 9.07 |
| OCP vs. SHE (mV) | −204.899 | −89.811 | +128.126 | +219.756 | +245.307 | +265.085 | |
| A36 | pH | 12.95 | 12.93 | 10.11 | 9.57 | 9.08 | 9.10 |
| OCP vs. SHE (mV) | −89.053 | −24.845 | +48.584 | +67.456 | +79.154 | +88.18 |
| Days/wt.% | Fe | Cr | Ni | O | Na | Ca | S | K | Si | C | Cl | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 7 | D1 | 5.93 | - | - | 30.88 | 7.69 | 5.20 | 8.49 | 16.95 | 24.86 | - | - |
| D2 | 4.11 | - | - | 34.08 | 18.65 | 1.52 | 21.98 | 7.85 | 11.80 | - | - | |
| D3 | 64.15 | 17.75 | 7.30 | 5.62 | 1.65 | - | - | - | 2.71 | 0.83 | - | |
| 30 | G1 | 2.42 | 0.74 | - | 50.98 | 10.24 | 1.15 | 2.06 | 6.40 | 25.08 | - | 0.91 |
| G2 | 2.38 | - | - | 51.86 | 14.81 | - | 6.49 | 6.96 | 17.50 | - | - | |
| G3 | 5.97 | 1.90 | - | 52.68 | 6.05 | - | 0.86 | 2.19 | 21.15 | 8.87 | 0.32 |
| Days/wt.% | Fe | O | Na | Ca | S | K | Si | C | Al | Cl | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 7 | B1 | 11.19 | 43.76 | 16.19 | 6.80 | 16.60 | 2.16 | 0.19 | 3.11 | - | - |
| B2 | 83.53 | 7.10 | 3.43 | 0.46 | 0.51 | - | 0.98 | 3.98 | - | - | |
| 30 | F1 | 8.29 | 50.18 | 1.04 | 18.19 | - | - | - | 12.30 | - | - |
| F2 | 1.20 | 45.18 | 16.07 | 6.25 | 13.74 | 12.51 | - | 2.63 | - | 1.76 |
| Steel/wt.% | Fe | Cr | Ni | Mn | Si | C | |
|---|---|---|---|---|---|---|---|
| 304 | 1 | 70.32 | 19.01 | 8.67 | 1.30 | 0.70 | - |
| 2 | 65.45 | 19.00 | 8.23 | 1.36 | 0.60 | 5.35 | |
| A36 | A | 98.15 | - | - | - | - | 1.85 |
| B | 95.33 | - | - | - | - | 2.54 | |
| Steel | Days | Rs | Rct | CPE | n | Rp | χ2 |
|---|---|---|---|---|---|---|---|
| 304 | 0 | 0.01 | 698 | 26.01 | 0.93 | 698 | 3.63 |
| 1 | 0.01 | 3155 | 20.88 | 0.93 | 3155 | 3.92 | |
| 7 | 0.01 | 9024 | 15.05 | 0.91 | 9024 | 2.82 | |
| 14 | 0.02 | 12,900 | 14.85 | 0.91 | 12,900 | 4.54 | |
| 21 | 0.02 | 12,360 | 14.40 | 0.91 | 12,360 | 6.10 | |
| 30 | 0.02 | 12,380 | 13.83 | 0.91 | 12,380 | 6.50 | |
| A36 | 0 | 0.01 | 262 | 73.95 | 0.91 | 262 | 1.96 |
| 1 | 0.02 | 1156 | 63.33 | 0.92 | 1156 | 2.06 | |
| 7 | 0.02 | 2226 | 56.45 | 0.92 | 2226 | 4.92 | |
| 14 | 0.03 | 4268 | 51.26 | 0.89 | 4268 | 10.44 | |
| 21 | 0.03 | 3268 | 59.07 | 0.89 | 3268 | 13.59 | |
| 30 | 0.04 | 3186 | 58.32 | 0.89 | 3186 | 12.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonfil, D.; Veleva, L.; Escalante-Garcia, J.I. Effect of a Hybrid Pumice–Portland Cement Extract on Corrosion Activity of Stainless Steel SS304 and Carbon Mild Steel A36. Materials 2024, 17, 2255. https://doi.org/10.3390/ma17102255
Bonfil D, Veleva L, Escalante-Garcia JI. Effect of a Hybrid Pumice–Portland Cement Extract on Corrosion Activity of Stainless Steel SS304 and Carbon Mild Steel A36. Materials. 2024; 17(10):2255. https://doi.org/10.3390/ma17102255
Chicago/Turabian StyleBonfil, David, Lucien Veleva, and Jose Ivan Escalante-Garcia. 2024. "Effect of a Hybrid Pumice–Portland Cement Extract on Corrosion Activity of Stainless Steel SS304 and Carbon Mild Steel A36" Materials 17, no. 10: 2255. https://doi.org/10.3390/ma17102255
APA StyleBonfil, D., Veleva, L., & Escalante-Garcia, J. I. (2024). Effect of a Hybrid Pumice–Portland Cement Extract on Corrosion Activity of Stainless Steel SS304 and Carbon Mild Steel A36. Materials, 17(10), 2255. https://doi.org/10.3390/ma17102255