
Role of Native Defects in Fe-Doped β-Ga2O3

 ,
,
Abstract
:1. Introduction
2. Calculation Methods
2.1. Computational Details
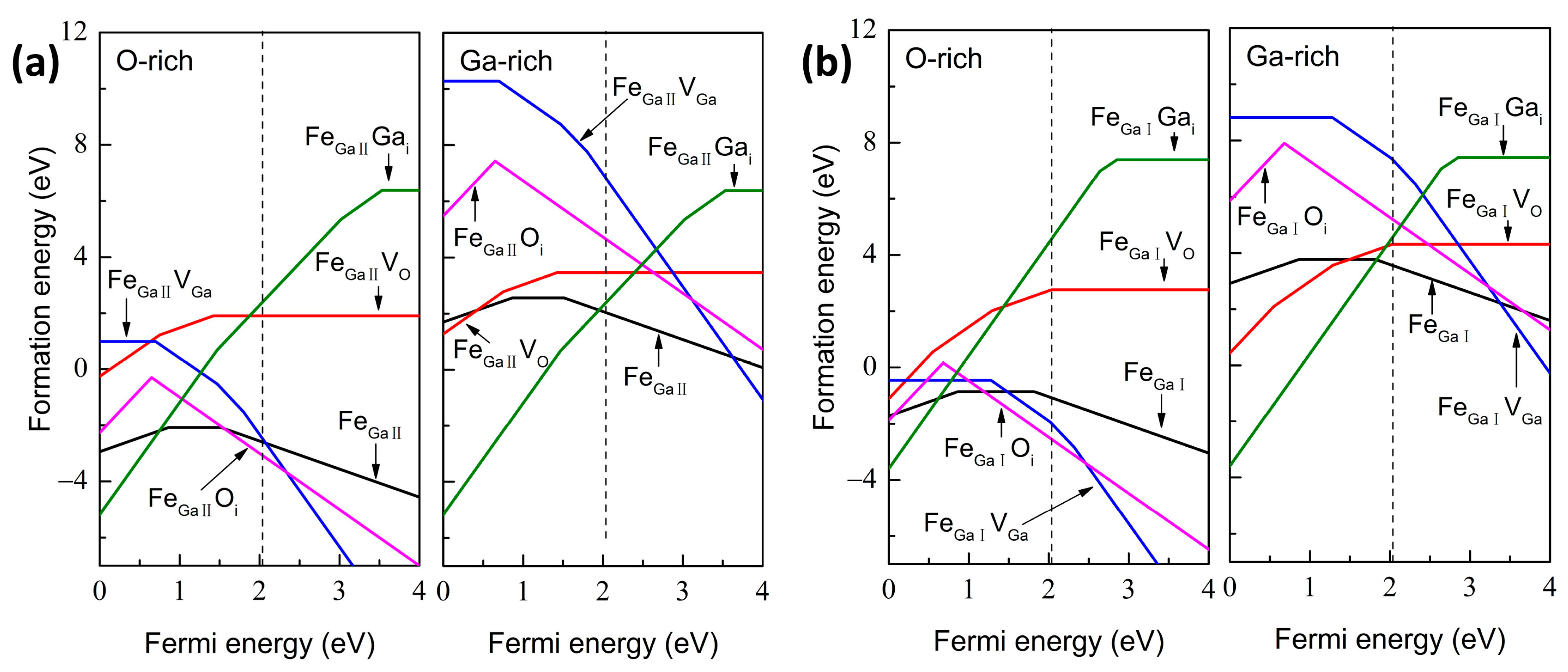
2.2. Formation Energies, Transitional Levels and Optical Calculations
3. Results and Discussions
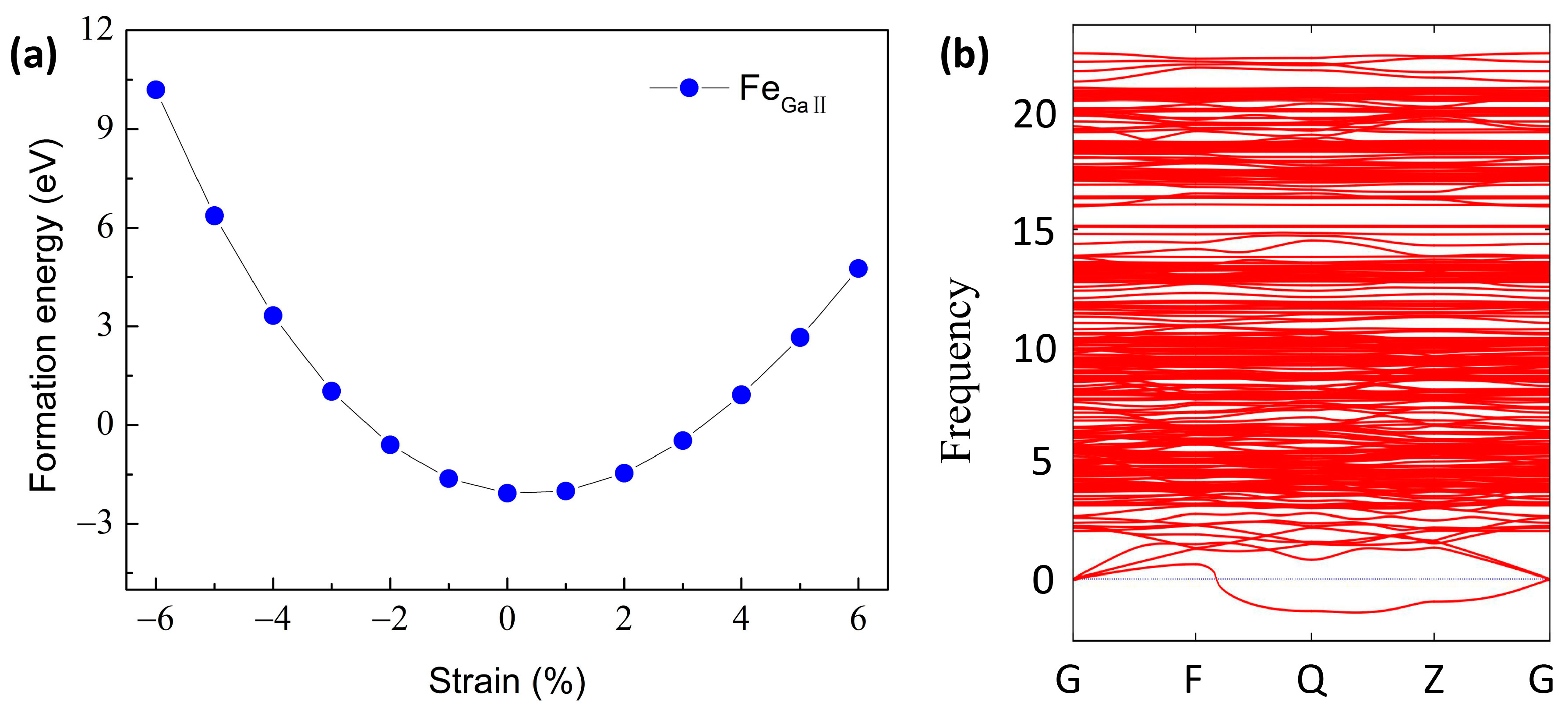
3.1. Structural Stability
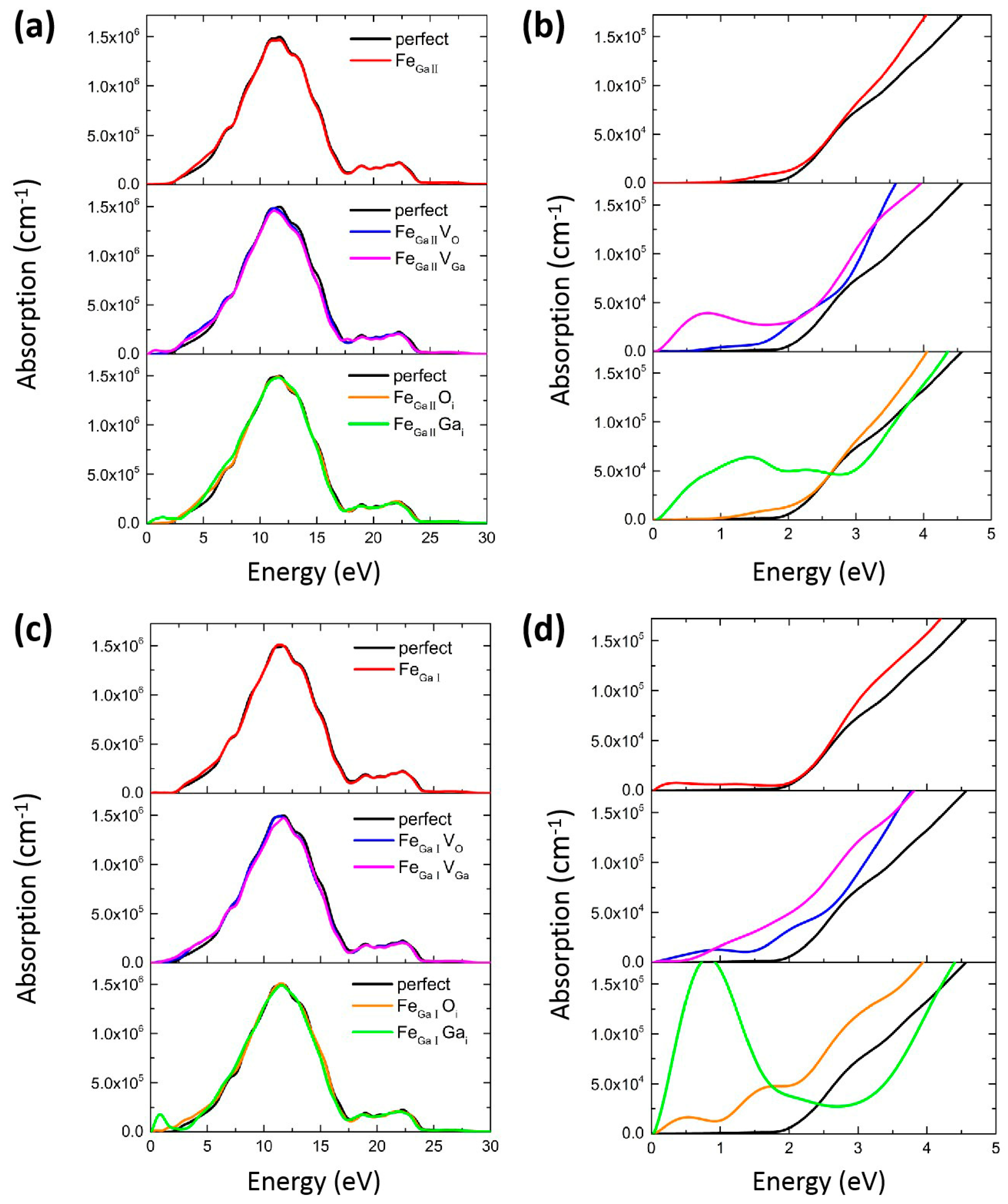
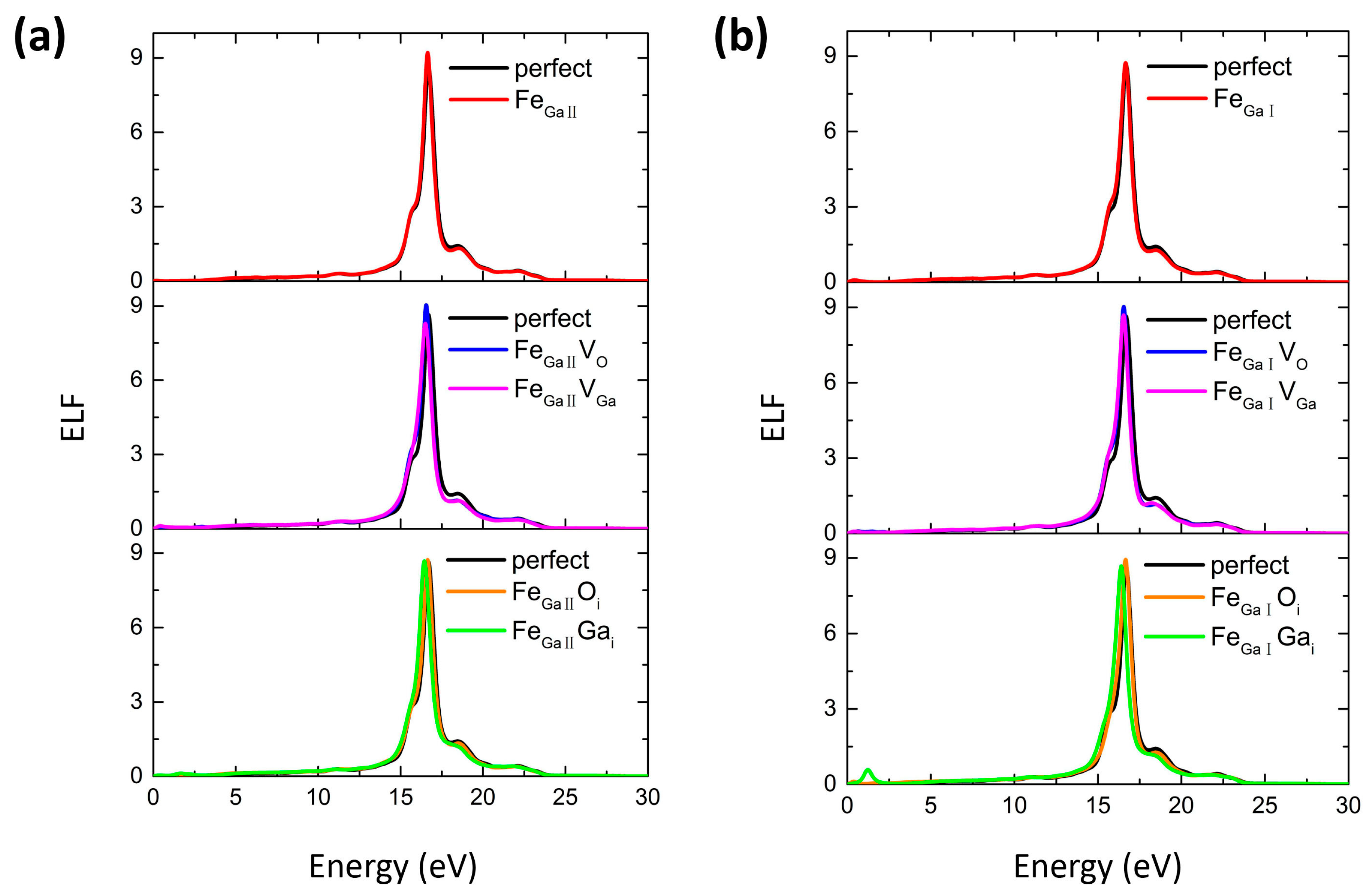
3.2. Optical Property
3.3. GGA + U
3.4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tang, R.; Li, G.; Li, C.; Li, J.; Zhang, Y.; Huang, K.; Ye, J.; Li, C.; Kang, J.Y.; Zhang, R.; et al. Localized surface plasmon enhanced Ga2O3 solar blind photodetectors. Opt. Express 2020, 28, 5731–5740. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.X.; Wu, Z.Y.; Ma, C.C.; Deng, J.N.; Zhang, H.; Xu, Y.; Ye, J.D.; Fang, Z.L.; Zhang, G.Q.; Kang, J.Y.; et al. P-type β-Ga2O3 metal-semiconductor-metal solar-blind photodetectors with extremely high responsivity and gain-bandwidth product. Mater. Today Phys. 2020, 14, 100226. [Google Scholar] [CrossRef]
- Tadjer, M.J. Toward gallium oxide power electronics. Science 2022, 378, 724–725. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Dong, P.; Dang, K.; Zhang, Y.; Yan, Q.; Xiang, H.; Su, J.; Liu, Z.; Si, M.; Gao, J.; et al. Ultra-wide bandgap semiconductor Ga2O3 power diodes. Nat. Commun. 2022, 13, 3900. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Ito, S.; Tsukazaki, A. Electric dipole effect in PdCoO2/β-Ga2O3 Schottky diodes for high-temperature operation. Sci. Adv. 2019, 5, eaax5733. [Google Scholar] [CrossRef]
- Harada, T.; Tsukazaki, A. Dynamic characteristics of PdCoO2/β-Ga2O3 Schottky junctions. Appl. Phys. Lett. 2020, 116, 232104. [Google Scholar] [CrossRef]
- Pang, R.; Teramura, K.; Morishita, M.; Asakura, H.; Hosokawa, S.; Tanaka, T. Enhanced CO evolution for photocatalytic conversion of CO2 by H2O over Ca modified Ga2O3. Commun. Chem. 2020, 3, 137. [Google Scholar] [CrossRef]
- Kyrtsos, A.; Matsubara, M.; Bellotti, E. Migration mechanisms and diffusion barriers of vacancies in Ga2O3. Phys. Rev. B 2017, 95, 245202. [Google Scholar] [CrossRef]
- Deák, P.; Ho, Q.D.; Seemann, F.; Aradi, B.; Lorke, M.; Frauenheim, T. Choosing the correct hybrid for defect calculations: A case study on intrinsic carrier trapping in β-Ga2O3. Phys. Rev. B 2017, 95, 075208. [Google Scholar] [CrossRef]
- Varley, J.B.; Weber, J.R.; Janotti, A.; Van de Walle, C.G. Oxygen vacancies and donor impurities in β-Ga2O3. Appl. Phys. Lett. 2010, 97, 142106. [Google Scholar] [CrossRef]
- Jiang, Q.; Meng, J.; Shi, Y.; Yin, Z.; Chen, J.; Zhang, J.; Wu, J.; Zhang, X. Electrical and optical properties of hydrogen plasma treated β-Ga2O3 thin films. J. Semicond. 2022, 43, 092802. [Google Scholar] [CrossRef]
- Simon, J.; Protasenko, V.; Lian, C.; Xing, H.; Jena, D. Polarization-Induced Hole Doping in Wide-Band-Gap Uniaxial Semiconductor Heterostructures. Science 2010, 327, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Werner, P.; Casula, M.; Miyake, T.; Aryasetiawan, F.; Millis, A.J.; Biermann, S. Satellites and large doping and temperature dependence of electronic properties in hole-doped BaFe2As2. Nat. Phys. 2012, 8, 331–337. [Google Scholar] [CrossRef]
- Zeng, H.; Wu, M.; Wang, H.Q.; Zheng, J.C.; Kang, J.Y. Tuning the Magnetism in Boron-Doped Strontium Titanate. Materials 2020, 13, 5686. [Google Scholar] [CrossRef] [PubMed]
- Euvrard, J.; Yan, Y.; Mitzi, D.B. Electrical doping in halide perovskites. Nat. Rev. Mater. 2021, 6, 531–549. [Google Scholar] [CrossRef]
- Zeng, H.; Wu, M.; Wang, H.Q.; Zheng, J.C.; Kang, J.Y. Tuning the magnetic and electronic properties of strontium titanate by carbon doping. Front. Phys. 2021, 16, 43501. [Google Scholar] [CrossRef]
- Lenyk, C.A.; Gustafson, T.D.; Halliburton, L.E.; Giles, N.C. Deep donors and acceptors in β-Ga2O3 crystals Determination of the Fe2+/3+ level by a noncontact method. J. Appl. Phys. 2019, 126, 245701. [Google Scholar] [CrossRef]
- Gunsser, W.; Rohwer, K. Determination of the correlation between the crystal field axis system and the crystallographic axes in chromium-doped β-Ga2O3 by EPR. Phys. Status Solidi B 1983, 116, 275–278. [Google Scholar] [CrossRef]
- Zhang, N.; Liu, H.; Sai, Q.; Shao, C.; Xia, C.; Wan, L.; Feng, Z.C.; Mohamed, H.F. Structural and electronic characteristics of Fe-doped β-Ga2O3 single crystals and the annealing effects. J. Mater. Sci. 2021, 56, 13178–13189. [Google Scholar] [CrossRef]
- Tbooster, J.M.; Dymanus, A. Mossbauer Mössbauer effect in Ga2-xFexO3 and related compounds. Phys. Status Solidi B 1967, 24, 487–499. [Google Scholar] [CrossRef]
- Büscher, R.; Lehmann, G. Correlation of zero-field splittings and site distortions. IX. Fe3+ and Cr3+ in β-Ga2O3. Z. Naturforsch. A 1987, 42, 67–71. [Google Scholar] [CrossRef]
- Bhandari, S.; Zvanut, M.E.; Varley, J.B. Optical absorption of Fe in doped Ga2O3. J. Appl. Phys. 2019, 126, 165703. [Google Scholar] [CrossRef]
- Zhou, L.; Chen, L.; Ruan, J.; Lu, X.; Liu, B.; Gao, R.; Li, Y.; Geng, L.; Ouyang, X. Pulsed X-ray detector based on Fe doped β-Ga2O3 single crystal. J. Phys. D Appl. Phys. 2021, 54, 274001. [Google Scholar] [CrossRef]
- Hany, I.; Yang, G.; Zhou, C.E.; Sun, C.; Gundogdu, K.; Seyitliyev, D.; Danilov, E.O.; Castellano, F.N.; Sun, D.; Vetter, E. Low temperature cathodoluminescence study of Fe-doped β-Ga2O3. Mater. Lett. 2019, 257, 126744. [Google Scholar] [CrossRef]
- Ingebrigtsen, M.E.; Varley, J.B.; Kuznetsov, A.Y.; Svensson, B.G.; Alfieri, G.; Mihaila, A.; Badstübner, U.; Vines, L. Iron and intrinsic deep level states in Ga2O3. Appl. Phys. Lett. 2018, 112, 04210. [Google Scholar] [CrossRef]
- Polyakov, A.Y.; Smirnov, N.B.; Schemerov, I.V.; Chernykh, A.V.; Yakimov, E.B.; Kochkova, A.I.; Tereshchenko, A.N.; Pearton, S.J. Electrical Properties, Deep Levels and Luminescence Related to Fe in Bulk Semi-Insulating β-Ga2O3 Doped with Fe. ECS J. Solid State Sci. Technol. 2019, 8, Q3091–Q3096. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 169–186. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Dong, L.; Jia, R.; Li, C.; Xin, B.; Zhang, Y. Ab initio study of N-doped β-Ga2O3 with intrinsic defects: The structural, electronic and optical properties. J. Alloys Compd. 2017, 712, 379–385. [Google Scholar] [CrossRef]
- Zacherle, T.; Schmidt, P.C.; Martin, M. Ab initio calculations on the defect structure of β-Ga2O3. Phys. Rev. B 2013, 87, 235206. [Google Scholar] [CrossRef]
- Dong, L.; Jia, R.; Xin, B.; Peng, B.; Zhang, Y. Effects of oxygen vacancies on the structural and optical properties of β-Ga2O3. Sci. Rep. 2017, 7, 40160. [Google Scholar] [CrossRef] [PubMed]
- Muratahan, A.; Wolverton, C. Local environment dependent GGA + Umethod for accurate thermochemistry of transition metal compounds. Phys. Rev. B 2014, 90, 115105. [Google Scholar]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Cryst. 2011, 4, 1272–1276. [Google Scholar] [CrossRef]
- Goyal, A.; Gorai, P.; Peng, H.; Lany, S.; Stevanović, V. A computational framework for automation of point defect calculations. Comput. Mater. Sci. 2017, 130, 1–9. [Google Scholar] [CrossRef]
- Kobayashi, T.; Gake, T.; Kumagai, Y.; Oba, F.; Matsushita, Y.-I. Energetics and electronic structure of native point defects in α-Ga2O3. Appl. Phys. Express 2019, 12, 091001. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Sun, D.; Gao, Y.; Xue, J.; Zhao, J. Defect stability and electronic structure of doped β-Ga2O3: A comprehensive ab initio study. J. Alloys Compd. 2019, 794, 374–384. [Google Scholar] [CrossRef]
- Mondal, A.K.; Mohamed, M.A.; Ping, L.K.; Mohamad Taib, M.F.; Samat, M.H.; Mohammad Haniff, M.A.S.; Bahru, R. First-principles studies for electronic structure and optical properties of p-type calcium doped α-Ga2O3. Materials 2021, 14, 604. [Google Scholar] [CrossRef]
- Kean Ping, L.; Mohamed, M.A.; Kumar Mondal, A.; Mohamad Taib, M.F.; Samat, M.H.; Berhanuddin, D.D.; Menon, P.S.; Bahru, R. First-Principles Studies for Electronic Structure and Optical Properties of Strontium Doped β-Ga2O3. Micromachines 2021, 12, 348. [Google Scholar] [CrossRef] [PubMed]
- Usseinov, A.; Platonenko, A.; Koishybayeva, Z.; Akilbekov, A.; Zdorovets, M.; Popov, A.I. Pair vacancy defects in β-Ga2O3 crystal: Ab initio study. Opt. Mater. X 2022, 16, 100200. [Google Scholar] [CrossRef]
- Mykhaylyk, V.B.; Kraus, H.; Kapustianyk, V.; Rudko, M. Low temperature scintillation properties of Ga2O3. Appl. Phys. Lett. 2019, 115, 081103. [Google Scholar] [CrossRef]
- Yan, H.; Guo, Y.; Song, Q.; Chen, Y.; Shi, Y. Electronic Structure and Magnetic Interactions in Ti-Doped and Ti-VO-Co-Doped β-Ga2O3 from First-Principles Calculations. J. Supercond. Nov. Magn. 2016, 29, 2607–2613. [Google Scholar] [CrossRef]
- Ao, L.; Pham, A.; Xiang, X.; Li, S.; Zu, X. Defect induced charge trapping in C-doped α-Al2O3. J. Appl. Phys. 2017, 122, 025702. [Google Scholar] [CrossRef]
- Goyal, A.; Gorai, P.; Toberer, E.S.; Stevanović, V. First-principles calculation of intrinsic defect chemistry and self-doping in PbTe. NPJ Comput. Mater. 2017, 3, 42. [Google Scholar] [CrossRef]
- Wu, Z.-j.; Zhao, E.-j.; Xiang, H.-p.; Hao, X.-f.; Liu, X.-j.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 2007, 76, 054115. [Google Scholar] [CrossRef]
- Osipov, A.V.; Grashchenko, A.S.; Kukushkin, S.A.; Nikolaev, V.I.; Osipova, E.V.; Pechnikov, A.I.; Soshnikov, I.P. Structural and elastoplastic properties of β-Ga2O3 films grown on hybrid SiC/Si substrates. Contin. Mech. Thermodyn. 2018, 30, 1059–1068. [Google Scholar] [CrossRef]
- Usseinov, A.; Koishybayeva, Z.; Platonenko, A.; Pankratov, V.; Suchikova, Y.; Akilbekov, A.; Zdorovets, M.; Purans, J.; Popov, A.I. Vacancy Defects in Ga2O3: First-Principles Calculations of Electronic Structure. Materials 2021, 14, 7384. [Google Scholar] [CrossRef]
- Miller, W.; Böttcher, K.; Galazka, Z.; Schreuer, J. Numerical Modelling of the Czochralski Growth of β-Ga2O3. Crystals 2017, 7, 26. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, T.; Zhang, Y.; Qi, C.; Luan, J.; Ma, G.; Tsai, H.-S.; Liu, C.; Huo, M. Effects of carbon related defects on opto-electronic properties of β-Ga2O3: The first principle calculation. J. Appl. Phys. 2020, 17, 103060. [Google Scholar] [CrossRef]
- Yan, H.; Guo, Y.; Song, Q.; Chen, Y. First-principles study on electronic structure and optical properties of Cu-doped β-Ga2O3. Phys. B 2014, 434, 181–184. [Google Scholar] [CrossRef]
- Pan, Y. First-principles investigation of the influence of point defect on the electronic and optical properties of α-Ga2O3. Int. J. Energy Res. 2022, 46, 13070–13078. [Google Scholar] [CrossRef]
- He, H.; Orlando, R.; Blanco, M.A.; Pandey, R.; Amzallag, E.; Baraille, I.; Rérat, M. First-principles study of the structural, electronic, and optical properties of Ga2O3 in its monoclinic and hexagonal phases. Phys. Rev. B 2006, 74, 195123. [Google Scholar] [CrossRef]
- Zachinskis, A.; Grechenkov, J.; Butanovs, E.; Platonenko, A.; Piskunov, S.; Popov, A.I.; Purans, J.; Bocharov, D. Ir impurities in α- and β-Ga2O3 and their detrimental effect on p-type conductivity. Sci. Rep. 2023, 13, 8522. [Google Scholar] [CrossRef]
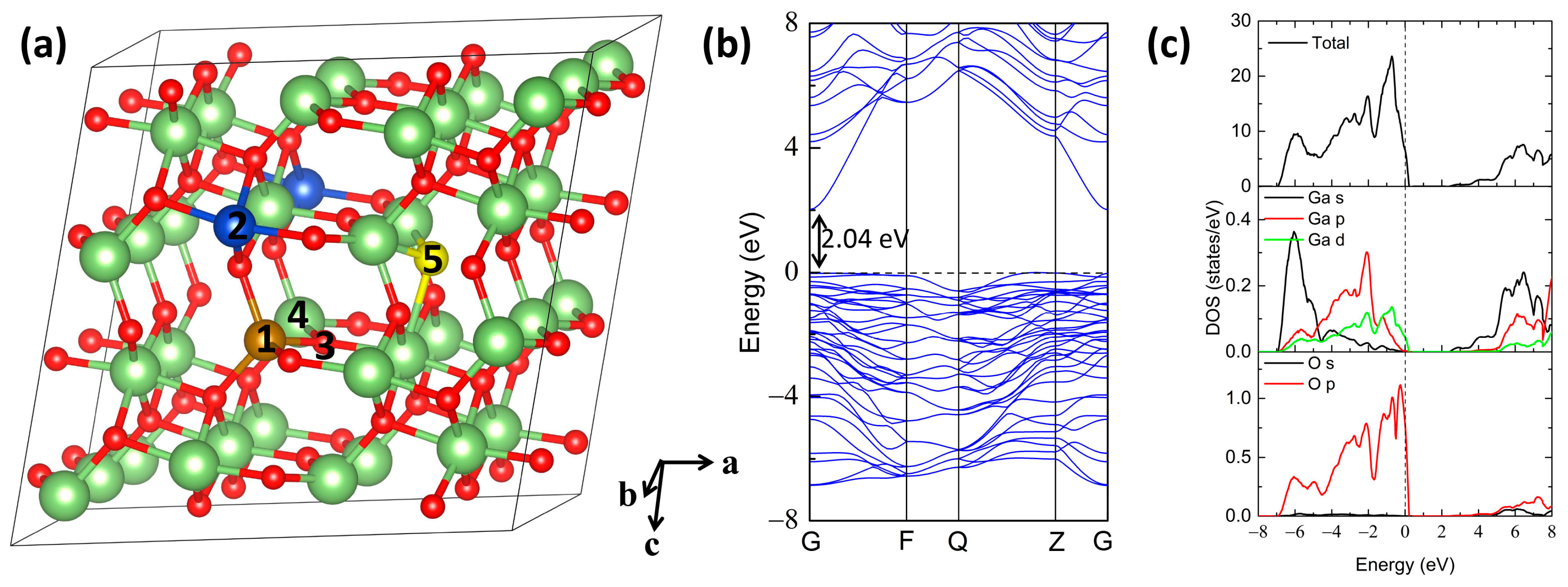
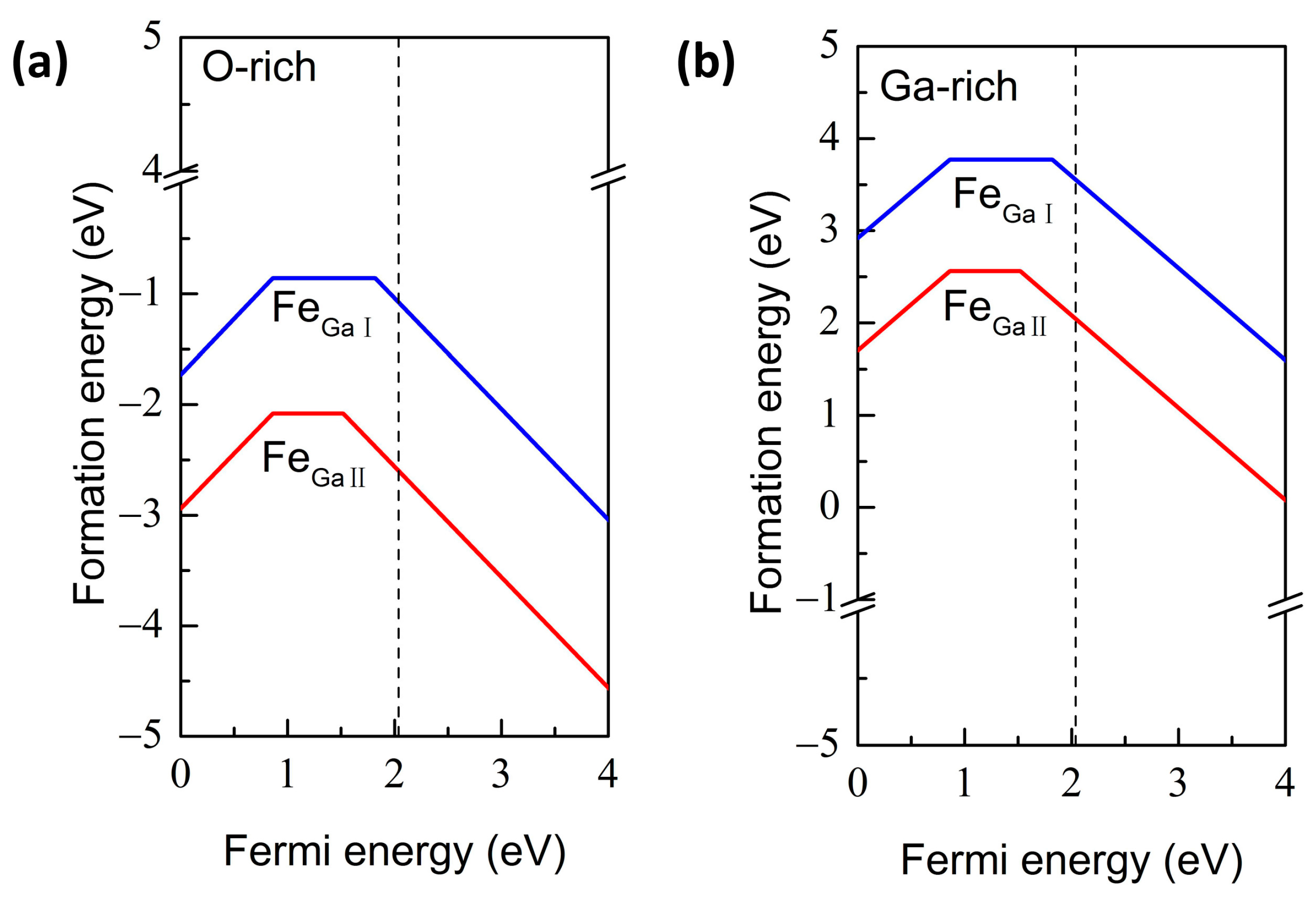

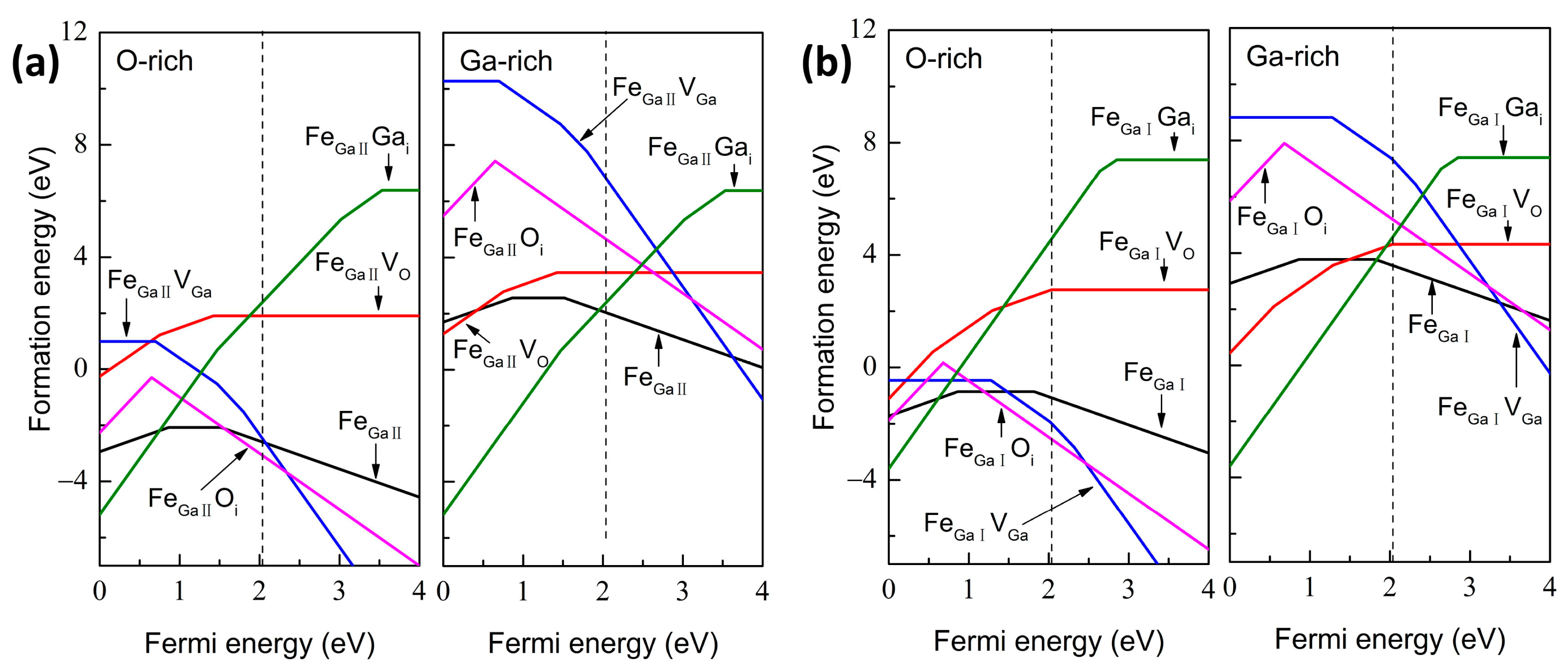
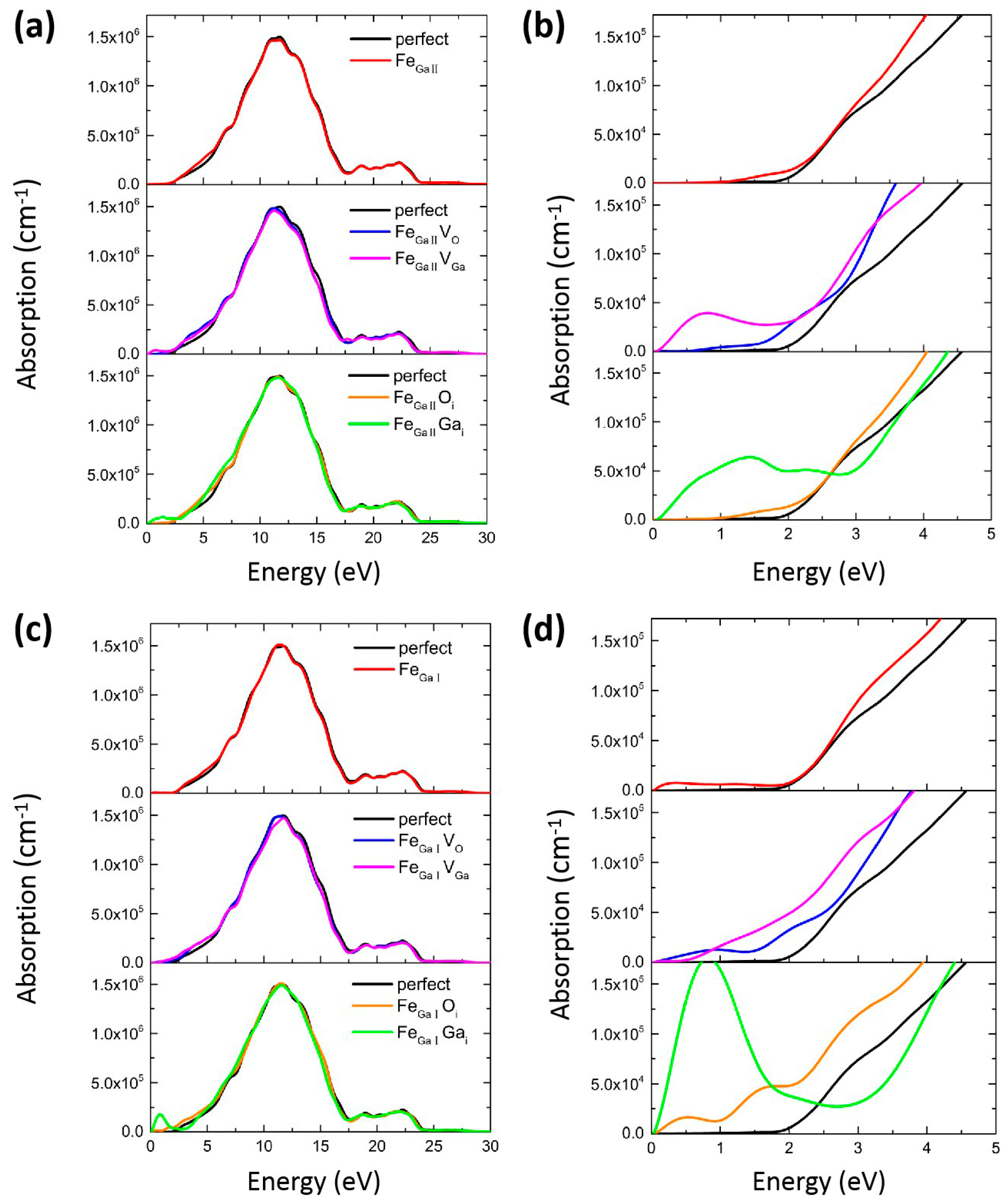
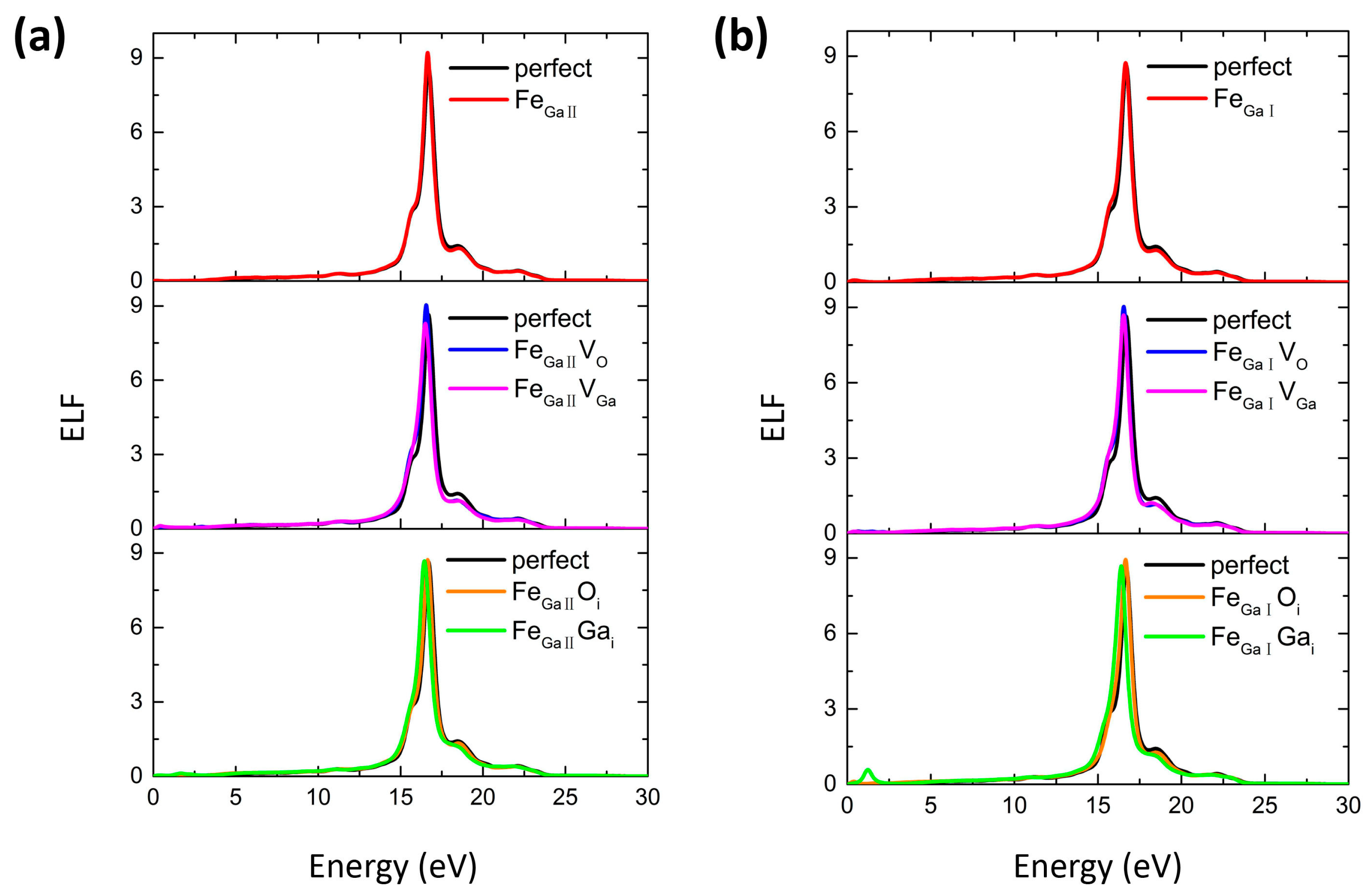

{kind=link}
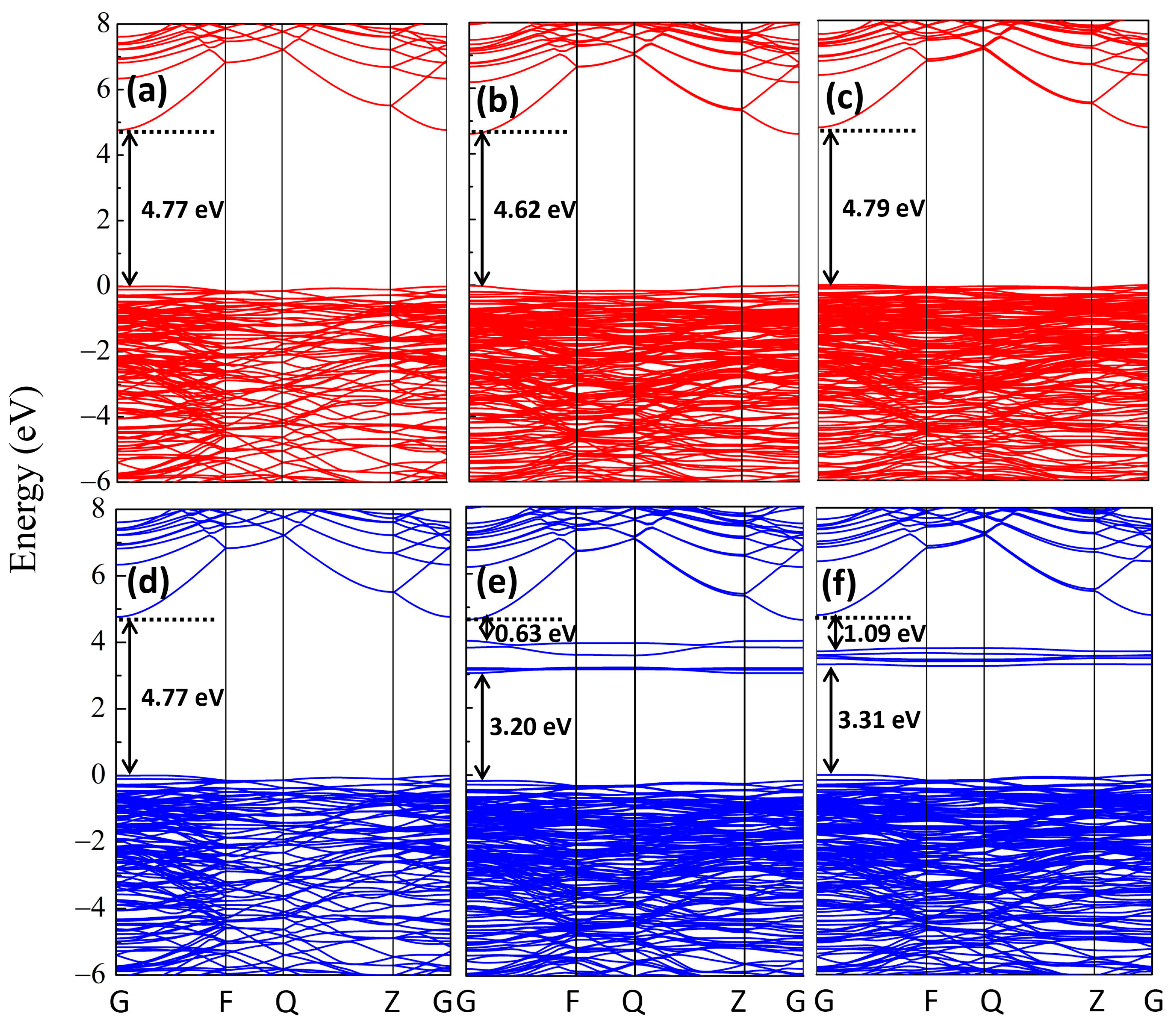
{kind=link}
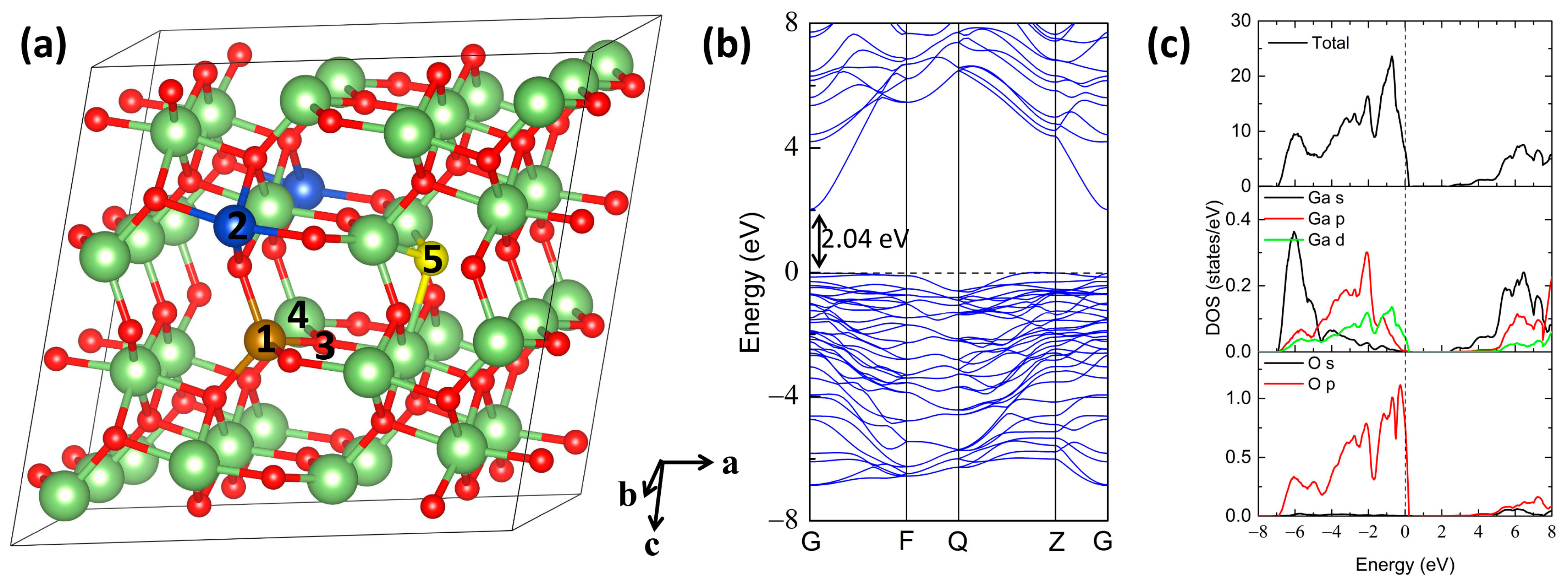
{kind=link}
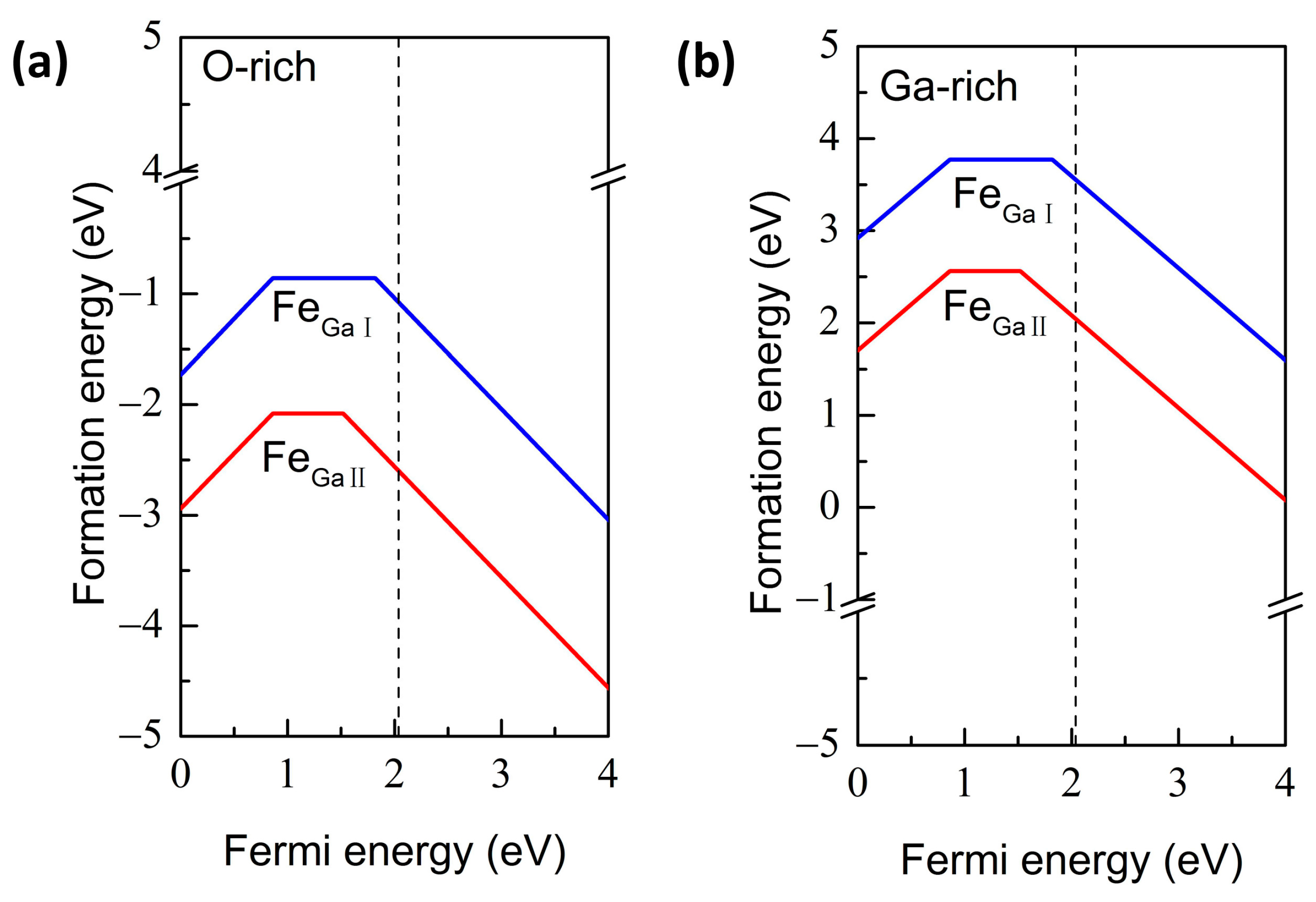
{kind=link}
{kind=link}
{kind=link}
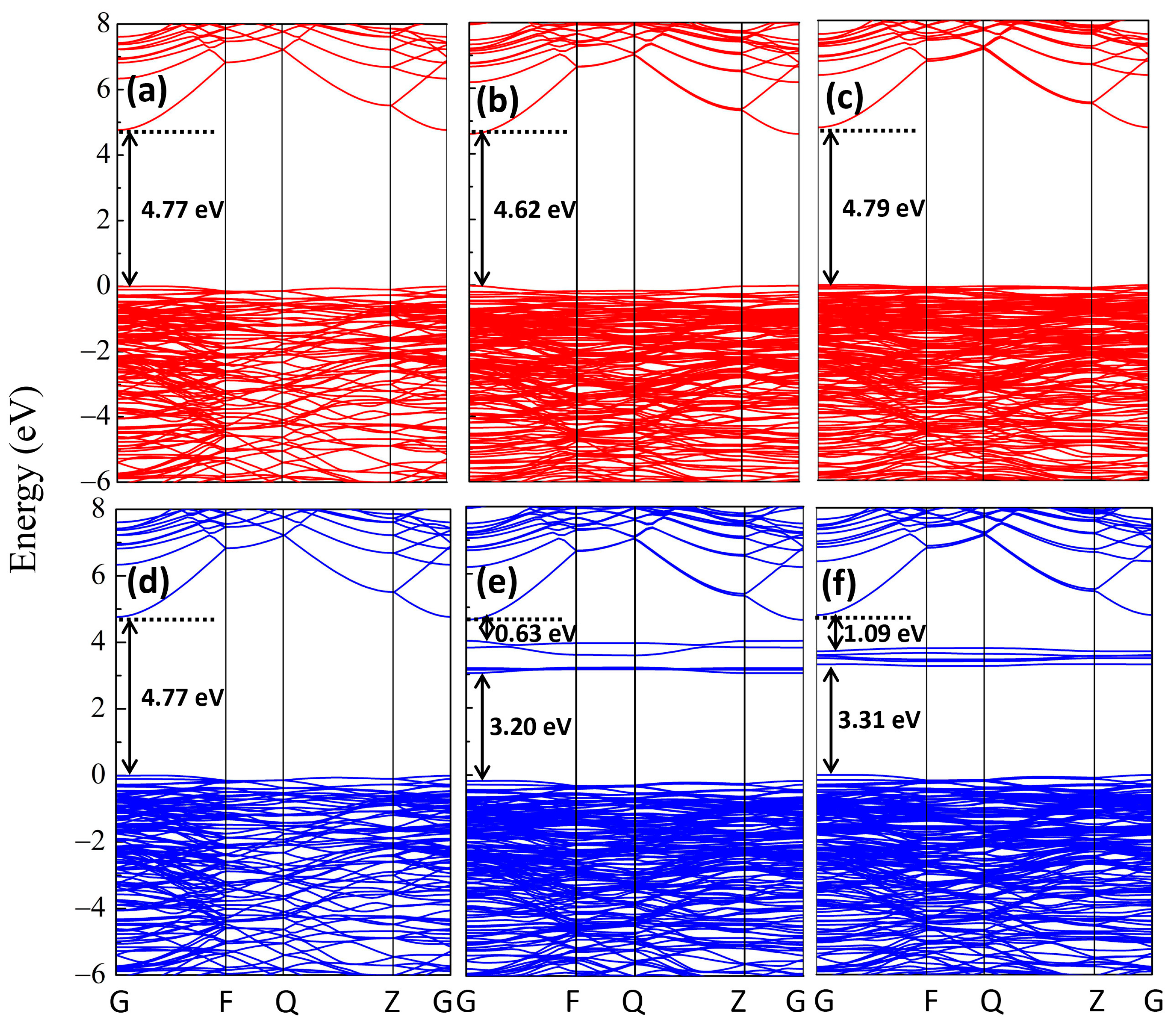{kind=link}
| Lattice Constants | Perfect (This Work) | Perfect (Literature) | FeGaI | FeGaΠ |
|---|---|---|---|---|
| a (Å) | 12.412 | 12.494 [41]/12.28 [42]/12.214 [43] | 12.387 (−0.20%) | 12.393 (−0.15%) |
| b (Å) | 3.076 | 3.096 [41]/3.05 [42]/3.037 [43] | 3.076 (0.02%) | 3.075 (−0.05%) |
| c (Å) | 5.872 | 5.898 [41]/5.82 [42]/5.798 [43] | 5.888 (0.28%) | 5.868 (−0.07%) |
| β (°) | 103.702 | 103.705 [41]/103.83 [43] | 103.835 (0.13%) | 103.700 (0%) |
| C11 | C12 | C13 | C15 | C22 | C23 | C25 | C33 | ||
|---|---|---|---|---|---|---|---|---|---|
| Perfect | This work (PBE) | 215 | 109 | 119 | –15 | 317 | 72 | 14 | 312 |
| PBE [48] | 208 | 118 | 146 | 0 | 335 | 83 | 0 | 318 | |
| B3LYP [49] | 235 | 124 | 138 | −13 | 357 | 76 | 7 | 357 | |
| Exp [50] | 238 | 130 | 152 | −4 | 359 | 78 | 2 | 346 | |
| FeGaΠ | 217 | 107 | 118 | −17 | 322 | 72 | 9 | 315 | |
| C35 | C44 | C46 | C55 | C66 | BH | EH | GH | ||
| Perfect | This work (PBE) | 6 | 48 | 14 | 64 | 94 | 159 | 188 | 72 |
| PBE [48] | 19 | 50 | 9 | 77 | 96 | 171 | 192 | 73 | |
| B3LYP [49] | 12 | 55 | 15 | 81 | 101 | 179 | 214 | 82 | |
| Exp [50] | 19 | 49 | 6 | 91 | 107 | 184 | 213 | 82 | |
| FeGaΠ | 7 | 51 | 18 | 67 | 90 | 160 | 190 | 73 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, H.; Wu, M.; Gao, H.; Wang, Y.; Xu, H.; Cheng, M.; Lin, Q. Role of Native Defects in Fe-Doped β-Ga2O3. Materials 2023, 16, 6758. https://doi.org/10.3390/ma16206758
Zeng H, Wu M, Gao H, Wang Y, Xu H, Cheng M, Lin Q. Role of Native Defects in Fe-Doped β-Ga2O3. Materials. 2023; 16(20):6758. https://doi.org/10.3390/ma16206758
Chicago/Turabian StyleZeng, Hui, Meng Wu, Haixia Gao, Yuansheng Wang, Hongfei Xu, Meijuan Cheng, and Qiubao Lin. 2023. "Role of Native Defects in Fe-Doped β-Ga2O3" Materials 16, no. 20: 6758. https://doi.org/10.3390/ma16206758
APA StyleZeng, H., Wu, M., Gao, H., Wang, Y., Xu, H., Cheng, M., & Lin, Q. (2023). Role of Native Defects in Fe-Doped β-Ga2O3. Materials, 16(20), 6758. https://doi.org/10.3390/ma16206758