
Prediction of Novel Ultrahard Phases in the B–C–N System from First Principles: Progress and Problems


Abstract
1. Introduction
2. Elements
3. Binary Compounds
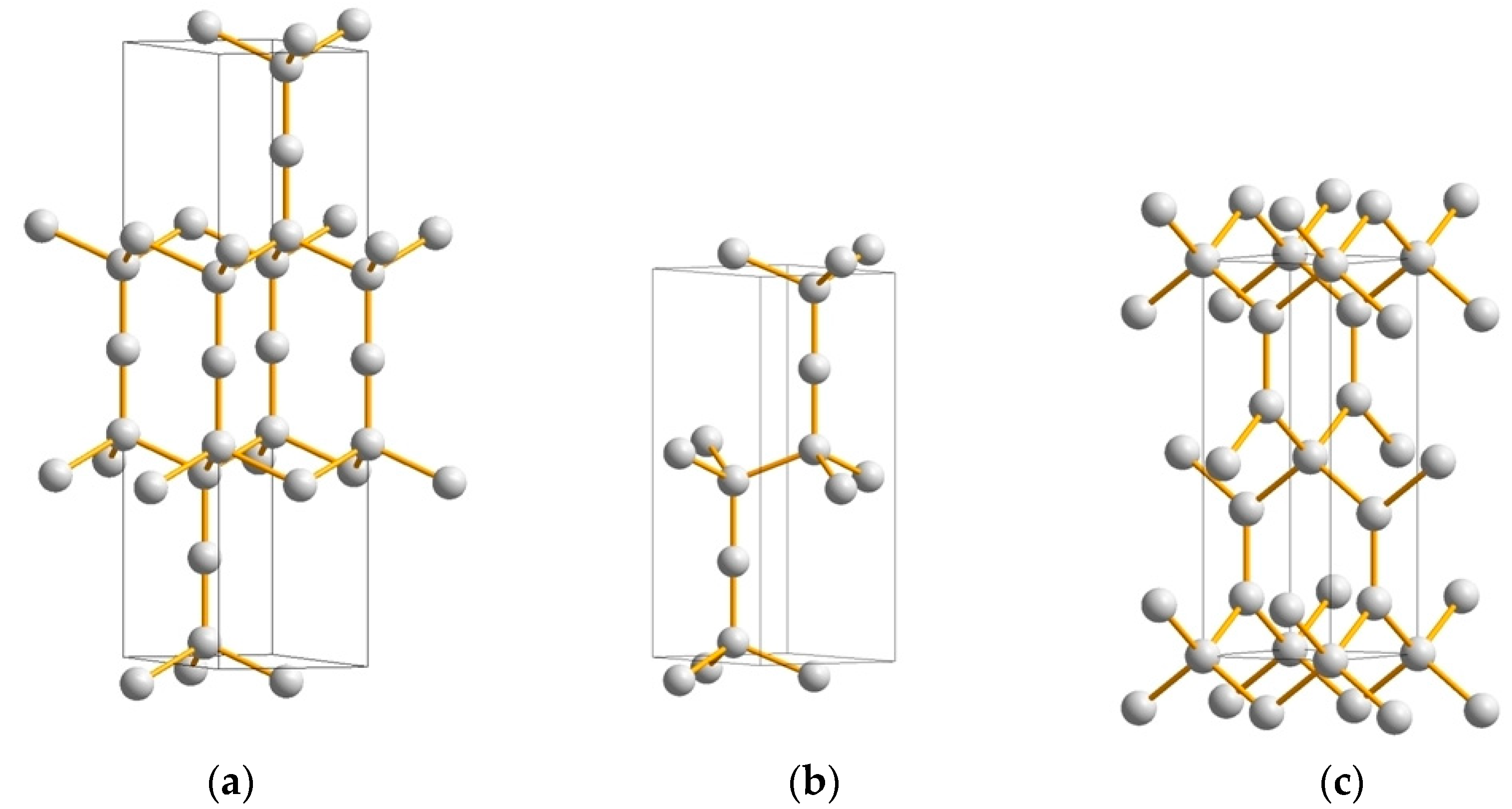
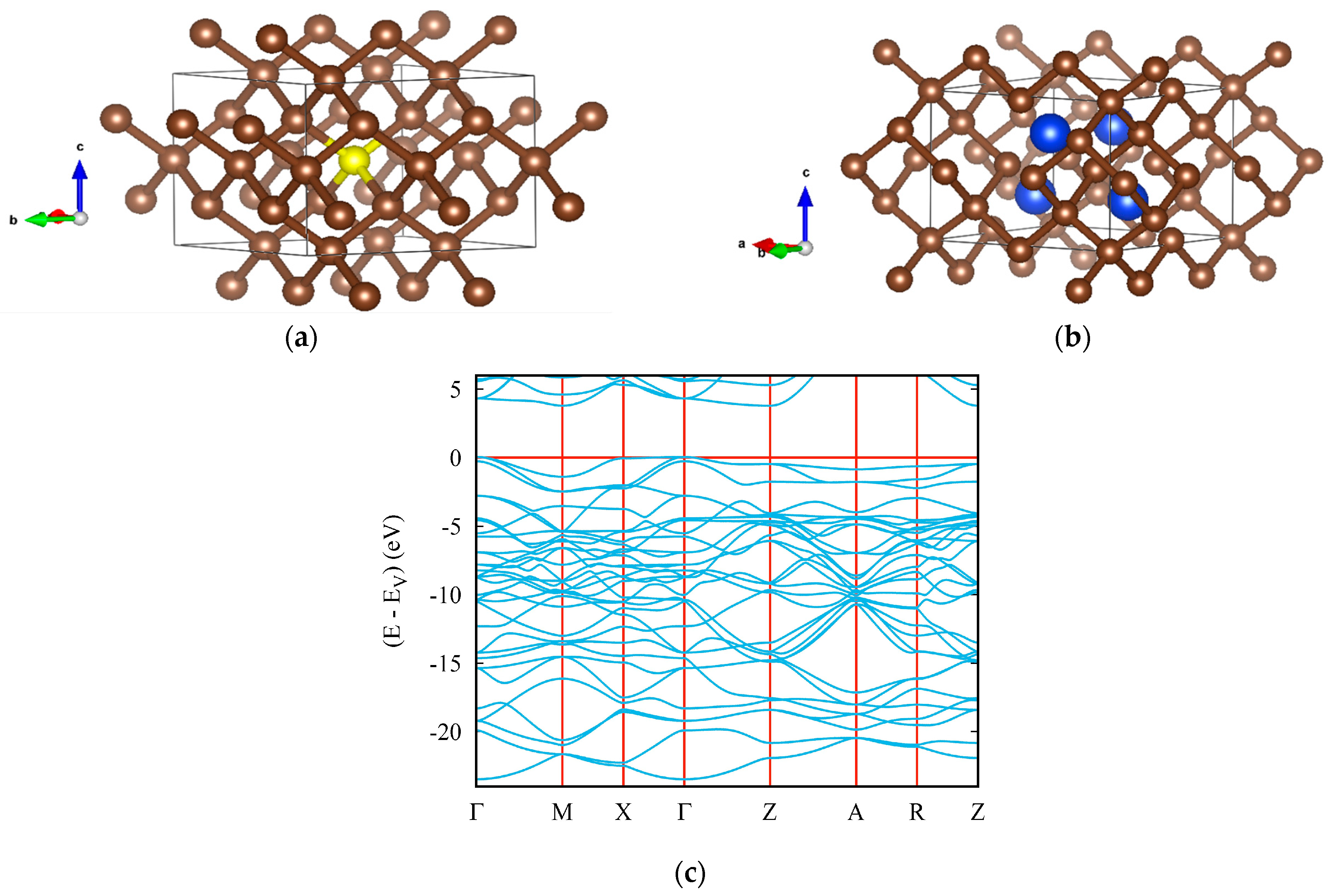
3.1. Carbon Nitrides
3.2. Boron Carbides
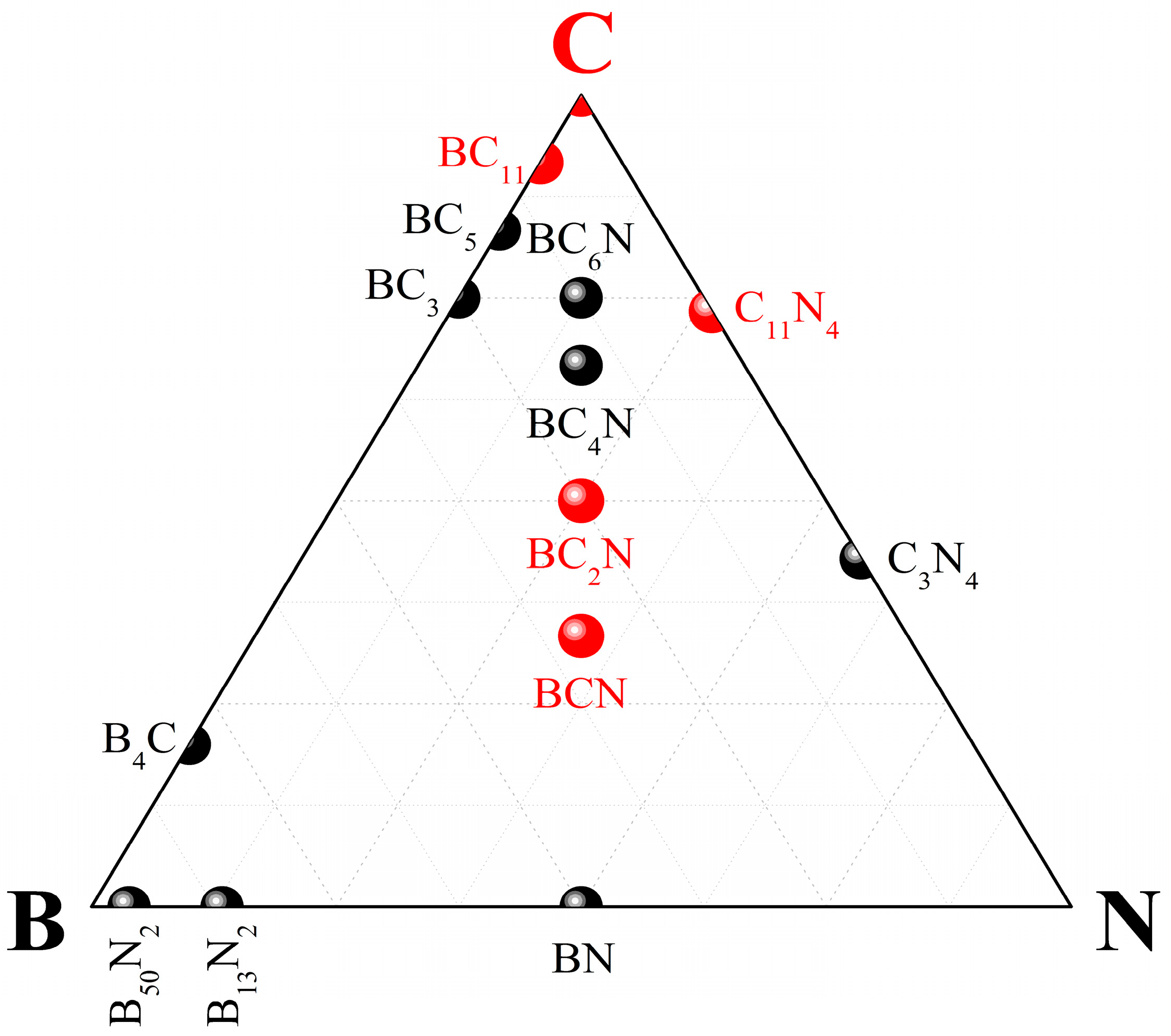
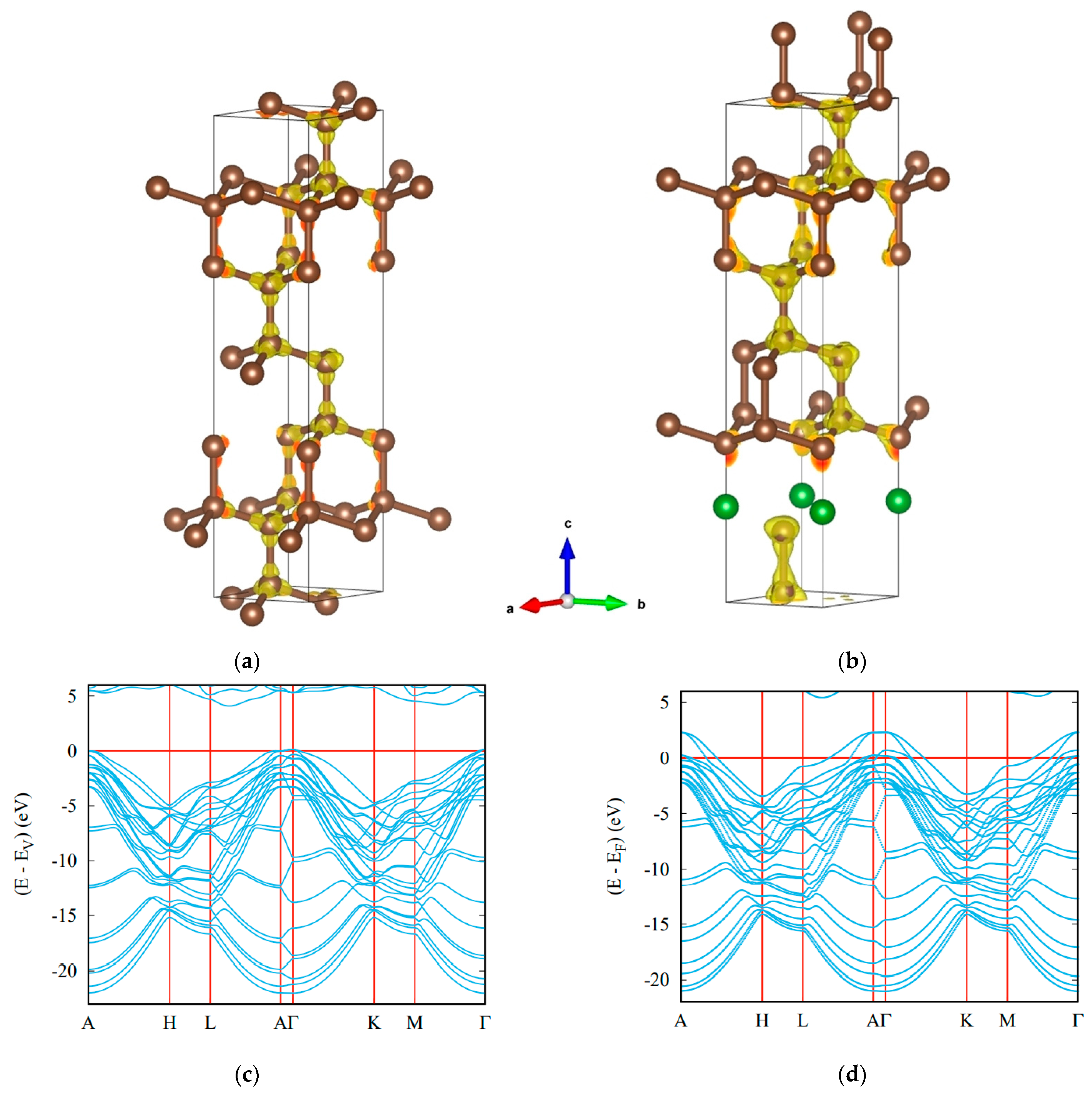
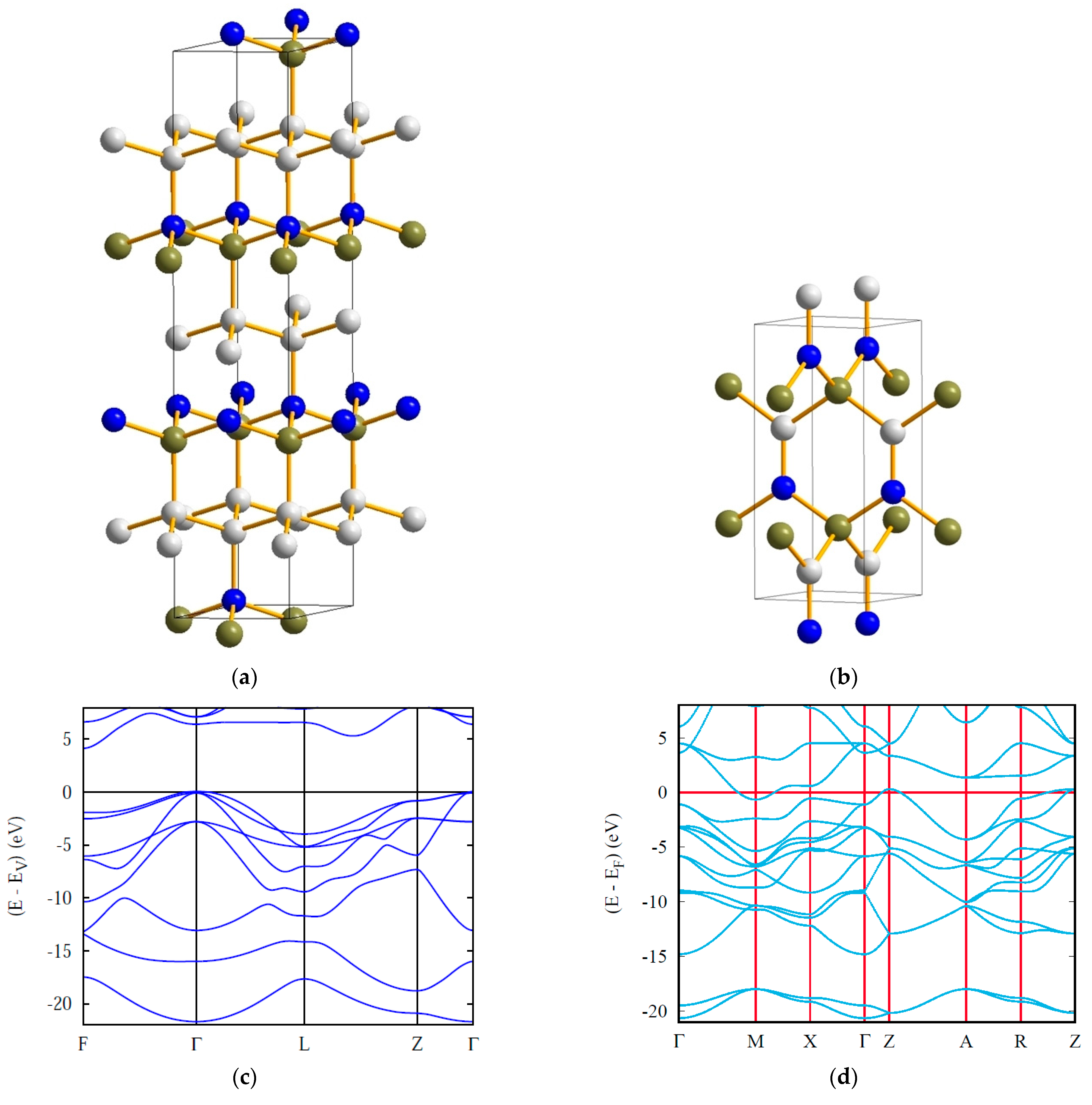
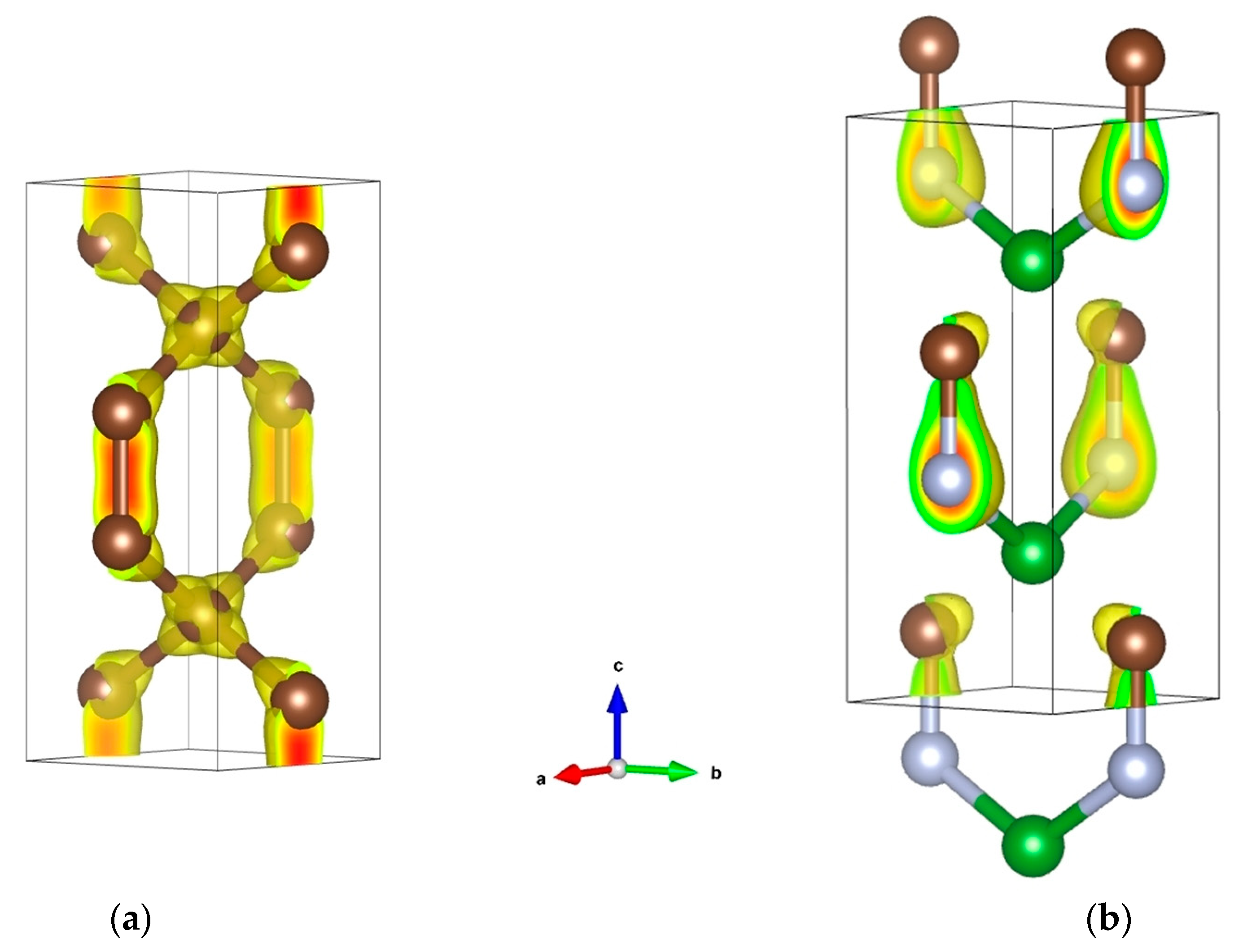
4. Ternary Compounds
5. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A. Computational Framework
References
- Bundy, F.; Hall, H.; Strong, H.; Wentorf, R.H. Man-made diamonds. Nature 1955, 176, 51–55. [Google Scholar] [CrossRef]
- Wentorf, R.H. Cubic form of boron nitride. J. Chem. Phys. 1957, 26, 956. [Google Scholar] [CrossRef]
- Solozhenko, V.L.; Andrault, D.; Fiquet, G.; Mezouar, M.; Rubie, D. Synthesis of superhard cubic BC2N. Appl. Phys. Lett. 2001, 78, 1385–1387. [Google Scholar] [CrossRef]
- Solozhenko, V.L.; Dub, S.; Novikov, N. Mechanical properties of cubic BC2N, a new superhard phase. Diam. Relat. Mater. 2001, 10, 2228–2231. [Google Scholar] [CrossRef]
- Solozhenko, V.L.; Kurakevych, O.; Le Godec, D.A.Y.; Mezouar, M. Ultimate metastable solubility of boron in diamond: Synthesis of superhard diamondlike BC5. Phys. Rev. Lett. 2009, 102, 015506. [Google Scholar] [CrossRef]
- Le Godec, Y.; Courac, A.; Solozhenko, V.L. High-pressure synthesis of superhard and ultrahard materials. J. Appl. Phys. 2019, 126, 151102. [Google Scholar] [CrossRef]
- Gao, F. Theoretical model of intrinsic hardness. Phys Rev. B 2006, 73, 132104. [Google Scholar] [CrossRef]
- Chen, X.; Niu, H.; Li, D.; Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 2011, 19, 1275–1281. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, B.; Zhao, Z. Microscopic theory of hardness and design of novel superhard crystals. Int. J. Refract. Met. Hard Mater. 2012, 33, 93–106. [Google Scholar] [CrossRef]
- Mazhnik, E.; Oganov, A. A model of hardness and fracture toughness of solids. J. Appl. Phys. 2019, 126, 125109. [Google Scholar] [CrossRef]
- Yao, H.; Ouyang, L.; Ching, W.-Y. Ab initio calculation of elastic constants of ceramic crystals. J. Am. Ceram. Soc. 2007, 90, 3194–3204. [Google Scholar] [CrossRef]
- Perger, W.; Criswell, J.; Civalleri, B.; Dovesi, R. Ab-initio calculation of elastic constants of crystalline systems with the CRYSTAL code. Comput. Phys. Commun. 2009, 180, 1753–1759. [Google Scholar] [CrossRef]
- Kvashnin, A.; Allahyari, Z.; Oganov, A. Computational discovery of hard and superhard materials. J. Appl. Phys. 2019, 126, 040901. [Google Scholar] [CrossRef]
- Avery, P.; Wang, X.; Oses, C.; Gossett, E.; Proserpio, D.; Toher, C.; Curtarolo, S.; Zurek, E. Predicting superhard materials via a machine learning informed evolutionary structure search. NPJ Comput. Mater. 2019, 5, 89. [Google Scholar] [CrossRef]
- Allahyari, Z.; Oganov, A. Coevolutionary search for optimal materials in the space of all possible compounds. NPJ Comput. Mater. 2020, 6, 55. [Google Scholar] [CrossRef]
- Zhang, S.; He, J.; Zhao, Z.; Yu, D.; Tian, Y. Discovery of superhard materials via CALYPSO methodology. Chin. Phys. B 2019, 28, 106104. [Google Scholar] [CrossRef]
- Oganov, A.; Glass, C. Crystal structure prediction using ab initio evolutionary techniques: Principles and applications. J. Chem. Phys. 2006, 124, 244704. [Google Scholar] [CrossRef]
- Glass, C.; Oganov, A.; Hansen, N. USPEX—Evolutionary crystal structure prediction. Comput. Phys. Commun. 2006, 175, 713–720. [Google Scholar] [CrossRef]
- Oganov, A.; Chen, J.; Gatti, C.; Ma, Y.; Ma, Y.; Glass, C.; Liu, Z.; Yu, T.; Kurakevych, O.; Solozhenko, V.L. Ionic high-pressure form of elemental boron. Nature 2009, 457, 863–867. [Google Scholar] [CrossRef]
- Oganov, A.; Solozhenko, V.L. Boron: A hunt for superhard polymorphs. J. Superhard Mater. 2009, 31, 285–291. [Google Scholar] [CrossRef]
- Németh, P.; Garvie, L.; Aoki, T.; Dubrovinskaia, N.; Dubrovinsky, L.; Buseck, P. Lonsdaleite is faulted and twinned cubic diamond and does not exist as a discrete material. Nat. Commun. 2014, 5, 5447. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.; Kabanov, A.; Golov, A.; Proserpio, D. Homo Citans and carbon allotropes: For an ethics of citation. Angew. Chem. Int. Ed. 2016, 55, 10962–10976, Samara Carbon Allotrope Database. Available online: http://sacada.sctms.ru (accessed on 8 December 2022). [CrossRef] [PubMed]
- Zhao, C.-X.; Niu, C.-Y.; Qin, Z.-J.; Ren, X.; Wang, J.-T.; Cho, J.-H.; Jia, Y. H18 carbon: A new metallic phase with sp2-sp3 hybridized bonding network. Sci. Rep. 2016, 6, 21879. [Google Scholar] [CrossRef] [PubMed]
- Tennyson, J. Molecules in Space. In Handbook of Molecular Physics and Quantum Chemistry; John Wiley & Sons: Chichester, UK, 2003; Volume 3, part III, p. 358. [Google Scholar]
- Matar, S.F.; Etourneau, J.; Solozhenko, V.L. First-principles investigations of tricarbon: From the isolated C3 molecule to a novel ultra-hard anisotropic solid. Carbon Trends 2021, 6, 100132. [Google Scholar] [CrossRef]
- Voigt, W. Über die Beziehung zwischen den beiden Elasticitätsconstanten isotroper Körper. Annal. Phys. 1889, 274, 573–587. [Google Scholar] [CrossRef]
- Mukhanov, V.; Kurakevych, O.; Solozhenko, V.L. The interrelation between hardness and compressibility of substances and their structure and thermodynamic properties. J. Superhard Mater. 2008, 30, 368–378. [Google Scholar] [CrossRef]
- Lyakhov, A.; Oganov, A. Evolutionary search for superhard materials: Methodology and applications to forms of carbon and TiO2. Phys. Rev. B 2011, 84, 092103. [Google Scholar] [CrossRef]
- Matar, S.F.; Solozhenko, V.L. Novel ultrahard sp2/sp3 hybrid carbon allotrope from crystal chemistry and first principles: Body-centered tetragonal C6 (′neoglitter′). ChemRxiv 2022. [Google Scholar] [CrossRef]
- Liu, A.; Wentzcovitch, R. Stability of carbon nitride solids. Phys. Rev. B 1994, 50, 10362–10365. [Google Scholar] [CrossRef]
- Teter, D.; Hemley, R. Low-compressibility carbon nitrides. Science 1996, 271, 53–55. [Google Scholar] [CrossRef]
- Yao, Y.; Tse, J.; Klug, D. Crystal and electronic structure of superhard BC5: First-principles structural optimizations. Phys. Rev. B 2009, 80, 094106. [Google Scholar] [CrossRef]
- Liu, H.; Li, Q.; Zhu, L.; Ma, Y. Superhard polymorphs of diamond-like BC7. Solid State Comm. 2011, 151, 716–719. [Google Scholar] [CrossRef]
- Matar, S.F.F. Novel trigonal BC11 as model structure of heavily doped diamond: Crystal chemistry rationale and first principles characterizations. Diam. Relat. Mater. 2022, 123, 108842. [Google Scholar] [CrossRef]
- Mattesini, M.; Matar, S.F. First-principles characterization of new ternary heterodiamond BC2N phases. Comput. Mater. Sci. 2001, 20, 107–119. [Google Scholar] [CrossRef]
- Matar, S.F.; Solozhenko, V.L. Crystal chemistry and ab initio prediction of ultrahard rhombohedral B2N2 and BC2N. Solid State Sci. 2021, 118, 106667. [Google Scholar] [CrossRef]
- Matar, S.F.; Solozhenko, V.L. Novel superhard tetragonal BCN from crystal chemistry and first principles. Materialia 2022, 26, 101581. [Google Scholar] [CrossRef]
- Ownby, P.; Yang, X.; Liu, J. Calculated X-ray diffraction data for diamond polytypes. J. Am. Ceram. Soc. 1992, 75, 1876–1883. [Google Scholar] [CrossRef]
- Bindzus, N.; Straasø, T.; Wahlberg, N.; Becker, J.; Bjerg, L.; Lock, N.; Dippel, A.-C.; Iversen, B. Experimental determination of core electron deformation in diamond. Acta Cryst. A 2014, 70, 39–48. [Google Scholar] [CrossRef]
- Brazhkin, V.; Solozhenko, V.L. Myths about new ultrahard phases: Why materials that are significantly superior to diamond in elastic moduli and hardness are impossible. J. Appl. Phys. 2019, 125, 130901. [Google Scholar] [CrossRef]
- Manyali, G.; Warmbier, R.; Quandt, A.; Lowther, J. Ab initio study of elastic properties of super hard and graphitic structures of C3N4. Comput. Mater. Sci. 2013, 69, 299–303. [Google Scholar] [CrossRef]
- Yang, X.; Liu, S.; Qin, J.Z.J.; Du, M.; Yang, D.; Li, X.; Liu, B.; Shan, C.-X. Orthorhombic C14 carbon: A novel superhard sp3 carbon allotrope. Carbon 2020, 156, 309–312. [Google Scholar] [CrossRef]
- Fan, Q.; Liu, H.; Jiang, L.; Yu, X.; Zhang, W.; Yun, S. Two orthorhombic superhard carbon allotropes: C16 and C24. Diam. Relat. Mater. 2021, 116, 108426. [Google Scholar] [CrossRef]
- Fan, Q.; Liu, H.; Yang, R.; Yu, X.; Zhang, W.; Yun, S. An orthorhombic superhard carbon allotrope: Pmma C24. J. Solid State Chem. 2021, 300, 122260. [Google Scholar] [CrossRef]
- Chen, J.; Ying, P.; Gao, Y.; Wei, X.; Li, B.; Huan, Q.; Luo, K. Orthorhombic C36: A sp2–sp3 carbon with pressure-induced metallization and superconductivity. J. Mater. Sci. 2021, 56, 17665–17673. [Google Scholar] [CrossRef]
- Solozhenko, V.L.; Le Godec, Y. A hunt for ultrahard materials. J. Appl. Phys. 2019, 126, 230401. [Google Scholar] [CrossRef]
- Wei, Q.; Zhang, Q.; Zhang, M.-G.; Yan, H.-Y.; Guo, L.-X.; Wei, B. A novel hybrid sp-sp2 metallic carbon allotrope. Front. Phys. 2018, 13, 136105. [Google Scholar] [CrossRef]
- Bucknum, M.; Hoffmann, R. A hypothetical dense 3,4-connected carbon net and related B2C and CN2 nets built from 1,4-cyclohexadienoid units. J. Am. Chem. Soc. 1994, 116, 11456–11464. [Google Scholar] [CrossRef]
- Kurakevych, O.; Solozhenko, V.L. Rhombohedral boron subnitride, B13N2, by X-ray powder diffraction. Acta Crystallogr. C 2007, 63, i80–i82. [Google Scholar] [CrossRef]
- Solozhenko, V.L.; Bushlya, V. Mechanical properties of superhard boron subnitride B13N2. J. Superhard Mater. 2017, 39, 422–426. [Google Scholar] [CrossRef]
- Cherednichenko, K.; Solozhenko, V.L. Structure and equation of state of tetragonal boron subnitride B50N2. J. Appl. Phys. 2017, 122, 155901. [Google Scholar] [CrossRef]
- Solozhenko, V.L.; Kurakevych, O.; Le Godec, Y. Creation of nanostuctures by extreme conditions: High-pressure synthesis of ultrahard nanocrystalline cubic boron nitride. Adv. Mater. 2012, 24, 1540–1544. [Google Scholar] [CrossRef] [PubMed]
- Solozhenko, V.L.; Bushlya, V.; Zhou, J. Mechanical properties of ultra-hard nanocrystalline cubic boron nitride. J. Appl. Phys. 2019, 126, 075107. [Google Scholar] [CrossRef]
- Liu, A.; Cohen, M. Prediction of new low compressibility solids. Science 1989, 245, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Matar, S.F.; Mattesini, M. Ab initio search of carbon nitrides, isoelectronic with diamond, likely to lead to new ultra-hard materials. Comptes Rendus De L’académie Des Sci. 2001, 4, 255–272. [Google Scholar] [CrossRef]
- Mattesini, M.; Matar, S.F. Density-functional theory investigation of hardness, stability, and electron-energy-loss spectra of carbon nitrides with C11N4 stoichiometry. Phys. Rev. B 2002, 65, 075110. [Google Scholar] [CrossRef]
- Betranhandy, E.; Matar, S.F. A model study for the breaking of cyanogen out of CNx within DFT. Diam. Relat. Mater. 2006, 15, 1609–1613. [Google Scholar] [CrossRef]
- Matar, S.F.; Solozhenko, V.L. The simplest dense carbon allotrope: Ultra-hard body-centered tetragonal C4. J. Solid State Chem. 2022, 314, 123424. [Google Scholar] [CrossRef]
- Werheit, H.; Leithe-Jasper, A.; Tanaka, T.; Rotter, H.; Schwetz, K. Some properties of single-crystal boron carbide. J. Solid State Chem. 2004, 177, 575–579. [Google Scholar] [CrossRef]
- Matar, S.F.; Solozhenko, V.L. Ultra-hard rhombohedral carbon by crystal chemistry and ab initio investigations. J. Solid State Chem. 2021, 302, 122354. [Google Scholar] [CrossRef]
- Liu, Z.; He, J.; Yang, J.; Guo, X.; Sun, H.; Wang, H.-T.; Wu, E.; Tian, Y. Prediction of a sandwichlike conducting superhard boron carbide: First-principles calculations. Phys. Rev. B 2006, 73, 172101. [Google Scholar] [CrossRef]
- Kim, E.; Pang, T.; Utsumi, W.; Solozhenko, V.L.; Zhao, Y. Cubic phases of BC2N: A first-principles study. Phys. Rev. B 2007, 75, 184115. [Google Scholar] [CrossRef]
- Fan, X.; Wu, H.; Shen, Z.; Kuo, J.-L. A first-principle study on the structure, stability and hardness of cubic BC2N. Diam. Relat. Mater. 2009, 18, 1278–1282. [Google Scholar] [CrossRef]
- Wang, H.; Qu, N.; Li, Q.; Li, Y.; Li, Z.; Gou, H.; Gao, F. First-principles calculations on two superhard BCN allotropes: P−3m1-BCN and I41md-BCN. Comput. Mater. Sci. 2020, 184, 109869. [Google Scholar] [CrossRef]
- Luo, X.; Zhou, X.-F.; Liu, Z.; He, J.; Xu, B.; Yu, D.; Wang, H.-T.; Tian, Y. Refined crystal structure and mechanical properties of superhard BC4N crystal: First-principles calculations. J. Phys. Chem. C 2008, 112, 9516–9519. [Google Scholar] [CrossRef]
- Qu, N.-R.; Wang, H.; Li, Q.; Li, Z.-P.; Gao, F.-M. An orthorhombic phase of superhard o-BC4N. Chin. Phys. Lett. 2019, 36, 036201. [Google Scholar] [CrossRef]
- Zhu, L.; Ma, M.; Gao, Q.; Li, B.; Wei, X.; Xiong, M.; Zhao, Z.; He, J. Prediction of a series of superhard BC4N structures. Diam. Relat. Mater. 2022, 127, 109192. [Google Scholar] [CrossRef]
- Luo, X.; Guo, X.; Liu, Z.; He, J.; Yu, D.; Tian, Y.; Wang, H.-T. Ground-state properties and hardness of high density BC6N phases originating from diamond structure. J. Appl. Phys. 2007, 101, 083505. [Google Scholar] [CrossRef]
- Qu, N.-R.; Wang, H.-C.; Li, Q.; Li, Y.-D.; Li, Z.-P.; Gou, H.-Y.; Gao, F.-M. Surperhard monoclinic BC6N allotropes: First-principles investigations. Chin. Phys. B 2019, 28, 096201. [Google Scholar] [CrossRef]
- Chen, W.-C.; Schmidt, J.; Yan, D.; Vohra, Y.; Chen, C.-C. Machine learning and evolutionary prediction of superhard B-C-N compounds. NPJ Comput. Mater. 2021, 7, 114. [Google Scholar] [CrossRef]
- Gao, F.; He, J.; Wu, E.; Liu, S.; Yu, D.; Li, D.; Zhang, S.; Tian, Y. Hardness of covalent crystals. Phys. Rev. Lett. 2003, 91, 015502. [Google Scholar] [CrossRef]
- Solozhenko, V.L.; Chernyshev, V.; Fetisov, G.; Rybakov, V.; Petrusha, I. Structure analysis of the cubic boron nitride crystals. J. Phys. Chem. Solids 1990, 51, 1011–1012. [Google Scholar] [CrossRef]
- Mazhnik, E.; Oganov, A. Application of machine learning methods for predicting new superhard materials. J. Appl. Phys. 2020, 128, 075102. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. B 1964, 136, 864–871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L. Self-consistent equations including exchange and correlation effects. Phys. Rev. A 1965, 140, 1133–1138. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, J. From ultrasoft pseudopotentials to the projector augmented wave. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.; Burke, K.; Ernzerhof, M. The Generalized Gradient Approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Klimeš, J.; Bowler, D.A. Michaelides, Van der Waals density functionals applied to solids. Phys. Rev. B 2011, 83, 195131. [Google Scholar] [CrossRef]
- Press, W.; Flannery, B.; Teukolsky, S.; Vetterling, W. Numerical Recipes, 2nd ed.; Cambridge University Press: New York, NY, USA, 1986. [Google Scholar]
- Blöchl, P.; Jepsen, O.; Anderson, O. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223–16233. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.; Pack, J. Special k-points for Brillouin Zone integration. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Eyert, V. Basic notions and applications of the augmented spherical wave method. Int. J. Quantum Chem. 2000, 77, 1007–1031. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Space Group | a = b (Å) | c (Å) | ρ (g/cm3) | HV (GPa) | B0 (GPa) | |
|---|---|---|---|---|---|---|
| Diamond | Fd−3m | 3.56661 ‡ | 3.517 | 98 pw | 445 § | |
| Lonsdaleite | P63/mmc | 2.5221 † | 4.1186 † | 3.516 | 97 pw | 443 pw |
| rh-C3#166 [25] | R−3m | 2.4900 | 10.4100 | 3.211 | 89 | 406 |
| hex-C6#194 [25] | P63/mmc | 2.4950 | 6.9610 | 3.189 | 87 | 404 |
| tet-C6#119 [29] | I−4m2 | 2.4666 | 6.4320 | 3.058 | 85 | 385 |
| c-C3N4#215 [30] | P−43m | 3.4300 | 3.788 | 71 pw | 421 pw | |
| c-C3N4#220 [31] | I−43d | 5.3973 | 3.889 | 73 pw | 430 pw | |
| tet-C11N4#115 [pw] | P−4m2 | 4.9526 | 3.5202 | 3.618 | 76 | 408 |
| tet-BC5#119 [32] | I−4m2 | 2.5250 | 11.3230 | 3.260 | 70 pw | 350 pw |
| c-BC7#215 [33] | P−43m | 3.6205 | 3.320 | 72 pw | 358 pw | |
| trig-BC11#156 [34] | P3m1 | 2.5381 | 12.5955 | 3.378 | 83 pw | 395 pw |
| o-BC2N #17 [35] | P2221 | 3.5536/3.5986 | 3.5528 | 3.570 | 73 pw | 381 pw |
| trig-BC2N #156 [35] | P3m1 | 2.4955 | 4.1923 | 3.587 | 74 pw | 384 pw |
| rh-BC2N #160 [36] | R3m | 2.5382 | 12.6054 | 3.460 | 75 | 426 pw |
| tet-BCN #105 [37] | P42mc | 2.7047 | 6.0073 | 2.783 | 65 | 282 |
| HV | B | GV | E ** | ν ** | KIc‡ | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| T * | LO † | MO ‡ | CN § | B0 * | BV | |||||
| GPa | MPa·m½ | |||||||||
| Diamond [pw] | 98 | 90 | 100 | 93 | 445 †† | 530 †† | 1138 | 0.074 | 6.4 | |
| Lonsdaleite [pw] | 97 | 90 | 99 | 94 | 443 | 432 | 521 | 1115 | 0.070 | 6.2 |
| rh-C3#166 [25] | 89 | 83 | 73 pw | 65 pw | 406 | 394 | 402 | 900 | 0.119 | 5.1 |
| hex-C6#194 [25] | 87 | 82 | 73 pw | 65 pw | 404 | 392 | 400 | 895 | 0.119 | 5.1 |
| tet-C6#119 [29] | 85 | 78 | 68 pw | 63 | 385 | 366 | 375 | 839 | 0.118 | 4.7 |
| c-C3N4#215 [30] | 71 pw | 63 pw | 68 pw | 58 pw | 421 pw | 425 | 397 *** | 908 pw | 0.144 pw | 7.1 pw |
| c-C3N4#220 [31] | 73 pw | 74 pw | 56 pw | 48 pw | 430 pw | 487 §§ | 393 §§ | 930 §§ | 0.18 §§ | 9.1 pw |
| tet-C11N4#115 [pw] | 76 | 78 | 87 | 82 | 408 | 406 | 461 | 1003 | 0.088 | 5.5 |
| tet-BC5#119 [32] | 70 pw | 62 pw | 68 pw | 62 pw | 350 pw | 376 | 379 | 851 pw | 0.123 pw | 4.9 pw |
| c-BC7#215 [33] | 72 pw | 59 pw | 75 pw | 70 pw | 358 pw | 375 | 403 | 890 pw | 0.104 pw | 4.8 pw |
| trig-BC11#156 [34] | 83 pw | 81 pw | 75 pw | 68 | 395 pw | 405 | 414 | 926 pw | 0.119 pw | 5.3 pw |
| o-BC2N #17 [35] | 73 pw | 69 pw | 88 pw | 76 pw | 381 pw | 459 | 482 | 1071 pw | 0.111 pw | 6.4 pw |
| trig-BC2N #156 [35] | 74 pw | 58 pw | –– | –– | 384 pw | 420 | –– | –– | –– | –– |
| rh-BC2N #160 [36] | 75 | 74 pw | 90 | 84 | 426 pw | 412 | 476 | 1031 | 0.083 | 5.7 |
| tet-BCN #105 [37] | 65 | 61 | 35 pw | 36 | 282 | 280 | 232 | 545 | 0.175 | 4.2 |
| HV (GPa) | |||
|---|---|---|---|
| Gao’s Model * | Tian’s Model † | Thermodynamic Model ‡ | |
| c-BC2N [63] | 79.7 | – | 75 pw |
| trig-BC4N (P3m1) [65] | 84.3 | – | 76 pw |
| o-BC4N (Imm2) [66] | 78.7 | – | 73 pw |
| trig-BC4N [67] | – | 87.5 | 73 pw |
| tet-BC6N (P−42m) [68] | 79.9 | – | 75 pw |
| rh-BC6N (R3m) [68] | 79.1 | – | 76 pw |
| m-BC6N (Pm) [69] | 77.4 | – | 72 pw |
| m-BC6N (Cm) [69] | 80.6 | – | 73 pw |
| trig-BC10N (P3m1) [70] | – | 87 | 75 pw |
| tet-BCN (I41md) [64] | 61.8 | – | 70 pw |
| trig-BCN (P−3m1) [64] | 68.5 | – | 69 pw |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solozhenko, V.L.; Matar, S.F. Prediction of Novel Ultrahard Phases in the B–C–N System from First Principles: Progress and Problems. Materials 2023, 16, 886. https://doi.org/10.3390/ma16020886
Solozhenko VL, Matar SF. Prediction of Novel Ultrahard Phases in the B–C–N System from First Principles: Progress and Problems. Materials. 2023; 16(2):886. https://doi.org/10.3390/ma16020886
Chicago/Turabian StyleSolozhenko, Vladimir L., and Samir F. Matar. 2023. "Prediction of Novel Ultrahard Phases in the B–C–N System from First Principles: Progress and Problems" Materials 16, no. 2: 886. https://doi.org/10.3390/ma16020886
APA StyleSolozhenko, V. L., & Matar, S. F. (2023). Prediction of Novel Ultrahard Phases in the B–C–N System from First Principles: Progress and Problems. Materials, 16(2), 886. https://doi.org/10.3390/ma16020886