Abstract
The synthesis of two series of monocyclic and bicyclic trifluoromethylated 4,5-dihydro-1,2,4-triazin-6(1H)-one derivatives based on (3+3)-annulation of methyl esters derived from natural α-amino acids with in situ generated trifluoroacetonitrile imines has been described. The devised protocol is characterized by a wide scope, easily accessible substrates, remarkable functional group tolerance, and high chemical yield. In reactions with chiral starting materials, no racemization at the stereogenic centers was observed and the respective enantiomerically pure products were obtained. Selected functional group interconversions carried out under catalytic hydrogenation and mild PTC oxidation conditions were also demonstrated.
1. Introduction
The functionalization of organic molecules with fluorine atom(s) and/or with fluoroalkyl groups has been recognized as an efficient method for the tuning of their physico-chemical behavior and biological activity [1,2,3,4]. The introduction of fluorine atoms into the parent non-fluorinated compound enables control on properties such as the metabolic stability, reactivity, acidity, oleophilicity, and conformational effects, among others, which are of general significance for the search of new advanced materials [5,6,7], and compounds of potential medicinal [8,9,10] and agrochemical applications [11]. For this reason, the development of new, efficient synthetic methods leading to fluorinated products, in particular, fluoromethylated N-heterocycles [12,13,14], is highly desirable.
Several synthetic strategies towards F-containing heterocycles, including cycloadditions and cyclocondensation reactions, functional group interconversions, and catalytic C–H functionalizations, have been developed in recent decades and are nicely summarized elsewhere [15,16,17,18]. In this context, there is increasing interest in the chemistry of fluoroalkylated 1,3-dipoles, which are recognized as highly useful building blocks for Huisgen (3+2)-cycloaddition reactions [19,20,21,22,23,24]. For example, despite some limitations and the difficult handling of 2,2,2-trifluorodiazoethane, this highly reactive intermediate has been extensively explored, not only as 1,3-dipolar reagent, but also as a valuable source of the respective carbene employed in (2+1)-annulation reactions [22,23,24].
On the other hand, a number of more recent publications reported on nitrile imines functionalized at the C-termini with either CF3 [25,26,27,28,29,30] or CF2H [31,32] groups. They were successfully applied for the synthesis of various 5-membered N-heterocyclic systems formed via (3+2)-cycloadditions, notably, with most of the cases proceeding in a fully regioselective manner. The presented protocols also revealed fluorinated nitrile imines as readily available building blocks, which can be generated in situ under mild conditions, i.e., via the base-mediated dehydrohalogenation of bench-stable hydrazonoyl halides (or pseudohalides).
For example, in 2018 we have demonstrated that the trapping of in situ generated trifluoroacetonitrile imines (1) with electron-rich vinyl ethers leads to 3-CF3-pyrazole derivatives 2 (Scheme 1a) [27]. In this case, the final product is produced via the spontaneous elimination of an alcohol molecule from the initially formed (3+2)-cycloadduct. Further studies on 1,3-dipolar cycloadditions of 1 onto C=C and C≡C bonds using nitroolefins [33], cyanoalkenes [34], enones [35,36,37], alkoxyallenes [28], and benzynes [30] as dipolarophiles also evidenced the high synthetic potential of CF3-nitrile imines in the synthesis of pyrazoline and pyrazole derivatives. The addition of 1 with hetero-dipolarophiles are also known (Scheme 1b); a series of thiocarbonyl compounds including (cyclo)aliphatic [26,38], aromatic, and ferrocenyl thioketones [25], as well as thiochalcones [39] and thioamides [40], smoothly reacted with trifluoroacetonitrile imines 1 to afford 1,3,4-thidiazole derivatives of type 3 formed as the exclusive products. More recently, Chen and Wang disclosed an elegant method employing imidates as suitable C=N dipolarophiles en route to 1,2,4-triazoles 4 (Scheme 1c) [41].
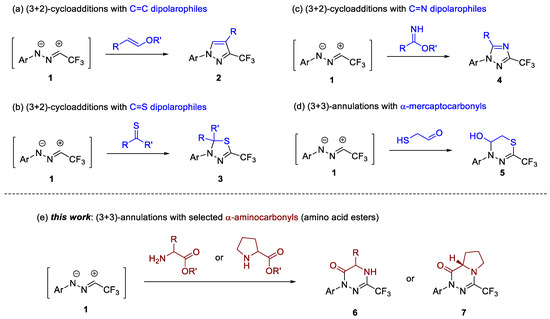
Scheme 1.
Exemplary applications of in situ generated trifluoroacetonitrile imines (1) in synthesis of: (a–c) 5-membered N-heterocycles available via (3+2)-cycloadditions with C=C and C=X dipolarophiles leading to pyrazole, 1,3,4-thiadiazole, and 1,2,4-triazole derivatives 2–4, respectively; (d) 1,3,4-thiadiazines 5 available through (3+3)-annulation with α-mercaptoacetaldehyde; (e) monocyclic and bicyclic 1,3,4-triazin-6(1H)-ones 6 and 7 reported herein.
Despite the remarkable progress in (3+2)-cycloaddition reactions in recent years, the chemistry of higher formal (3+n)-cycloadditions of fluorinated nitrile imines, such as 1 with bifunctional reagents, are explored to a limited extent. Some time ago, we demonstrated that nitrile imines 1 can be smoothly trapped with α-mercaptoacetaldehyde, and also with other α-mercaptocarbonyl compounds, to give 1,3,4-thiadiazolines 5 as the exclusive products (Scheme 1d) [42]. Nevertheless, to the best of our knowledge, no reports on cyclocondesations of 1 with other bifunctional compounds, such as α-aminocarbonyls, have been published thus far. Hence, we turned the attention to methyl esters derived from natural amino acids as easily available substrates for the preparation of hitherto unknown trifluoromethylated 1,3,4-triazin-6(1H)-one derivatives 6 and 7 (Scheme 1e).
In recent years, monocyclic and bicyclic 1,2,4-triazine derivatives, including their oxo analogues, attracted considerable attention, as the compounds of that type exhibit a wide range of pharmacological properties, in particular antimicrobial and anticancer activity [43,44]. For example, in 2010 Krauth et al. reported on 1,2,4-triazin-5-ones of type 8, which showed distinct antiproliferative effects against the chronic myeloid leukemia cell line (K-562) combined with remarkably low cytotoxicity (Figure 1) [45]. Later on, Khan et al. evidenced that the introduction of fluorine atom into aryl substituent improves the biological activity of the resulting 1,2,4-triazinones (such as 9), which were recognized as potent CDK2 and anti-HIV-1 inhibitors [46]. Furthermore, a promising thioredoxin reductase (TrxR) inhibition at submicromolar concentration by trifluoromethylated bicyclic triazinone 10 has also been discovered [47]. Thus, despite the reported progress in the synthesis and biological evaluation of fluorinated 1,2,4-triazinones, further development of synthetic protocols and the evaluation of biological properties of this group of N-heterocycles is of general importance.
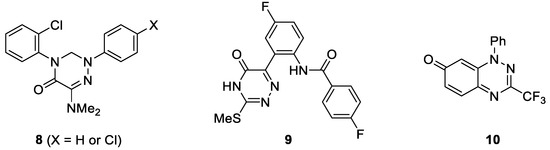
Figure 1.
Structures of selected biologically active halogenated 1,2,4-triazinones.
Here we report on our results aimed at the application of nitrile imines 1 in the synthesis of a series of monocyclic and bicyclic 1,2,4-triazin-6(1H)-ones 6 and 7, respectively, by using selected natural amino acid esters as suitable reaction partners. Particularly, achiral (glycine) and the selected chiral substrates bearing either primary or secondary amino group (proline) were examined. Furthermore, the subsequent transformations of the target products, particularly the oxidations of the core heterocyclic ring and interconversions of selected functional group under catalytic hydrogenation conditions, were studied. Finally, taking into account the well-documented biological activity of some fluorinated and non-fluorinated 1,2,4-triazinones and related 1,2,4-triazine-based analogues [43,44,45,46,47,48,49,50,51], the cytotoxic properties of representative final compounds were examined against MCF-7 and HL-60 cancer cell lines.
2. Materials and Methods
2.1. General Information
All commercially available chemicals (solvents, reagents) were used as received. If not stated otherwise, reactions were performed in flame dried flasks under the atmosphere of inert gas with the addition of the reactants using a syringe; subsequent manipulations were conducted in the air. NMR spectra were measured with Bruker AVIII instrument (1H NMR (600 MHz); 13C NMR (151 MHz); 19F NMR (565 MHz) Bruker BioSpin AG, Fällanden, Switzerland); chemical shifts are given relative to the residual undeuterated solvent peaks (for CDCl3: 1H NMR δ = 7.16, 13C NMR δ = 77.16; for CD3OD: 1H NMR δ = 3.31, 13C NMR δ = 49.00; for DMSO-d6: 1H NMR δ = 2.50, 13C NMR δ = 39.52) or to CFCl3 (19F NMR δ = 0.00) used as the external standard. Integrals in accordance with the assignments and coupling constants J are given in Hz. For detailed peak assignments, 2D spectra were measured (i.e., COSY, HMQC). Mass spectra (ESI) were performed with a Varian 500-MS LC Ion Trap (Varian Inc., Palo Alto, CA, USA); high resolution measurements were performed with a Waters Synapt G2-Si mass spectrometer (Waters Corporation, Milford, MA, USA). IR spectra were obtained with a Cary 630 FTIR (Agilent Technologies, Santa Clara, CA, USA) spectrometer, in neat. Elemental analyses were performed with a Vario EL III (Elementar Analysensysteme GmbH, Langenselbold, Germany) instrument. The melting points were determined in the capillaries with a Melt-Temp II (Laboratory Devices, Holliston, MA, USA) apparatus or with a polarizing optical microscope (Opta-Tech, Warsaw, Poland), and they are uncorrected. The ball-milling apparatus was a MM 400 mixer mill (Retsch GmbH, Haan, Germany). The mechanochemical reactions were performed in 5 mL stainless steel jars, at 25 Hz, with three stainless steel balls (ø 7 mm). The optical rotations were determined with a MCP 500 (Anton Paar, Graz, Austria) polarimeter at the temperatures indicated. The enantiopurity was analyzed with 1260 Infinity HPLC (Agilent Technology, Germany) using a column with chiral support (CHIRALPAK AD-H). The required known nitrile imine precursors, i.e., hydrazonoyl bromides 11, were prepared starting with readily available trifluoroacetaldehyde arylhydrazones 14 [52], by NBS-mediated bromination of the latter, as described [25].
2.2. Synthetic Protocols
Synthesis of 1,2,4-triazin-6(1H)-ones 6 and 7: An excess Et3N (8.0 mmol, 1.12 mL) was added under inert atmosphere to a suspension of amino ester hydrochloride 12 (1.0 mmol) in dry THF (3.0 mL). Then, a solution of hydrazonoyl bromide 11 (1.1 mmol) in dry THF (3.0 mL) was added, and the stirring was continued overnight (the consumption of 11 was confirmed by TLC). The resulting solution was filtered and the precipitate was washed with Et2O (2 × 4.0 mL). After the filtrates were combined and the solvents were removed under reduced pressure, the crude product 6 or 7 was purified by standard column chromatography (CC). In certain cases of glycine derivatives, the resulting material was additionally recrystallized from hexane-dichloromethane mixtures by the slow evaporation of the solvents.
1-(4-Nitrophenyl)-3-trifluoromethyl-4,5-dihydro-1,2,4-triazin-6(1H)-one (6a): CC (SiO2, CH2Cl2 gradient CH2Cl2/EtOAc 9:1), 216 mg (75%). Colorless solid, m.p. 224–225 °C (CH2Cl2/hexanes). 1H NMR (CDCl3, 600 MHz): δ 4.30 (dbr, J ≈ 1.5 Hz, 2 H, CH2), 5.31 (sbr, H, NH), 7.92, 8.27 (2 dbr, J ≈ 9.2 Hz, 2 H each). 13C NMR (CDCl3, 151 MHz): δ 44.0 (t, CH2), 118.2 (q, 1JC-F = 275.2 Hz, CF3), 123.7, 124.2 (2 d, 4 CH), 137.3 (q, 2JC-F = 37.8 Hz, C(3)), 145.0, 145.5 (2 s, 2 i-C), 158.1 (s, C=O). 19F NMR (CDCl3, 565 MHz): δ −70.6 (s, CF3). IR (neat): ν 3295 (NH), 1685 (C=O), 1584, 1487, 1312, 1144 (CF3), 1059, 854 cm−1. ESI-MS (m/z): 311.1 (100, [M+Na]+), 289.2 (31, [M+H]+). C10H7F3N4O3 (288.0): calcd. C 41.68, H 2.45, N 19.44; found: C 41.50, H 2.46, N 19.61.
1-(3-Nitrophenyl)-3-trifluoromethyl-4,5-dihydro-1,2,4-triazin-6(1H)-one (6b): CC (SiO2, 3% MeOH in CH2Cl2), 248 mg (86%). Yellow crystals, m.p. 144–146 °C (CH2Cl2/hexanes). 1H NMR (CD3OD, 600 MHz): δ 4.19 (s, 2 H, CH2), 4.61 (sbr, 1 H, NH), 7.63 (t, J = 8.2 Hz, 1H), 8.04 (ddd, J = 1.0, 2.2, 8.2 Hz, 1 H), 8.12 (ddd, J = 1.0, 2.2, 8.2 Hz, 1 H), 8.53 (t, J = 2.2 Hz, 1 H). 13C NMR (CD3OD, 151 MHz): δ 44.2 (t, CH2), 119.9 (q, 1JC-F = 274.4 Hz, CF3), 119.8, 121.9, 130.5, 130.8 (4 d, 4 CH), 139.7 (q, 2JC-F = 37.1 Hz, C(3)), 142.6, 149.4 (2 s, 2 i-C), 160.8 (s, C=O). 19F NMR (CD3OD, 565 MHz): δ −72.2 (s, CF3). IR (neat): ν 3290 (NH), 1682 and 1647 (C=O), 1536, 1472, 1349, 1271, 1197–1129 (CF3), 977 cm−1. ESI-MS (m/z): 289.2 (100, [M+H]+). C10H7F3N4O3 (288.0): calcd. C 41.68, H 2.45, N 19.44; found: C 41.44, H 2.36, N 19.48.
4-(3-Trifluoromethyl-4,5-dihydro-6(1H)-oxo-1,2,4-triazin-1-yl)benzonitrile (6c): CC (SiO2, 4% MeOH in CH2Cl2), 247 mg (92%). Colorless solid, m.p. 212–214 °C. 1H NMR (DMSO-d6, 600 MHz): δ 4.17 (s, 2 H, CH2), 7.79, 7.88 (2 dbr, J ≈ 8.8 Hz, 2 H each), 8.67 (sbr, 1 H, NH). 13C NMR (DMSO-d6, 151 MHz): δ 43.0 (t, CH2), 108.1 (s, CN), 118.4 (q, 1JC-F = 275.2 Hz, CF3), 118.7 (s, i-C), 123.8, 132.7 (2 d, 4 CH), 137.4 (q, 2JC-F = 36.2 Hz, C(3)), 143.8 (s, i-C), 159.2 (s, C=O). 19F NMR (DMSO-d6, 565 MHz): δ −69.3 (s, CF3). IR (neat): ν 3261 (NH), 2236 (CN), 1700 and 1681 (C=O), 1334, 1200–1126 (CF3), 1066, 839 cm−1. ESI-MS (m/z): 291.2 (100, [M+Na]+), 269.2 (12, [M+H]+). C11H7F3N4O (268.2): calcd. C 49.26, H 2.63, N 20.89; found: C 49.36, H 2.89, N 20.61.
1-(4-Chlorophenyl)-3-trifluoromethyl-4,5-dihydro-1,2,4-triazin-6(1H)-one (6d): CC (SiO2, 4% MeOH in CH2Cl2), 233 mg (84%). Yellow crystals, m.p. 159–160 °C (CH2Cl2/hexanes). 1H NMR (CDCl3, 600 MHz): δ 4.18 (dbr, J ≈ 1.5 Hz, 2 H, CH2), 5.39 (sbr, 1 H, NH), 7.36–7.38, 7.51–7.53 (2 m, 2 H each). 13C NMR (CDCl3, 151 MHz): δ 43.8 (t, CH2), 118.2 (q, 1JC-F = 275.2 Hz, CF3), 125.7, 128.9 (2 d, 4 CH), 132.7 (s, i-C), 136.9 (q, 2JC-F = 37.5 Hz, C(3)), 138.5 (s, i-C), 157.8 (s, C=O). 19F NMR (CDCl3, 565 MHz): δ −70.6 (s, CF3). IR (neat): ν 3294 (NH), 1689 and 1655 (C=O), 1491, 1349, 1316, 1194–1133 (CF3), 1085, 828 cm−1. ESI-MS (m/z): 278.2 (100, [M+H]+), 244.2 (16, [M–Cl+H]+). C10H7ClF3N3O (277.6): calcd. C 43.26, H 2.54, N 15.14; found: C 43.30, H 2.53, N 15.24.
1-(2,4-Dichlorophenyl)-3-trifluoromethyl-4,5-dihydro-1,2,4-triazin-6(1H)-one (6e): The crude reaction mixture was additionally refluxed for 2h in order to accomplish the second cyclization step (see the text), and the product was isolated after the standard work-up; CC (SiO2, CH2Cl2/EtOAc 95:5), 289 mg (93%). Colorless solid, m.p. 138–139 °C. 1H NMR (CDCl3, 600 MHz): δ 4.24 (sbr, 2 H, CH2), 5.33 (sbr, 1 H, NH), 7.34, 7.51 (2 mc, 2 H, 1 H). 13C NMR (CDCl3, 151 MHz): δ 43.7 (t, CH2), 118.2 (q, 1JC-F = 275.2 Hz, CF3), 128.3, 130.3, 130.4 (3 d, 3 CH), 133.4, 135.6, 136.3 (3 s, 3 i-C), 136.8 (q, 2JC-F = 37.6 Hz, C(3)), 157.9 (s, C=O). 19F NMR (CDCl3, 565 MHz): δ −70.5 (s, CF3). IR (neat): ν 3284 (NH), 1685 and 1665 (C=O), 1525, 1402, 1357, 1323, 1189–1129 (CF3), 1103, 1050 cm−1. ESI-MS (m/z): 312.1 (100, [M+H]+). C10H6Cl2F3N3O (312.1): calcd. C 38.49, H 1.94, N 13.47; found: C 38.72, H 2.19, N 13.54.
Amidrazone 13e: The spectroscopically pure sample of intermediate 13e (75 mg, 22%; as ca. 87:13 mixture of E/Z-isomers) was obtained in reaction of 11e with 12a and was isolated from the mother liquor by preparative thin layer chromatography (PTLC; SiO2, petroleum ether/CH2Cl2 3:2) followed by recrystallisation from a hexanes/CH2Cl2 mixture. Colorless solid, m.p. 79–81 °C. 1H NMR (CDCl3, 600 MHz); major isomer: δ 3.82 (s, 3 H, Me), 4.05 (d, J = 5.7 Hz, 2 H), 4.57 (tbr, J ≈ 5.7 Hz, 1 H, NH), 7.20 (dd, J = 2.3, 8.8 Hz, 1 H), 7.21 (sbr, 1 H, NH), 7.29 (d, J = 2.3 Hz, 1 H), 7.39 (d, J = 8.8 Hz, 1 H); diagnostic signals for minor isomer: δ 3.80 (s, 3 H, Me), 4.00 (d, J = 5.2 Hz, 2 H), 4.95 (tbr, J ≈ 5.2 Hz, 1 H, NH), absorptions in the aromatic region could not be seen due to overlap of the signals. 13C NMR (CDCl3, 151 MHz); major isomer: δ 44.8 (t, CH2), 53.0 (q, Me), 116.4 (d, CH), 119.20 (q, 1JC-F = 274.5 Hz, CF3), 119.23, 125.7 (2 s, 2 i-C), 128.2, 128.7 (2 d, 2 CH), 136.7 (q, 2JC-F = 34.9 Hz), 140.4 (s, i-C), 170.8 (s, C=O); diagnostic signals for minor isomer: δ 43.8 (t, CH2), 52.6 (q, Me), 114.1 (d, CH), 117.8, 123.8 (2 s, 2 i-C), 128.0, 128.6 (2 d, 2 CH), 140.7 (s, i-C), 170.4 (s, C=O); absorptions of the CF3 group and of the neighboring C atom could not be found due to overlap and low intensity, respectively.
1-Phenyl-3-trifluoromethyl-4,5-dihydro-1,2,4-triazin-6(1H)-one (6f): CC (SiO2, 4% MeOH in CH2Cl2), 192 mg (79%). Colorless crystals, m.p. 146–148 °C (CH2Cl2/hexanes). 1H NMR (CDCl3, 600 MHz): δ 4.13 (dbr, J ≈ 1.6 Hz, 2 H, CH2), 5.39 (sbr, 1 H, NH), 7.28–7.31, 7.40–7.43, 7.52–7.54 (3 m, 1 H, 2 H, 2 H). 13C NMR (CDCl3, 151 MHz): δ 43.8 (t, CH2), 118.3 (q, 1JC-F = 275.1 Hz, CF3), 124.7, 127.5, 128.9 (3 d, 5 CH), 136.7 (q, 2JC-F = 37.6 Hz, C(3)), 140.0 (s, i-C), 157.9 (s, C=O). 19F NMR (CDCl3, 565 MHz): δ −70.6 (s, CF3). IR (neat): ν 3288 (NH), 1689 and 1655 (C=O), 1495, 1394, 1353, 1320, 1193–1137 (CF3), 1092, 1057 cm−1. ESI-MS (m/z): 244.2 (100, [M+H]+). C10H8F3N3O (243.2): calcd. C 49.39, H 3.32, N 17.28; found: C 49.23, H 3.47, N 17.30.
1-(4-Tolyl)-3-trifluoromethyl-4,5-dihydro-1,2,4-triazin-6(1H)-one (6g): CC (SiO2, 5% MeOH in CH2Cl2), 195 mg (76%). Colorless crystals, m.p. 153–154 °C (CH2Cl2/hexanes). 1H NMR (CDCl3, 600 MHz): δ 2.35 (s, 3 H, Me), 4.14 (dbr, J ≈ 1.5 Hz, 2 H, CH2), 5.37 (sbr, 1 H, NH), 7.21, 7.39 (2 dbr, J ≈ 8.2 Hz, 2 H each). 13C NMR (CDCl3, 151 MHz): δ 21.2 (q, Me), 43.8 (t, CH2), 118.3 (q, 1JC-F = 275.0 Hz, CF3), 124.7, 129.5 (2 d, 4 CH), 136.6 (q, 2JC-F = 37.4 Hz, C(3)), 137.45, 137.53 (2 s, 2 i-C), 157.8 (s, C=O). 19F NMR (CDCl3, 565 MHz): δ −70.6 (s, CF3). IR (neat): ν 3243 (NH), 1681 and 1648 (C=O), 1513, 1402, 1357, 1316, 1189–1133 (CF3), 1085, 820 cm−1. ESI-MS (m/z): 258.2 (100, [M+H]+). C11H10F3N3O (257.2): calcd. C 51.37, H 3.92, N 16.34; found: C 51.33, H 3.93, N 16.80.
1-(4-Benzyloxyphenyl)-3-trifluoromethyl-4,5-dihydro-1,2,4-triazin-6(1H)-one (6h): CC (SiO2, 2% MeOH in CH2Cl2), 286 mg (82%). Light orange crystals, m.p. 130–132 °C (CH2Cl2/hexanes). 1H NMR (CDCl3, 600 MHz): δ 4.15 (dbr, J ≈ 1.4 Hz, 2 H, NCH2), 5.07 (s, 2 H, OCH2), 5.33 (sbr, 1 H, NH), 6.98–7.01, 7.32–7.34, 7.37–7.43 (3 m, 2 H, 1 H, 6 H). 13C NMR (CDCl3, 151 MHz): δ 43.8 (t, NCH2), 70.3 (t, OCH2), 115.1 (d, 2 CH), 118.3 (q, 1JC-F = 275.1 Hz, CF3), 126.3, 127.6, 128.2, 128.7 (4 d, 7 CH), 133.3 (s, i-C), 136.5 (q, 2JC-F = 37.3 Hz, C(3)), 136.8, 157.8, 157.9 (3 s, 2 i-C, C=O). 19F NMR (CDCl3, 565 MHz): δ −70.5 (s, CF3). IR (neat): ν 3276 (NH), 1692–1651 (C=O), 1506, 1349, 1327, 1185–1141 (CF3), 1081, 1051 cm−1. ESI-MS (m/z): 372.3 (100, [M+Na]+), 350.4 (52, [M+H]+). C17H14F3N3O2 (349.3): calcd. C 58.45, H 4.04, N 12.03; found: C 58.23, H 4.12, N 12.20.
(S)-5-Methyl-1-(4-nitrophenyl)-3-trifluoromethyl-4,5-dihydro-1,2,4-triazin-6(1H)-one (6i): CC (SiO2, CH2Cl2), 233 mg (77%). Light orange solid, m.p. 132–133 °C. [α]D20 = −31.7 (c 0.26, CHCl3). 1H NMR (CD3OD, 600 MHz): δ 1.51 (d, J = 6.8 Hz, 3 H, CH3), 4.33 (q, J = 6.8 Hz, 1 H, 5-H), 7.91–7.93, 8.26–8.28 (2 m, 2 H each). 13C NMR (CD3OD, 151 MHz): δ 19.2 (q, Me), 50.8 (d, C(5)), 120.0 (q, 1JC-F = 274.5 Hz, CF3), 124.8, 125.0 (2 d, 4 CH), 139.7 (q, 2JC-F = 37.2 Hz, C(3)), 146.5, 147.0 (2 s, 2 i-C), 164.2 (s, C=O). 19F NMR (CD3OD, 565 MHz): δ −68.5 (s, CF3). IR (neat): ν 3247 (NH), 1685 (C=O), 1588, 1517, 1331, 1197, 1140–1082 (CF3), 855 cm−1. ESI-MS (m/z): 325.2 (100, [M+Na]+). C11H9F3N4O3 (302.2): calcd. C 43.72, H 3.00, N 18.54; found: C 43.60, H 3.17, N 18.66.
(S)-5-(1-Methylethyl)-1-(4-nitrophenyl)-3-trifluoromethyl-4,5-dihydro-1,2,4-triazin-6(1H)-one (6j): CC (SiO2, petroleum ether/EtOAc 4:1), 271 mg (82%). Pale yellow solid, m.p. 143–144 °C. [α]D20 = −213.4 (c 0.27, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 1.02 (d, J = 6.8 Hz, 3 H, CH3), 1.07 (d, J = 7.0 Hz, 3 H, CH3), 2.36–2.44 (m, 1 H), 4.14 (ddbr, J ≈ 2.4, 3.9 Hz, 1 H, 5-H), 5.44 (sbr, 1 H, NH), 7.88–7.90, 8.24–8.27 (2 m, 2 H each). 13C NMR (CDCl3, 151 MHz): δ 16.7, 18.3 (2 q, 2 Me), 33.2 (d, CHMe2), 59.5 (d, C(5)), 117.4 (q, 1JC-F = 275.4 Hz, CF3), 123.9, 124.2 (2 d, 4 CH), 137.4 (q, 2JC-F = 37.5 Hz, C(3)), 145.3, 145.4 (2 s, 2 i-C), 160.9 (s, C=O). 19F NMR (CDCl3, 565 MHz): δ −70.8 (s, CF3). IR (neat): ν 3284 (NH), 1689 and 1659 (C=O), 1524, 1495, 1334, 1193, 1148–1111 (CF3), 1081, 850 cm−1. ESI-MS (m/z): 353.2 (100, [M+Na]+), 331.3 (20, [M+H]+). C13H13F3N4O3 (330.3): calcd. C 47.28, H 3.97, N 16.96; found: C 47.38, H 4.12, N 16.83.
(S)-5-(2-Methylpropyl)-1-(4-nitrophenyl)-3-trifluoromethyl-4,5-dihydro-1,2,4-triazin-6(1H)-one (6k): CC (SiO2, petroleum ether/CH2Cl2 1:3), 186 mg (54%). Light orange solid, m.p. 107–108 °C (hexanes). [α]D20 = −190.6 (c 0.23, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 0.98, 1.01 (2 d, J = 6.1 Hz, 3 H each, 2 CH3), 1.69–1.83 (m, 3 H), 4.27 (ddd, J = 2.2, 4.6, 8.4 Hz, 1 H, 5-H), 5.59 (sbr, 1 H, NH), 7.88–7.90, 8.23–8.25 (2 m, 2 H each). 13C NMR (CDCl3, 151 MHz): δ 21.8, 22.9 (2 q, 2 Me), 24.2 (d, CHMe2), 41.9 (t, CH2), 52.7 (d, C(5)), 118.2 (q, 1JC-F = 275.3 Hz, CF3), 123.6, 124.2 (2 d, 4 CH), 137.4 (q, 2JC-F = 37.5 Hz, C(3)), 145.2, 145.4 (2 s, 2 i-C), 161.7 (s, C=O). 19F NMR (CDCl3, 565 MHz): δ −70.8 (s, CF3). IR (neat): ν 3295 (NH), 1685 and 1659 (C=O), 1521, 1331, 1193–1123 (CF3), 1081, 849 cm−1. ESI-MS (m/z): 345.3 (100, [M+H]+). C14H15F3N4O3 (344.3): calcd. C 48.84, H 4.39, N 16.27; found: C 48.83, H 4.40, N 16.53.
(S)-1-(4-Nitrophenyl)-5-phenyl-3-trifluoromethyl-4,5-dihydro-1,2,4-triazin-6(1H)-one (6l): CC (SiO2, CH2Cl2/hexanes 3:1), 211 mg (58%). Pale yellow solid, m.p. 116–117 °C. [α]D20 = +20.7 (c 0.28, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 5.32 (dbr, J ≈ 2.0 Hz, 1 H, 5-H), 5.83 (sbr, 1 H, NH), 7.41–7.46, 7.86–7.89, 8.21–8.24 (3 m, 5 H, 2 H, 2 H each). 13C NMR (CDCl3, 151 MHz): δ 58.2 (d, C(5)), 118.3 (q, 1JC-F = 275.5 Hz, CF3), 123.8, 124.2, 126.8, 129.6, 129.8 (5 d, 9 CH), 136.8 (q, 2JC-F = 37.7 Hz, C(3)), 137.2, 145.2, 145.5 (3 s, 3 i-C), 159.6 (s, C=O). 19F NMR (CDCl3, 565 MHz): δ −70.4 (s, CF3). IR (neat): ν 3291 (NH), 1688 (C=O), 1592, 1517, 1320, 1193–1141 (CF3), 1081, 854 cm−1. ESI-MS (m/z): 387.2 (100, [M+Na]+), 365.4 (20, [M+H]+). C16H11F3N4O3 (364.3): calcd. C 52.75, H 3.04, N 15.38; found: C 52.69, H 3.13, N 15.21.
(S)-5-(Hydroxymethyl)-1-(4-nitrophenyl)-3-trifluoromethyl-4,5-dihydro-1,2,4-triazin-6(1H)-one (6m): CC (SiO2, CH2Cl2/EtOAc 6:1), 239 mg (75%). Yellow solid, m.p. 127–128 °C. [α]D20 = −96.5 (c 0.28, MeCN). 1H NMR (CD3OD, 600 MHz): δ 3.72 (dd, J = 2.8, 11.6 Hz, 1 H, CH2), 4.08 (dd, J = 3.1, 11.6 Hz, 1 H, CH2), 4.33 (pseudo-t, J ≈ 2.9 Hz, 5-H), 7.93–7.95, 8.25–8.27 (2 m, 2 H each). 13C NMR (CD3OD, 151 MHz): δ 57.8 (d, C(5)), 64.7 (t, CH2), 119.9 (q, 1JC-F = 274.5 Hz, CF3), 124.7, 125.2 (2 d, 4 CH), 139.7 (q, 2JC-F = 36.9 Hz, C(3)), 146.5, 147.1 (2 s, 2 i-C), 162.5 (s, C=O). 19F NMR (CD3OD, 565 MHz): δ −72.1 (s, CF3). IR (neat): ν 3411 (OH), 3198 (NH), 1692 and 1659 (C=O), 1513, 1327, 1193–1088 (CF3), 1036 cm−1. ESI-MS (m/z): 341.1 (100, [M+Na]+), 319.2 (13, [M+H]+). C11H9F3N4O4 (318.1): calcd. C 41.52, H 2.85, N 17.61; found: C 41.60, H 3.04, N 17.33.
(S)-5-[2-(Methylthio)ethyl]-1-(4-nitrophenyl)-3-trifluoromethyl-4,5-dihydro-1,2,4-triazin-6(1H)-one (6n): CC (SiO2, petroleum ether/EtOAc 3:1), 329 mg (91%). Thick yellow oil. [α]D20 = −93.0 (c 0.25, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 2.14 (s, 3 H, Me), 2.15–2.20, 2.32–2.37, 2.68–2.79 (3 m, 1 H, 1 H, 2 H), 4.45 (ddd, J = 1.9, 4.1, 7.9 Hz, 1 H, 5-H), 6.07 (sbr, 1 H, NH), 7.88–7.90, 8.24–8.27 (2 m, 2 H each). 13C NMR (CDCl3, 151 MHz): δ 15.3 (q, Me), 30.4, 31.2 (2 t, 2 CH2), 54.0 (d, C(5)), 118.2 (q, 1JC-F = 275.5 Hz, CF3), 123.8, 124.2 (2 d, 4 CH), 137.3 (q, 2JC-F = 37.5 Hz, C(3)), 145.2, 145.4 (2 s, 2 i-C), 161.0 (s, C=O). 19F NMR (CDCl3, 565 MHz): δ −70.8 (s, CF3). IR (neat): ν 3302 (NH), 1689 and 1657 (C=O), 1592, 1517, 1327, 1193–1108 (CF3), 1081, 854 cm−1. (-)-ESI-MS (m/z): 360.9 (100, [M–H]−), 350.1 (13). C13H13F3N4O3S (362.1): calcd. C 43.09, H 3.62, N 15.46, S 8.85; found: C 42.99, H 3.73, N 15.67, S 8.78.
Methyl (S)-2-[1-(4-nitrophenyl)-3-trifluoromethyl-6(1H)-oxo-4,5-dihydro-1,2,4-triazin-5-yl]acetate (6o): CC (SiO2, petroleum ether/CH2Cl2 2:1), 335 mg (93%). Yellow solid, m.p. 114–115 °C (hexanes). [α]D20 = −82.0 (c 0.24, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 2.90 (dd, J = 10.3, 17.6 Hz, 1 H, CH2), 3.24 (dd, J = 2.8, 17.6 Hz, 1 H, CH2), 3.79 (s, 3 H, Me), 4.66 (ddd, J = 1.8, 2.8, 10.3 Hz, 1 H, 5-H), 6.19 (sbr, 1 H, NH), 7.90, 8.26 (2 dbr, J ≈ 9.2 Hz, 2 H each). 13C NMR (CDCl3, 151 MHz): δ 37.1 (t, CH2), 50.8 (d, C(5)), 52.8 (q, Me), 118.1 (q, 1JC-F = 275.4 Hz, CF3), 123.8, 124.2 (2 d, 4 CH), 137.3 (q, 2JC-F = 37.8 Hz, C(3)), 145.0, 145.6 (2 s, 2 i-C), 159.8, 171.6 (2 s, 2 C=O). 19F NMR (CDCl3, 565 MHz): δ −70.8 (s, CF3). IR (neat): ν 3396 (NH), 1715 (C=O), 1681 (C=O), 1588, 1521, 1491, 1390, 1323, 1193–1139 (CF3), 1107, 857 cm−1. ESI-MS (m/z): 383.2 (100, [M+Na]+). C13H11F3N4O5 (360.2): calcd. C 43.34, H 3.08, N 15.55; found: C 43.07, H 2.95, N 15.61.
(S)-5-[(Indol-3-yl)methyl]-1-(4-nitrophenyl)-3-trifluoromethyl-4,5-dihydro-1,2,4-triazin-6(1H)-one (6p): CC (SiO2, EtOAc), 346 mg (83%). Pale yellow solid, m.p. 140–141 °C. [α]D20 = −143.6 (c 0.24, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 3.24 (dd, J = 9.4, 14.6 Hz, 1 H, CH2), 3.53 (dd, J = 3.4, 14.6 Hz, 1 H, CH2), 4.54 (ddd, J = 1.7, 3.4, 9.4 Hz, 1 H, 5-H), 5.32 (sbr, 1 H, NH), 7.13–7.17, 7.25–7.28 (2 m, 2 H, 1 H), 7.43 (dbr, J ≈ 8.2 Hz, 1 H), 7.63 (dbr, J ≈ 7.9 Hz, 1 H), 7.77–7.80, 8.22–8.24 (2 m, 2 H each) °8.24 (sbr, 1 H, NH). 13C NMR (CDCl3, 151 MHz): δ 30.1 (t, CH2), 54.4 (d, C(5)), 108.5 (s, i-C), 111.7 (d, CH), 118.2 (q, 1JC-F = 275.3 Hz, CF3), 118.6, 120.4, 123.1, 123.8, 123.9, 124.1 (6 d, 8 CH), 126.7, 136.6 (2 s, 2 i-C), 137.0 (q, 2JC-F = 37.6 Hz, C(3)), 145.2, 145.4 (2 s, 2 i-C), 161.39 (s, C=O). 19F NMR (CDCl3, 565 MHz): δ −70.6 (s, CF3). IR (neat): ν 3351 (NH), 1689 and 1662 (C=O), 1592, 1517, 1327, 1200–1144 (CF3), 1085, 854 cm−1. (-)-ESI-MS (m/z): 416.0 (88, [M–H]−). C19H14F3N5O3 (417.3): calcd. C 54.68, H 3.38, N 16.78; found: C 54.43, H 3.49, N 16.74.
(S)-5-Phenyl-1-(4-tolyl)-3-trifluoromethyl-4,5-dihydro-1,2,4-triazin-6(1H)-one ((S)-6q): CC (SiO2, CH2Cl2), 250 mg (75%). Colorless crystals, m.p. 153–154 °C. [α]D20 = +171.6 (c 0.28, CHCl3). 1H NMR (CD3OD, 600 MHz): δ 2.35 (s, 3 H, CH3), 5.28 (sbr, 1 H, NH), 7.22, 7.32 (dbr, J ≈ 8.3 Hz, 2 H each), 7.37–7.40, 7.42–7.45 (2 m, 1 H, 4 H). 13C NMR (CD3OD, 151 MHz): δ 21.1 (q, Me), 58.9 (d, C(5)), 120.1 (q, 1JC-F = 274.4 Hz, CF3), 126.1, 127.9, 129.9, 130.1, 130.2 (5 d, 9 CH), 138.5 (s, i-C), 138.6 (q, 2JC-F = 37.0 Hz, C(3)), 139.4, 140.7 (2 s, 2 i-C), 161.6 (s, C=O). 19F NMR (CD3OD, 565 MHz): δ −72.1 (s, CF3). IR (neat): ν 3336 (NH), 1677 and 1655 (C=O), 1510, 1338, 1189–1144 (CF3), 1084, 820 cm−1. ESI-MS (m/z): 372.1 (57, [M+K]+), 356.1 (47, [M+Na]+), 334.2 (100, [M+H]+). C17H14F3N3O (333.3): calcd. C 61.26, H 4.23, N 12.61; found: C 61.08, H 4.37, N 12.86. A sample of rac-6q (276 mg, 83%) was prepared in an analogous manner starting with hydrazonoyl bromide 11g and racemic methyl phenylglycinate (rac-12e). The obtained 1H and 13C NMR data perfectly matched those obtained for (S)-6q.
(S)-2-(4-Nitrophenyl)-4-trifluoromethyl-6,7,8,8a-tetrahydropyrrolo [1,2-d][1,2,4]-triazin-1(2H)-one (7a): CC (SiO2, CH2Cl2), 269 mg (82%). Yellow solid, m.p. 90–92 °C (CH2Cl2/hexanes). [α]D20 = +79.2 (c 0.17, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 2.08–2.14 (m, 2 H, 7-H2), 2.28–2.34 (m, 1 H, 8-H), 2.46–2.51 (m, 1 H, 8-H), 3.75 (tbr, J ≈ 7.4 Hz, 2 H, 6-H2), 4.17 (dd, J = 6.9, 8.9 Hz, 1 H, 8a-H), 7.92–7.94, 8.24–8.26 (2 m, 2 H each). 13C NMR (CDCl3, 151 MHz): δ 24.1 (t, C(7)), 28.3 (t, C(8)), 48.8 (q, 4JC-F = 2.3 Hz, C(6)), 58.4 (d, C(8a)), 118.5 (q, 1JC-F = 276.0 Hz, CF3), 123.5, 124.2 (2 d, 4 CH), 138.1 (q, 2JC-F = 36.5 Hz, C(4)), 145.2* (s, 2 i-C), 161.5 (s, C=O); *higher intensity. 19F NMR (CDCl3, 565 MHz): δ −68.6 (s, CF3). IR (neat): ν 1698 (C=O), 1519, 1452, 1344, 1191, 1135 (CF3) cm−1. ESI-MS (m/z): 329.3 (100, [M+H]+). C13H11F3N4O3 (328.2): calcd. C 47.57, H 3.38, N 17.07; found: C 47.78, H 3.48, N 17.19.
(S)-2-(3-Nitrophenyl)-4-trifluoromethyl-6,7,8,8a-tetrahydropyrrolo [1,2-d][1,2,4]-triazin-1(2H)-one (7b): CC (SiO2, CH2Cl2), 213 mg (65%). Thick yellow oil. [α]D20 = +27.4 (c 0.15, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 2.06–2.14 (m, 2 H, 7-H2), 2.27–2.33 (m, 1 H, 8-H), 2.45–2.51 (m, 1 H, 8-H), 3.71–3.77 (m, 2 H, 6-H2), 4.17 (dd, J = 6.9, 8.9 Hz, 1 H, 8a-H), 7.55 (t, J = 8.2 Hz, 1H), 8.01 (ddd, J = 1.0, 2.2, 8.2 Hz, 1 H), 8.10 (ddd, J = 1.0, 2.2, 8.2 Hz, 1 H), 8.56 (t, J = 2.2 Hz, 1 H). 13C NMR (CDCl3, 151 MHz): δ 24.1 (t, C(7)), 28.3 (t, C(8)), 48.8 (q, 4JC-F = 2.4 Hz, C(6)), 58.3 (d, C(8a)), 118.5 (q, 1JC-F = 276.0 Hz, CF3), 119.0, 121.2, 129.4, 129.6 (4 d, 4 CH), 138.0 (q, 2JC-F = 36.5 Hz, C(4)), 141.0, 148.4 (2 s, 2 i-C), 161.3 (s, C=O). 19F NMR (CDCl3, 565 MHz): δ −68.5 (s, CF3). IR (neat): ν 1696 and 1648 (C=O), 1528, 1454, 1349, 1193–1118 (CF3) cm−1. ESI-MS (m/z): 329.3 (64, [M+H]+), 327.3 (100, [M–H]+), 299.2 (36). C13H11F3N4O3 (328.2): calcd. C 47.57, H 3.38, N 17.07; found: C 47.75, H 3.43, N 16.84.
(S)-4-[4-Trifluoromethyl-1(2H)-oxo-6,7,8,8a-tetrahydropyrrolo [1,2-d][1,2,4]triazin-2-yl]benzonitrile (7c): CC (SiO2, CH2Cl2), 286 mg (93%). Light orange solid, m.p. 103–105 °C. [α]D20 = +68.3 (c 0.16, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 2.07–2.13 (m, 2 H, 7-H2), 2.26–2.33 (m, 1 H, 8-H), 2.45–2.50 (m, 1 H, 8-H), 3.71–3.76 (m, 2 H, 6-H2), 4.16 (dd, J = 6.9, 8.9 Hz, 1 H, 8a-H), 7.66–7.69, 7.84–7.86 (2 m, 2 H each). 13C NMR (CDCl3, 151 MHz): δ 24.1 (t, C(7)), 28.3 (t, C(8)), 48.8 (q, 4JC-F = 2.3 Hz, C(6)), 58.4 (d, C(8a)), 109.7 (s, CN), 118.5 (q, 1JC-F = 275.8 Hz, CF3), 118.8 (s, i-C), 123.8, 132.7 (2 d, 4 CH), 138.0 (q, 2JC-F = 36.3 Hz, C(4)), 143.7 (s, i-C), 161.3 (s, C=O). 19F NMR (CDCl3, 565 MHz): δ −68.5 (s, CF3). IR (neat): ν 2223 (CN), 1690 and 1655 (C=O), 1603, 1452, 1315, 1126 (CF3) cm−1. ESI-MS (m/z): 331.3 (29, [M+Na]+), 309.3 (100, [M+H]+). C14H11F3N4O (308.3): calcd. C 54.55, H 3.60, N 18.18; found: C 54.61, H 3.73, N 18.13.
(S)-2-(4-Chlorophenyl)-4-trifluoromethyl-6,7,8,8a-tetrahydropyrrolo [1,2-d][1,2,4]triazin-1(2H)-one (7d): Reaction time: 2d; CC (SiO2, petroleum ether/EtOAc 4:1), 219 mg (69%). Pale yellow solid, m.p. 86–87 °C. [α]D20 = +35.1 (c 0.19, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 2.03–2.12 (m, 2 H, 7-H2), 2.24–2.31 (m, 1 H, 8-H), 2.43–2.48 (m, 1 H, 8-H), 3.68–3.75 (m, 2 H, 6-H2), 4.13 (dd, J = 6.9, 8.9 Hz, 1 H, 8a-H), 7.34–7.36, 7.53–7.55 (2 m, 2 H each). 13C NMR (CDCl3, 151 MHz): δ 24.1 (t, C(7)), 28.4 (t, C(8)), 48.7 (q, 4JC-F = 2.3 Hz, C(6)), 58.3 (d, C(8a)), 118.6 (q, 1JC-F = 275.7 Hz, CF3), 125.5, 128.8 (2 d, 4 CH), 132.3 (s, i-C), 137.6 (q, 2JC-F = 36.2 Hz, C(4)), 138.7 (s, i-C), 160.9 (s, C=O). 19F NMR (CDCl3, 565 MHz): δ −68.5 (s, CF3). IR (neat): ν 1692 and 1644 (C=O), 1491, 1446, 1331, 1193, 1122 (CF3) cm−1. ESI-MS (m/z): 320.3 (31, [M{37Cl}+H]+), 318.2 (100, [M{35Cl}+H]+). C13H11ClF3N3O (317.7): calcd. C 49.15, H 3.49, N 13.23; found: C 49.22, H 3.68, N 13.10.
(S)-2-(2,4-Dichlorophenyl)-4-trifluoromethyl-6,7,8,8a-tetrahydropyrrolo [1,2-d][1,2,4]triazin-1(2H)-one (7e): In order to accelerate the second cyclization step, the crude reaction mixture was heated in an oil bath (60 °C) for three days; CC (SiO2, petroleum ether/EtOAc 4:1), 176 mg (50%). Yellow solid, m.p. 107–108 °C. [α]D20 = −6.8 (c 0.13, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 2.04–2.12 (m, 2 H, 7-H2), 2.25–2.32 (m, 1 H, 8-H), 2.43–2.48 (m, 1 H, 8-H), 3.68–3.78 (m, 2 H, 6-H2), 4.17 (pseudo-t, J ≈ 7.9 Hz, 1 H, 8a-H), 7.32 (mc, 2 H), 7.47 (t, J = 1.2 Hz, 1 H). 13C NMR (CDCl3, 151 MHz): δ 24.0 (t, C(7)), 28.2 (t, C(8)), 48.8 (q, 4JC-F = 2.5 Hz, C(6)), 58.1 (d, C(8a)), 118.5 (q, 1JC-F = 275.7 Hz, CF3), 128.2, 130.2, 130.4 (3 d, 3 CH), 133.3, 135.2, 136.5 (3 s, 3 i-C), 137.6 (q, 2JC-F = 36.2 Hz, C(4)), 161.0 (s, C=O). 19F NMR (CDCl3, 565 MHz): δ −68.4 (s, CF3). IR (neat): ν 1689 and 1640 (C=O), 1480, 1443, 1228, 1189, 1160–1118 (CF3) cm−1. ESI-MS (m/z): 353.5 (24), 352.4 (100). C13H10Cl2F3N3O (352.1): calcd. C 44.34, H 2.86, N 11.93; found: C 44.42, H 2.96, N 12.04.
(S)-2-Phenyl-4-trifluoromethyl-6,7,8,8a-tetrahydropyrrolo [1,2-d][1,2,4]triazin-1(2H)-one (7f): CC (SiO2, petroleum ether/EtOAc 4:1), 170 mg (60%). Yellow solid, m.p. 61–63 °C. [α]D20 = +22.6 (c 0.18, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 2.02–2.11 (m, 2 H, 7-H2), 2.25–2.32 (m, 1 H, 8-H), 2.42–2.47 (m, 1 H, 8-H), 3.67–3.75 (m, 2 H, 6-H2), 4.14 (dd, J = 6.9, 8.8 Hz, 1 H, 8a-H), 7.25–7.28, 7.38–7.41, 7.54–7.56 (3 m, 1 H, 2 H, 2 H). 13C NMR (CDCl3, 151 MHz): δ 24.1 (t, C(7)), 28.4 (t, C(8)), 48.7 (q, 4JC-F = 2.3 Hz, C(6)), 58.2 (d, C(8a)), 118.7 (q, 1JC-F = 275.6 Hz, CF3), 124.5, 127.0, 128.7 (3 d, 5 CH), 137.3 (q, 2JC-F = 36.1 Hz, C(4)), 140.2 (s, i-C), 160.9 (s, C=O). 19F NMR (CDCl3, 565 MHz): δ −68.4 (s, CF3). IR (neat): ν 1685 and 1640 (C=O), 1457, 1312, 1199, 1133 (CF3), 1102 cm−1. ESI-MS (m/z): 284.2 (32, [M+H]+), 205.2 (100, [M–Ph]+). C13H12F3N3O (283.2): calcd. C 55.12, H 4.27, N 14.84; found: C 55.41, H 4.44, N 14.59.
(S)-2-(4-Tolyl)-4-trifluoromethyl-6,7,8,8a-tetrahydropyrrolo [1,2-d][1,2,4]triazin-1(2H)-one (7g): CC (SiO2, petroleum ether/CH2Cl2 1:3), 166 mg (56%). Light gray solid, m.p. 78–80 °C. [α]D20 = +22.7 (c 0.16, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 2.03–2.11 (m, 2 H, 7-H2), 2.25–2.32 (m, 1 H, 8-H), 2.35 (s, 3 H, Me), 2.42–2.47 (m, 1 H, 8-H), 3.67–3.76 (m, 2 H, 6-H2), 4.13 (dd, J = 6.9, 8.8 Hz, 1 H, 8a-H), 7.18–7.20, 7.39–7.41 (2 m, 2 H each). 13C NMR (CDCl3, 151 MHz): δ 21.2 (q, Me), 24.1 (t, C(7)), 28.5 (t, C(8)), 48.7 (q, 4JC-F = 2.4 Hz, C(6)), 58.2 (d, C(8a)), 118.7 (q, 1JC-F = 275.6 Hz, CF3), 124.5, 129.4 (2 d, 4 CH), 137.0 (s, i-C), 137.2 (q, 2JC-F = 36.1 Hz, C(4)), 137.7 (s, i-C), 160.9 (s, C=O). 19F NMR (CDCl3, 565 MHz): δ −68.4 (s, CF3). IR (neat): ν 1687 and 1644 (C=O), 1511, 1444, 1198, 1120 (CF3), 820 cm−1. ESI-MS (m/z): 299.2 (100, [M+2H]+). C14H14F3N3O (297.1): calcd. C 56.56, H 4.75, N 14.14; found: C 56.67, H 4.84, N 13.90.
(S)-2-(4-Benzyloxyphenyl)-4-trifluoromethyl-6,7,8,8a-tetrahydropyrrolo [1,2-d][1,2,4]triazin-1(2H)-one (7h): CC (SiO2, petroleum ether/EtOAc 4:1), 183 mg (47%). Colorless solid, m.p. 97–99 °C. [α]D20 = +35.0 (c 0.21, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 2.02–2.10 (m, 2 H, 7-H2), 2.24–2.31 (m, 1 H, 8-H), 2.42–2.47 (m, 1 H, 8-H), 3.66–3.74 (m, 2 H, 6-H2), 4.13 (dd, J = 6.9, 8.9 Hz, 1 H, 8a-H), 5.07 (s, 2 H, OCH2), 6.97–7.00, 7.31–7.34, 7.37–7.39, 7.41–7.44 (4 m, 2 H, 1 H, 2 H, 4 H). 13C NMR (CDCl3, 151 MHz): δ 24.1 (t, C(7)), 28.5 (t, C(8)), 48.7 (q, 4JC-F = 2.6 Hz, C(6)), 58.2 (d, C(8a)), 70.3 (t, OCH2), 115.0 (d, 2 CH), 118.7 (q, 1JC-F = 275.6 Hz, CF3), 126.1, 127.6, 128.1, 128.7 (4 d, 7 CH), 133.5, 137.0 (2 s, 2 i-C), 137.2 (q, 2JC-F = 36.2 Hz, C(4)), 157.6 (s, i-C), 160.9 (s, C=O). 19F NMR (CDCl3, 565 MHz): δ −68.4 (s, CF3) ppm. IR (neat): ν 1685 and 1644 (C=O), 1506, 1450, 1338, 1241, 1133 (CF3), 1018 cm−1. ESI-MS (m/z): 412.4 (77, [M+Na]+), 390.4 (100, [M+H]+). C20H18F3N3O2 (389.4): calcd. C 61.69, H 4.66, N 10.79; found: C 61.50, H 4.83, N 10.56.
Synthesis of (S)-4-Methyl-5-phenyl-1-(4-tolyl)-3-trifluoromethyl-4,5-dihydro-1,2,4-triazin-6(1H)-one ((S)-6r)): A solution of 1,2,4-triazin-6(1H)-one (S)-6q (0.5 mmol, 167 mg) in anhydrous MeOH (5.0 mL) was added dropwise to a vigorously stirred solution of sodium methoxide (5.0 mmol, 270 mg) in dry MeOH (25 mL) under argon, at room temperature. Then MeI (5.0 mmol, 705 mg) was added to the resulting mixture and the stirring was continued for 24 h. After the solvents were removed in vacuo, the residue was washed with EtOAc (3 × 20 mL). The organic layers were combined, the solvent was removed, and the crude product was purified by CC (SiO2, DCM) to give (S)-6r (101 mg, 58%). Colorless solid, m.p. 92–93 °C. [α]D20 = +5.2 (c 0.24, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 2.34 (s, 3 H, Me), 3.05 (s, 3 H, NMe), 4.94 (s, 1 H, 5-H), 7.17–7.19, 7.36–7.44 (2 m, 2 H, 7 H). 13C NMR (CDCl3, 151 MHz): δ 21.2 (q, Me), 36.2* (q, JC-F = 3.4 Hz, NMe), 66.0 (d, C(5)), 118.9 (q, 1JC-F = 275.9 Hz, CF3), 124.4, 126.9, 129.36, 129.43, 129.5 (5 d, 9 CH), 135.9 (s), 136.5 (q, 2JC-F = 34.3 Hz, C(3)), 137.1, 137.6 (2 s), 158.6 (s, C=O); *through-space C–F coupling observed. 19F NMR (CDCl3, 565 MHz): δ −66.1 (s, CF3) ppm. IR (neat): ν 1681 and 1655 (C=O), 1513, 1416, 1364, 1238, 1182, 1126 (CF3), 1077 cm−1. ESI-MS (m/z): 370.1 (100, [M+Na]+), 348.1 (80, [M+H]+), 318.2 (94). C18H16F3N3O (347.1): calcd. C 62.24, H 4.64, N 12.10; found: C 61.99, H 4.51, N 12.08. A sample of rac-6r (111 mg, 64%; 0.5 mmol scale) was obtained in an analogous manner starting with rac-6q. Colorless crystals, m.p. 93–95 °C. The NMR spectra (1H and 13C) of rac-6r were in accordance with those of (S)-6r.
Synthesis of 1-(4-tolyl)-3-trifluoromethyl-1,2,4-triazin-6(1H)-one (6s): A mixture of 4,5-dihydro-1,2,4-triazinone 6g (0.5 mmol, 128.5 mg), K3Fe(CN)6 (3.0 mmol, 987 mg), aqueous solution of Na2CO3 (0.5M, 10 mL), and Et4NBr (15 mol%) in CH2Cl2 (10 mL) was vigorously stirred at room temperature for 4 h (monitored on TLC). The resulting mixture was extracted with CH2Cl2 (3 × 10 mL), the combined organic layers were dried over anh. Na2SO4, filtered and the solvents were removed under reduced pressure. The crude product was purified by standard CC (SiO2, CH2Cl2) to give 6s (99 mg, 78%). Colorless solid, m.p. 80–82 °C. 1H NMR (CDCl3, 600 MHz): δ 2.36 (s, 3 H, Me), 7.25, 7.57 (2 dbr, J ≈ 8.3 Hz, 2 H each), 8.47 (s, 1 H, 5-H). 13C NMR (CDCl3, 151 MHz): δ 21.4 (q, Me), 119.2 (q, 1JC-F = 273.6 Hz, CF3), 123.9, 129.9 (2 d, 4 CH), 136.6, 140.2 (2 s, 2 i-C), 140.6 (q, 2JC-F = 39.0 Hz, C(3)), 152.6 (s, C=O), 161.0 (d, C(5)). 19F NMR (CDCl3, 565 MHz): δ −69.7 (s, CF3). IR (neat): ν 1674 (C=O), 1391, 1346, 1156, 1088 (CF3) cm−1. ESI-MS (m/z): 256.1 (100, [M+H]+). C11H8F3N3O (255.1): calcd. C 51.77, H 3.16, N 16.47; found: C 51.59, H 3.23, N 16.43.
General procedure for catalytic hydrogenation reactions: A solution of the corresponding triazinone (7a or 7h, 0.5 mmol) in EtOH (10 mL) was added Pd/C (5.0 mmol), and the resulting mixture was vigorously shaken in the atmosphere of H2 (3 atm) for the required time. The mixture was filtered through Celite, washed with EtOH (5 mL), and the solvents were removed under reduced pressure. The resulting mixture was filtered through a short plug of silica (CC) to give the spectroscopically pure product.
(S)-2-(4-Aminophenyl)-4-trifluoromethyl-6,7,8,8a-tetrahydropyrrolo [1,2-d][1,2,4]-triazin-1(2H)-one (7i): Reaction time: 3 h; CC (SiO2, silica was washed with 5% Et3N in EtOAc prior to use; petroleum ether/EtOAc 4:1), 132 mg (89%). Thick light orange oil. [α]D20 = +43.1 (c 0.15, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 2.03–2.10 (m, 2 H, 7-H2), 2.23–2.32 (m, 1 H, 8-H), 2.41–2.46 (m, 1 H, 8-H), 3.65–3.74 (m, 2 H, 6-H2), 3.70 (sbr, 2 H, NH2), 4.12 (dd, J = 6.9, 8.8 Hz, 1 H, 8a-H), 6.66–6.96, 7.24–7.27 (2 m, 2 H each). 13C NMR (CDCl3, 151 MHz): δ 24.1 (t, C(7)), 28.5 (t, C(8)), 48.7 (q, 4JC-F = 2.1 Hz, C(6)), 58.2 (d, C(8a)), 115.1 (d, 2 CH), 118.7 (q, 1JC-F = 275.9 Hz, CF3), 126.1 (d, 2 CH), 131.5 (s, i-C), 137.0 (q, 2JC-F = 36.1 Hz, C(4)), 145.6 (s, i-C), 160.8 (s, C=O). 19F NMR (CDCl3, 565 MHz): δ −68.3 (s, CF3). IR (neat): ν 3362 (NH), 1674 and 1633 (C=O), 1513, 1446, 1193, 1140–1095 (CF3) cm−1. ESI-MS (m/z): 299.3 (100, [M+H]+), 279.3 (30).
(S)-2-(4-Hydroxyphenyl)-4-trifluoromethyl-6,7,8,8a-tetrahydropyrrolo [1,2-d][1,2,4]-triazin-1(2H)-one (7j): Reaction time: 16 h; CC (SiO2, hexanes/EtOAc 2:3), 100 mg (67%). Colorless solid, m.p. 94–96 °C. [α]D20 = +23.2 (c 0.18, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 2.02–2.10 (m, 2 H, 7-H2), 2.24–2.31 (m, 1 H, 8-H), 2.42–2.47 (m, 1 H, 8-H), 3.67–3.76 (m, 2 H, 6-H2), 4.15 (dd, J = 6.9, 8.9 Hz, 1 H, 8a-H), 5.69 (sbr, 1 H, OH), 6.74–6.77, 7.28–7.30 (2 m, 2 H each). 13C NMR (CDCl3, 151 MHz): δ 24.1 (t, C(7)), 28.5 (t, C(8)), 48.7 (q, 4JC-F = 2.5 Hz, C(6)), 58.2 (d, C(8a)), 115.8 (d, 2 CH), 118.7 (q, 1JC-F = 275.6 Hz, CF3), 126.5 (d, 2 CH), 133.0 (s, i-C), 137.4 (q, 2JC-F = 36.1 Hz, C(4)), 155.0 (s, i-C), 161.1 (s, C=O). 19F NMR (CDCl3, 565 MHz): δ −68.4 (s, CF3). IR (neat): ν 3317 (OH), 1666 and 1636 (C=O), 1513, 1446, 1341, 1189–1122 (CF3), 835 cm−1. ESI-MS (m/z): 300.2 (100, [M+H]+), 298.2 (59).
General procedure for synthesis of hydrazonoyl bromides 11: Following the general literature protocol [25], arylhydrazone 14 (1.0 mmol) was dissolved in dry DMF (3 mL), the solution was cooled to 0 °C, then solid NBS (1.05 mmol, 187 mg) was added and stirring was continued at this temperature. After the starting hydrazone was fully consumed (TLC monitoring, typically ca. 2 h), the resulting mixture was extracted with H2O/Et2O 1:1 mixture (20 mL), the organic layer was washed with H2O (3 × 10 mL), dried over anh. Na2SO4, filtered and the solvents were removed in vacuo. Crude products were purified by column chromatography.
N-(3-Nitrophenyl)-trifluoroacetohydrazonoyl bromide (11b): reaction time: 2h; CC (SiO2, petroleum ether/CH2Cl2 3:2), 265 mg (85%). Yellow solid, m.p. 110–111 °C. 1H NMR (CDCl3, 600 MHz): δ 7.49–7.52, 7.87–7.91, 7.99–8.01 (3 m, 2 H, 1 H, 1 H), 8.24 (sbr, 1 H, NH). 13C NMR (CDCl3, 151 MHz): δ 107.1 (q, 2JC-F = 44.0 Hz, =CCF3), 109.2, 117.7 (2 d, 2 CH), 118.2 (q, 1JC-F = 272.1 Hz, CF3), 120.0, 130.7 (2 d, 2 CH), 142.7, 149.4 (2 s, 2 i-C). 19F NMR (CDCl3, 565 MHz): δ −66.6 (s, CF3). IR (neat): ν 3258 (NH), 1614, 1524, 1346, 1304, 1238, 1121–1075 (CF3) cm−1. (-)-ESI-MS (m/z): 311.9 (100, [M{81Br}–H]−), 309.9 (99, [M{79Br}–H]−). C8H5BrF3N3O2 (312.0): calcd. C 30.79, H 1.62, N 13.47; found: C 30.95, H 1.88, N 13.65.
N-(2,4-Dichlorophenyl)-trifluoroacetohydrazonoyl bromide (11e): Reaction time: 3 h; CC (SiO2, hexanes), 332 mg (99%). Thick yellow oil. 1H NMR (CDCl3, 600 MHz): δ 7.24 (dd, J = 2.3, 8.8 Hz, 1 H), 7.35 (d, J = 2.3 Hz, 1 H), 7.42 (d, J = 8.8 Hz, 1 H), 8.52 (sbr, 1 H, NH). 13C NMR (CDCl3, 151 MHz): δ 107.8 (q, 2JC-F = 43.9 Hz, =CCF3), 116.5 (d, CH), 118.3 (q, 1JC-F = 271.9 Hz, CF3), 119.2, 127.8 (2 s, 2 i-C), 128.6, 129.2 (2 d, 2 CH), 136.6 (s, i-C). 19F NMR (CDCl3, 565 MHz): δ −66.6 (s, CF3). IR (neat): ν 3314 (NH), 1595, 1506, 1327, 1282, 1124, 1208, 1133 (CF3), 969 cm−1. (-)-ESI-MS (m/z): 336.7 (38), 335.6 (11), 334.7 (100), 332.8 (63). C8H4BrCl2F3N2 (335.9): calcd. C 28.60, H 1.20, N 8.34; found: C 28.42, H 1.36, N 8.02.
General procedure for synthesis of trifluoroacetaldehyde arylhydrazones 14: Following the literature protocol [52], a mixture of arylhydrazine hydrochloride (1.0 mmol), excess fluoral hydrate (ca. 3.0 mmol), and freshly activated powdered molecular sieves 4Å (ca. 450 mg) in MeOH (3.5 mL) was heated in a closed ampoule in an oil bath (75 °C) overnight. The solution was cooled to room temperature and filtered through a short pad of Celite, which was washed with several portions of CH2Cl2 (4 × 5 mL). The combined organic layers were washed with H2O (10 mL), then with 5%-aqueous solution of NaHCO3 (10 mL), and dried over Na2SO4. The solid inorganics were filtered off and the solvents were removed under reduced pressure (cold bath). The crude products were purified by standard CC to give spectroscopically pure materials, which were used for the next step without further purification.
Trifluoroacetaldehyde 3-nitrophenylhydrazone (14b): CC (SiO2, petroleum ether/CH2Cl2 3:2), 152 mg (65%). Yellow solid, m.p. 156–158 °C. 1H NMR (CDCl3, 600 MHz): δ 7.08 (qd, JH-H = 1.4 Hz, JH-F = 3.9 Hz, 1 H, =CHCF3), 7.43 (ddd, J = 1.2, 2.2, 8.2 Hz, 1 H), 7.47 (tbr, J ≈ 8.0 Hz, 1 H), 7.83 (ddd, J = 1.2, 2.2, 7.9 Hz, 1 H), 7.91 (t, J = 2.2 Hz, 1 H), 8.18 (sbr, 1 H, NH). 13C NMR (CDCl3, 151 MHz): δ 108.3, 116.9, 119.3 (3 d, 3 CH), 120.7 (q, 1JC-F = 269.8 Hz, CF3), 125.2 (q, 2JC-F = 39.6 Hz, =CCF3), 130.5 (d, CH), 144.0, 149.4 (2 s, 2 i-C). 19F NMR (CDCl3, 565 MHz): δ −65.9 (d, JH-F = 3.9 Hz, CF3). IR (neat): ν 3302 (NH), 1610, 1558, 1342, 1290, 1245, 1118 (CF3), 1074 cm−1. (-)-ESI-MS (m/z): 231.8 (100, [M–H]−). C8H6F3N3O2 (233.0): calcd. C 41.21, H 2.59, N 18.02; found: C 41.21, H 2.72, N 18.03.
Trifluoroacetaldehyde 2,4-dichlorophenylhydrazone (14e): CC (SiO2, petroleum ether/CH2Cl2 3:2), 202 mg (79%). Light orange oil. 1H NMR (CDCl3, 600 MHz): δ 7.12 (qd, JH-H = 1.4 Hz, JH-F = 3.8 Hz, 1 H, =CHCF3), 7.22 (dd, J = 2.3, 8.8 Hz, 1 H), 7.31 (d, J = 2.3 Hz, 1 H), 7.46 (d, J = 8.8 Hz, 1 H), 8.33 (sbr, 1 H, NH). 13C NMR (CDCl3, 151 MHz): δ 115.9 (d, CH), 118.2 (s, i-C), 120.8 (q, 1JC-F = 269.7 Hz, CF3), 125.6 (q, 2JC-F = 39.3 Hz, =CCF3), 126.8 (s, i-C), 128.5, 129.0 (2 d, 2 CH), 137.7 (s, i-C). 19F NMR (CDCl3, 565 MHz): δ −65.9 (d, JH-F = 3.8 Hz, CF3). IR (neat): ν 3354 (NH), 1591, 1521, 1357, 1279, 1234, 1115 (CF3), 1051, 913, 816 cm−1. (-)-ESI-MS (m/z): 254.8 (100, [M–H]−). C8H5Cl2F3N2 (256.0): calcd. C 37.38, H 1.96, N 10.90; found: C 37.36, H 2.20, N 10.97.
3. Results and Discussion
Based on our experience in the chemistry of (3+2)-cycloaddition reactions of nitrile imines 1, the first experiments were carried out in dry THF solutions, under inert atmosphere, at room temperature, using hydrazonoyl bromides 11 as the source of nitrile imines and excess Et3N as a base triggering the dehydrohalogenation reaction [25,26,27,28]. For a test experiment, N-(4-nitrophenyl)-trifluoroacetohydrazonoyl bromide (11a) and methyl glycinate (12a, as hydrochloride) were selected as reaction partners, and after the addition of a base (3.0 equiv.), the slow consumption of the nitrile imine precursor was observed (according to TLC monitoring) to give after 16 h the expected (3+3)-cycloadduct 6a, which was isolated by flash chromatography in a fair 59% yield (Scheme 2). Brief optimization of the reaction conditions revealed that increasing the amount of Et3N (8.0 equiv.) enhanced the yield of isolated product 6a (75%) formed as the only product. On the other hand, neither the change of the base (Cs2CO3, pyridine, DBU), nor the type of solvent used (CH2Cl2, toluene) resulted in remarkable change of the chemical yield of the studied reaction. Prompted by the often-observed positive effects of mechanochemical activation on reaction outcomes, (e.g., remarkable shortening of reaction times, higher yields, etc.) [53,54,55], the (3+3)-annulation reaction of 11a and 12a was also tested under ball-mill conditions (steel balls, ø 7 mm, 25 Hz). However, in this case, the formation of viscous mixture was observed upon the reaction progress, which enabled effective ball-milling and the reaction could not be completed. Moreover, the use of three-fold excess of glycinate 12a (with respect to bromide 11a) was necessary to obtain the desired product 6a in a comparable yield (77%) and the remarkably shorter reaction time of 2 h, and for these reasons we waved on the mechanochemical approach.
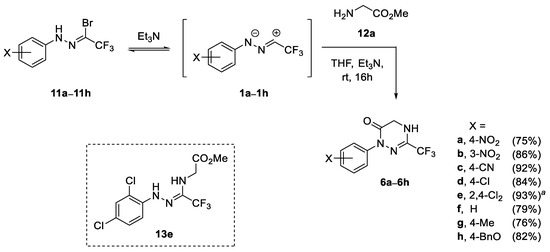
Scheme 2.
Synthesis of 1,2,4-triazin-6(1H)-ones 6a–6h derived from methyl glycinate (scope of nitrile imines 1) and the structure of intermediate 13e; a crude reaction mixture was refluxed for 2 h.
The structure of the isolated product 6a was confirmed based on NMR methods; for example, in 1H NMR spectrum, a broadened doublet (J ≈ 1.5 Hz) attributed to 5-H2 was found at δ 4.30, along with absorption (sbr) of the NH (δ 5.31), and the characteristic set of signals (two dbr located at δ 7.92 and 8.27, J ≈ 9.2 Hz each) of the C6H4NO2 group. Furthermore, two diagnostic quartets attributed to the CF3 group, and the C-3 atom were found at δ 118.2 (1JC-F = 275.2 Hz) and δ 137.3 (2JC-F = 37.8 Hz), respectively, whereas the absorption (s) of the amide-type C=O group was found at δ 158.1. Finally, the 19F NMR indicated the presence of a single CF3 group (singlet at δ −70.6), while the ESI-MS, supplemented by combustion analysis, confirmed the molecular formula of C10H7F3N4O3. Based on the collected data, the structure of isolated product was established as the hitherto unknown 1-(4-nitrophenyl)-3-trifluoromethyl-4,5-dihydro-1,2,4-triazin-6(1H)-one (6a).
With the optimized reaction conditions in hand, a series of variously substituted nitrile imines 1b–1h were checked in the reaction with glycinate 12a (Scheme 2). The required nitrile imine precursors of type 11 were prepared according to the general literature protocols by condensation of arylhydrazines with fluoral hydrate [52], followed by NBS-mediated bromination of the first formed hydrazones 14 (Scheme 3) [25]. As shown in Scheme 2, the expected products were formed in high yields (74–93%), irrespective of the electronic nature of the substituent X. These observations nicely correspond to the previous results reported by Dalloul for reactions of some non-fluorinated nitrile imines with methyl glycinate and other amino acid esters [56,57]. Notably, in the case of highly electron-deficient nitrile imine 1e functionalized at the N-termini with 2,4-dichlorophenyl group, the formation of a mixture of the target material 6e and the first formed acyclic adduct 13e in ca. 3:2 ratio was observed, which nicely evidenced the stepwise character of the studied (3+3)-annulation reaction. Therefore, in the next attempt, the resulting crude mixture was additionally refluxed for 2 h in order to accelerate the second ring-closure step, and after standard work-up, the product 6e was isolated in excellent yield (93%).
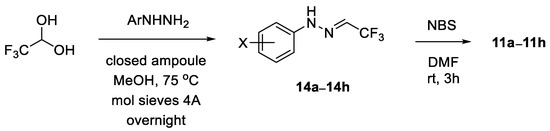
Scheme 3.
Synthesis of hydrazonoyl bromides 11a–11h.
Next, a series of enantiopure α-substituted (S)-amino esters 12b–12i derived from alanine, valine, leucine, phenylglycine, serine, methionine, aspartic acid, and tryptophan, respectively, were examined in reaction with selected hydrazonoyl bromides 11a and 11g to afford the expected optically active products 6i–6q (Scheme 4 and Scheme 5). Thus, along with simple alkyl and aryl substituents in 6i–6l, functional groups such as hydroxy (6m), thioether (6n) and secondary amine (6p) could also be efficiently introduced. Notably, in the case of methyl aspartate (12h), the presence of the additional CO2Me group did not interfere with the subsequent cyclisation step, and the 6-membered product 6o was formed exclusively in excellent yield (93%).
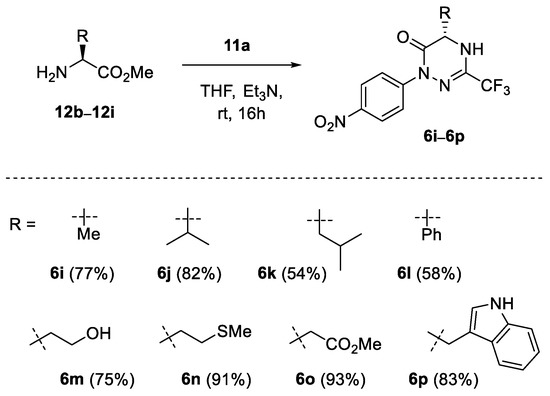
Scheme 4.
Synthesis of chiral 1,2,4-triazin-6(1H)-ones 6i–6q derived from nitrile imine 1a; scope of α-amino esters.
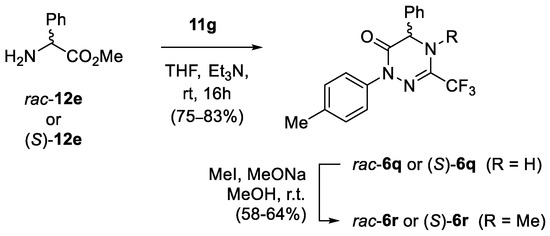
Scheme 5.
Synthesis of racemic and enantiopure phenylglycine-derived 1,2,4-triazin-6-ones rac-6q and (S)-6q, and their transformation into N-methylated analogues 6r.
In order to check the optical purity of the resulting products, phenylglycine-derived product 6q was selected for a more detailed examination, and a sample of racemic 1,2,4-triazinone rac-6q was prepared as a reference compound (Scheme 5). Unfortunately, neither chiral-HPLC analysis of pure samples of 6q nor 1H NMR measurements of their diastereomeric 1:1 mixtures with (+)-(S)-mandelic acid and with (+)-(R)-(tert-butyl)(phenyl)phosphonothioic acid [58] selected as chiral solvating agents was successful. In addition, the attempted derivatization of 1,2,4-triazin-6-ones 6q using enantiopure acid chloride derived from (S)-naproxen was in vain. Nevertheless, the treatment of 6q with strongly electrophilic MeI under basic conditions enabled fully regioselective methylation at N(4) to afford the expected products (S)-6r and rac-6r isolated in 58% and 64% yield, respectively. Subsequent HPLC analysis of both samples proved the optical purity of the product derived from chiral methyl phenylglycinate (S)-12e (for details, see Supporting Information). Hence, taking into account the high susceptibility for epimerization of the phenylglycine-derived compounds under basic conditions, we assumed that all the other products in the series (6i–6q) were optically pure as well.
The 13C NMR spectrum of N-methylated product 6r also deserves a brief comment. Along with the expected diagnostic quartets located at δ 118.9 (1JC-F = 275.9 Hz) and δ 136.5 (2JC-F = 34.3 Hz) attributed to the CF3 group and the C(3) atom, respectively, an additional quartet of the N-Me at δ 36.2 (JC-F = 3.4 Hz) was found. Apparently, the close proximity of the CF3 and the Me groups results in the observed C–F through-space coupling.
In addition to a series of experiments performed with the primary α-amino esters 12a–12i, (S)-proline methyl ester (12j) was also involved in the study as a model secondary amino-compound. As shown in Scheme 6, (3+3)-annulation of 12j with nitrile imines of type 1 carried out under the analogous reaction conditions provided the expected bicyclic products 7a–7h, which were generally isolated in fair yield (47–93%). Similar to the cyclo-condensation of 2,4-dichlorophenyl-functionalized nitrile imine 1e with methyl glycinate (Scheme 1), the reaction of 12j with 1e also provided the respective amidrazone intermediate, which required prolonged heating in THF (3 d, 60 °C) to afford the target bicyclic material 7e (50%).
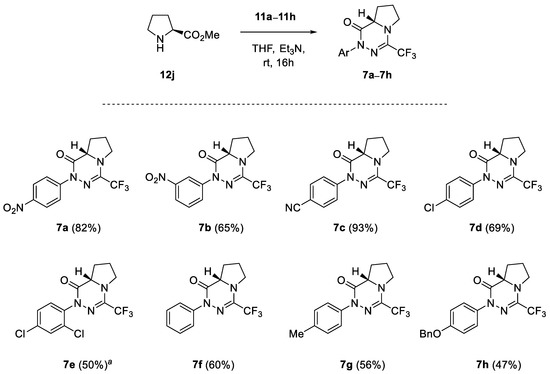
Scheme 6.
Synthesis of l-proline-derived bicyclic 1,2,4-triazinones 7a–7h; crude reaction mixture was heated at 60 °C for 3 d.
Having in hand two series of 3-trifluoromethylated 1,2,4-triazin-6-ones 6 and 7, selected functional group interconversions were carried out to demonstrate the robustness of the core heterocycle. Thus, NH2 and OH groups were easily accessed by medium-pressure (3 atm) catalytic hydrogenation of the NO2 and BnO groups in 7a and 7h, respectively, and the expected products 7i (89%) and 7j (67%) were isolated in high yields (Scheme 7). On the other hand, with the treatment of the 4,5-dihydro-1,2,4-triazine derivative 6g with an aqueous solution of K3Fe(CN)6, in the presence of tetraethylammonium bromide as a phase transfer catalyst, the smooth oxidation of the N(4)–C(5) bond was observed to yield fairly stable 1,2,4-triazin-6-one 6s (78%) identified as the only product. The structure of 6s was confirmed based on 1H and 13C NMR spectra supplemented by 2D measurements (HMQC); particularly, characteristic low-field shifted absorptions attributed to C(5)-H unit, i.e., a singlet at δ 8.47 in 1H NMR and a signal at δ 161.0 in 13C NMR, were observed.
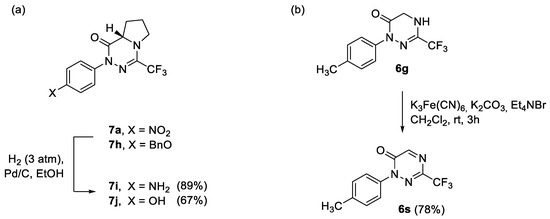
Scheme 7.
Selected transformations of title heterocycles: (a) synthesis of amino- and hydroxy-functionalized products 7i and 7j through catalytic hydrogenation of 7a and 7h, respectively; (b) selective oxidation of 6g with K3Fe(CN)6 under PTC-conditions leading to 1,2,4-triazin-6-one 6s.
Finally, prompted by recent reports on anticancer properties of some fluorinated and non-fluorinated 1,2,4-triazinone-based materials [43,44,45,46,47,48,49,50,51], the cytotoxicity of 3-CF3-1,2,4-triazin-6(1H)-ones 6a–6q and 7a–7h obtained in this study was checked against human promyelocytic leukemia (HL-60) and breast cancer adenocarcinoma (MCF-7) cell lines (for details, see Supplementary Materials). However, most of the examined samples showed no mentionable cytotoxicity; for example, all compounds in glycine-derived series 6a–6h proved inactive irrespective of the type of substituent X, whereas in the case of chiral representatives 6i–6q only for the analogue functionalized with H-donor/acceptor 2-hydroxyethyl group (compound 6m), a moderate (IC50 = 23.0 μM) activity against HL-60 line was observed. For the proline-derived analogues 7a–7h, the best result was noticed for benzonitrile-functionalized analogue 7c with only low cytotoxicity of IC50 = 45.4 μM against HL-60 cell line.
4. Conclusions
In the presented study, the synthesis of a series of 4,5-dihydro-1,2,4-triazin-6(1H)-ones functionalized with the CF3 group is reported. The devised protocol is based on the (3+3)-annulation of methyl esters derived from natural α-amino acids with in situ generated trifluoroacetonitrile imines applied as reactive 1,3-dipolar reaction partners. Notably, starting with chiral α-amino esters, no racemization occurred under the optimized reaction conditions, and the expected enantiopure materials were isolated as the only products. Furthermore, with the application of methyl l-prolinate as a model secondary amino ester, the respective fused 1,2,4-triazinones were obtained. The selected functional group interconversions performed under catalytic hydrogenation or mild PTC-oxidation conditions demonstrated remarkable stability of the core heterocycle. Thus, the presented method offers straightforward access to the desired heterocyclic system functionalized not only with simple alkyl and aryl substituents, but also with such functional groups as nitro, cyano, hydroxy, amino, methoxycarbonyl, and sulfide, as well as 1H-indol-3-yl and halogen(s). Taking into account the easy accessibility of the starting materials and the exceptionally mild reaction conditions, the presented approach can be recommended for the synthesis of title 3-trifluoromethylated heterocycles, and nicely supplements previous reports on the synthesis of 1,2,4-triazin-6(1H)-ones exploiting amino acids and their derivatives as key building blocks [56,57,59,60,61,62,63,64].
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ma16020856/s1: Copies of 1H and 13C NMR spectra of all new compounds, HPLC analyses, technical details, and results on biological activity screening. Ref. [65] cited in Supplementary Materials.
Author Contributions
Conceptualization, M.J.; methodology, M.J. and K.G.-J.; investigation, A.K., M.C., K.Ś., G.U.-J. and A.J.; writing—original draft preparation, M.J. and K.G.-J.; writing—review and editing, M.J. and K.G.-J.; supervision, M.J.; funding acquisition, M.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the University of Lodz in the framework of the IDUB grant (M.J.; Grant No. 3/IDUB/DOS/2021).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Raw electronic experimental NMR data (FIDs), as well as samples of all new compounds, are available from the authors.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev. 2008, 37, 308–319. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar] [CrossRef]
- Nenajdenko, V. (Ed.) Fluorine in Heterocyclic Chemistry, Vol. 1: 5-Membered Heterocycles and Macrocycles; Springer International Publishing: Manhattan, NY, USA, 2014. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, L.; Wang, H.; Wang, H.; Jiao, W.; Chen, G.; Zhang, P.; Hui, D.; Jian, X. A brief review for fluorinated carbon: Synthesis. Properties and applications. Nanotechnol. Rev. 2019, 8, 573–586. [Google Scholar] [CrossRef]
- Lemoine, K.; Hémon-Ribaud, A.; Leblanc, M.; Lhoste, J.; Tarascon, J.-M.; Maisonneuve, V. Fluorinated materials as positive electrodes for Li- and Na-ion batteries. Chem. Rev. 2022, 122, 14405–14439. [Google Scholar] [CrossRef]
- Chen, X.; Fan, K.; Liu, Y.; Liu, X.; Feng, W.; Wang, X. Recent advances in fluorinated graphene from synthesis to applications: Critical review on functional chemistry and structure engineering. Adv. Mater. 2022, 34, 2101665. [Google Scholar] [CrossRef]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in pharmaceutical industry: Fluorine-containing drugs introduced to the market in the last decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yan, K.; Fu, C.; Peng, H.; Hawker, C.J.; Whittaker, A.K. Biological utility od fluorinated compounds: From materials design to molecular imaging, therapeutics and environmental remediation. Chem. Rev. 2022, 122, 167–208. [Google Scholar] [CrossRef]
- Tiz, D.B.; Bagnoli, L.; Rosati, O.; Marini, F.; Sancineto, L.; Santi, C. New halogen-containing drugs approved by FDA in 2021: An overview on their syntheses and pharmaceutical use. Molecules 2022, 27, 1643. [Google Scholar] [CrossRef]
- Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. Current contributions of organofluorine compounds to the agrochemical industry. iScience 2020, 23, 101467. [Google Scholar] [CrossRef]
- Gakh, A.A.; Shermolovich, Y. Trifluoromethylated heterocycles. Curr. Top. Med. Chem. 2014, 14, 952–965. [Google Scholar] [CrossRef]
- Meyer, F. Trifluoromethyl nitrogen heterocycles: Synthetic aspects and potential biological targets. Chem. Commun. 2016, 52, 3077–3094. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Sameem, B. Fluorinated N-heterocycles as conformationally diverse bioactives for drug discovery. In Targets in Heterocyclic Systems; Italian Chemical Society: Rome, Italy, 2021; Volume 25, pp. 502–516. [Google Scholar] [CrossRef]
- Petrov, V.A. (Ed.) Fluorinated Heterocyclic Compound: Synthesis, Chemistry, and Applications; J. Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009. [Google Scholar]
- Luzzio, F.A. Synthesis and reactivity of fluorinated heterocycles. In Advances in Heterocyclic Chemistry; Academic Press: Cambridge, MA, USA, 2020; Volume 132, pp. 1–84. [Google Scholar] [CrossRef]
- Sap, J.B.I.; Meyer, C.F.; Straathof, N.J.W.; Iwumene, N.; am Ende, C.W.; Trabanco, A.A.; Governeur, V. Late-stage difluoromethylation: Concepts, developments and perspective. Chem. Soc. Rev. 2021, 50, 8214–8247. [Google Scholar] [CrossRef] [PubMed]
- Mlostoń, G.; Shermolovich, Y.; Heimgartner, H. Synthesis of fluorinated and fluoroalkylated heterocycles containing at least one sulfur atom via cyclocondensation reactions. Materials 2022, 15, 7244. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Zhu, S.Z. Preparation of fluorine-containing heterocyclic compounds via cycloaddition reactions. Mini-Rev. Org. Chem. 2004, 4, 417–435. [Google Scholar] [CrossRef]
- Jamieson, C.; Livingstone, K. The Nitrile Imine 1,3-Dipole; Properties, Reactivity and Applications; Springer Nature: Cham, Switzerland, 2020. [Google Scholar]
- Deepthi, A.; Acharjee, N.; Sruthi, S.L.; Meenakshy, C.B. An overview of nitrile imine based [3+2] cycloadditions over half a decade. Tetrahedron 2022, 116, 132812. [Google Scholar] [CrossRef]
- Mykhailiuk, P.K. 2,2,2-Trifluorodiazoethane (CF3CHN2): A long journey since 1943. Chem. Rev. 2020, 120, 12718–12755. [Google Scholar] [CrossRef]
- Kumar, A.; Khan, W.A.; Ahamad, S.; Mohanan, K. Trifluorodiazoethane: A versatile building block to access trifluoromethylated heterocycles. J. Heterocycl. Chem. 2022, 59, 607–632. [Google Scholar] [CrossRef]
- Chandrasekharan, S.P.; Dhami, A.; Kumar, S.; Mohanan, K. Recent advances in pyrazole synthesis employing diazo compounds and synthetic analogues. Org. Biomol. Chem. 2022, 20, 8787–8817. [Google Scholar] [CrossRef]
- Mlostoń, G.; Urbaniak, K.; Utecht, G.; Lentz, D.; Jasiński, M. Trifluoromethylated 2,3-dihydro-1,3,4-thiadiazoles via the regioselective [3+2]-cycloadditions of fluorinated nitrile imines with aryl, hetaryl, and ferrocenyl thioketones. J. Fluor. Chem. 2016, 192, 147–154. [Google Scholar] [CrossRef]
- Utecht, G.; Sioma, J.; Jasiński, M.; Mlostoń, G. Expected and unexpected results in reactions of fluorinated nitrile imines with (cyclo)aliphatic thioketones. J. Fluor. Chem. 2017, 201, 68–75. [Google Scholar] [CrossRef]
- Utecht, G.; Fruziński, A.; Jasiński, M. Polysubstituted 3-trifluoromethylpyrazoles: Regioselective (3+2)-cycloaddition of trifluoroacetonitrile imines with enol ethers and functional group transformations. Org. Biomol. Chem. 2018, 16, 1252–1257. [Google Scholar] [CrossRef] [PubMed]
- Utecht, G.; Mlostoń, G.; Jasiński, M. A straightforward access to trifluoromethylated spirobipyrazolines through a double (3+2)-cycloaddition of fluorinated nitrile imines with alkoxyallenes. Synlett 2018, 29, 1753–1758. [Google Scholar] [CrossRef]
- Tian, Y.-C.; Li, J.-K.; Zhang, F.-G.; Ma, J.-A. Regioselective decarboxylative cycloaddition route to fully substituted 3-CF3-pyrazoles from nitrilimines and isoxazolidinediones. Adv. Synth. Catal. 2021, 363, 2093–2097. [Google Scholar] [CrossRef]
- Kowalczyk, A.; Utecht-Jarzyńska, G.; Mlostoń, G.; Jasiński, M. A straightforward access to 3-trifluoromethyl-1H-indazoles via (3+2)-cycloaddition of arynes with nitrile imines derived from trifluoroacetonitrile. J. Fluor. Chem. 2021, 241, 109691. [Google Scholar] [CrossRef]
- Han, T.; Wang, K.-H.; Yang, M.; Zhao, P.; Wang, F.; Wang, J.; Huang, D.; Hu, Y. Synthesis of difluoromethylated pyrazoles by the [3+2] cycloaddition reaction of difluoroacetohydrazonoyl bromides. J. Org. Chem. 2022, 87, 498–511. [Google Scholar] [CrossRef]
- Ren, Y.; Ma, R.; Feng, Y.; Wang, K.-H.; Wang, J.; Huang, D.; Lv, X.; Hu, Y. Synthesis of difluoromethyl pyrazolines and pyrazoles by [3+2] cycloaddition reaction of difluoroacetohydrazonoyl bromides with electron-deficient olefins. Asian J. Org. Chem. 2022, 11, e202200438. [Google Scholar] [CrossRef]
- Wang, K.-H.; Liu, H.; Liu, X.; Bian, C.; Wang, J.; Su, Y.; Huang, D.; Hu, Y. Regioselective synthesis of 3-trifluoromethyl 4-subtituted pyrazoles by [3+2]-cycloaddition of trifluoroacetonitrile imines and nitroalkenes. Asian J. Org. Chem. 2022, 11, e202200103. [Google Scholar] [CrossRef]
- Zhou, Y.; Gao, C.-F.; Ma, H.; Nie, J.; Ma, J.-A.; Zhang, F.-G. Quadruple functionalized pyrazole pharmacophores by one-pot regioselective [3+2]-cycloaddition of fluorinated nitrile imines and dicyanoalkenes. Chem. Asian J. 2022, 17, e202200436. [Google Scholar] [CrossRef]
- Utecht-Jarzyńska, G.; Nagła, K.; Mlostoń, G.; Heimgartner, H.; Palusiak, M.; Jasiński, M. A straightforward conversion of 1,4-quinones into polycyclic pyrazoles via [3+2]-cycloaddition with fluorinated nitrile imines. Beilstein J. Org. Chem. 2021, 17, 1509–1517. [Google Scholar] [CrossRef]
- Kowalczyk, A.; Utecht-Jarzyńska, G.; Mlostoń, G.; Jasiński, M. Trifluoromethylated pyrazoles via sequential (3+2)-cycloaddition of fluorinated nitrile imines with chalcones and solvent-dependent deacylative oxidation reactions. Org. Lett. 2022, 24, 2499–2503. [Google Scholar] [CrossRef]
- Utecht-Jarzyńska, G.; Kowalczyk, A.; Jasiński, M. Fluorinated and non-fluorinated 1,4-diarylpyrazoles via MnO2-mediated mechanochemical deacylative oxidation of 5-acylpyrazolines. Molecules 2022, 27, 8446. [Google Scholar] [CrossRef]
- Utecht-Jarzyńska, G.; Jasiński, M.; Świątek, K.; Mlostoń, G.; Heimgartner, H. Novel trifluoromethylated spiro-1,3,4-thiadiazoles via [3+2]-cycloadditions of 2,3-diphenylcyclopropenethione with selected in situ-generated nitrile imines derived from trifluoroacetonitrile. Heterocycles 2020, 101, 251–262. [Google Scholar] [CrossRef]
- Grzelak, P.; Utecht, G.; Jasiński, M.; Mlostoń, G. First (3+2)-cycloadditions of thiochalcones as C=S dipolarophiles: Efficient synthesis of 1,3,4-thiadiazoles via reactions with fluorinated nitrile imines. Synthesis 2017, 49, 2129–2137. [Google Scholar] [CrossRef]
- Utecht-Jarzyńska, G.; Mykhaylychenko, S.S.; Rusanov, E.B.; Shermolovich, Y.G.; Jasiński, M.; Mlostoń, G. Highly fluorinated 2,3-dihydro-1,3,4-thiadiazole derivatives via [3+2]-cycloadditions of tertiary thioamides with nitrile imines derived from trifluoroacetonitrile. J. Fluor. Chem. 2021, 242, 109702. [Google Scholar] [CrossRef]
- Zhang, Y.; Zeng, J.-L.; Chen, Z.; Wang, R. Base-promoted (3+2)-cycloaddition of trifluoroacetohydrazonoyl chlorides with imidates en route to trifluoromethyl-1,2,4-triazoles. J. Org. Chem. 2022, 87, 14514–14522. [Google Scholar] [CrossRef] [PubMed]
- Utecht-Jarzyńska, G.; Michalak, A.; Banaś, J.; Mlostoń, G.; Jasiński, M. Trapping of trifluoroacetonitrile imines with mercaptoacetaldehyde and mercaptocarboxylic acids: An access to fluorinated 1,3,4-thiadiazine derivatives via (3+3)-annulation. J. Fluor. Chem. 2019, 222–223, 8–14. [Google Scholar] [CrossRef]
- Kumar, R.; Sirohi, T.S.; Singh, H.; Yadav, R.; Roy, R.K.; Chaudhary, A.; Pandeya, S.N. 1,2,4-Triazine analogs as novel class of therapeutic agents. Mini-Rev. Med. Chem. 2014, 14, 168–207. [Google Scholar] [CrossRef]
- Cascioferro, S.; Parrino, B.; Spaño, V.; Carbone, A.; Montalbano, A.; Barraja, P.; Diana, P.; Cirrincione, G. An overview on the recent developments of 1,2,4-triazine derivatives as anticancer compounds. Eur. J. Med. Chem. 2017, 142, 328–375. [Google Scholar] [CrossRef]
- Krauth, F.; Dahse, H.-M.; Rüttinger, H.-H.; Frohberg, P. Synthesis and characterization of novel 1,2,4-triazine derivatives with antiproliferative activity. Bioorg. Med. Chem. 2010, 18, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Makki, M.S.I.; Abdel-Rahman, R.M.; Khan, K.A. Fluorine substituted 1,2,4-triazinones as potential ani-HIV-1 and CDK2 inhibitors. J. Chem. 2014, 2014, 430573. [Google Scholar] [CrossRef]
- Sweeney, M.; Coyle, R.; Kavanagh, P.; Berezin, A.A.; Lo Re, D.; Zissimou, G.A.; Koutentis, P.A.; Carty, M.P.; Aldabbagh, F. Discovery of anti-cancer activity for benzo[1,2,4]triazin-7-ones: Very strong correlation to pleurotin and thioredoxin reductase inhibition. Bioorg. Med. Chem. 2016, 24, 3565–3570. [Google Scholar] [CrossRef] [PubMed]
- Srinivasa Rao, D.; Pavan Kumar, G.V.; Pooja, B.; Harika, G.; Anil Kumar, Y.; Sadasiva Rao, G. An extensive review on 1,2,3 and 1,2,4-triazines scaffold-valuable lead molecules with potent and diverse pharmacological activities. Chem. Sin. 2016, 7, 101–130. [Google Scholar]
- Zaki, I.; Abdelhameid, M.K.; El-Deen, I.M.; Abdel Wahab, A.H.A.; Ashmawy, A.M.; Mohamed, K.O. Design, synthesis and screening of 1,2,4-triazinone derivatives as potential antitumor agents with apoptosis inducing activity on MCF-7 breast cancer cell line. Eur. J. Med. Chem. 2018, 156, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Saloutina, L.V.; Zapevalov, A.Y.; Kodess, M.I.; Slepukhin, P.A.; Ganebnykh, I.N.; Saloutin, V.I.; Chupakhin, O.N. Trifluoromethyl-containing 1,2,4-triazines. Synthesis on the base of perfluorobiacetyl and reactions with thiosemicarbazide and thiourea. J. Fluor. Chem. 2019, 227, 109362. [Google Scholar] [CrossRef]
- Al-Otaibi, F.A.; Bakhotmah, D.A. Synthesis and biological evaluation of new fluorine compounds bearing 4-amino-1,2,4-triazino[4,3-b]-1,2,4-triazin-8-one and the related derivatives as CDK2 inhibitors of tumor cell. Polycycl. Aromat. Comp. 2020, 42, 623–634. [Google Scholar] [CrossRef]
- Wojciechowska, A.; Jasiński, M.; Kaszyński, P. Tautomeric equilibrium in trifluoroacetaldehyde arylhydrazones. Tetrahedron 2015, 71, 2349–2356. [Google Scholar] [CrossRef]
- Howard, J.L.; Cao, Q.; Browne, D.L. Mechanochemistry as an emerging tool for molecular synthesis: What can it offer? Chem. Sci. 2018, 9, 3080–3094. [Google Scholar] [CrossRef]
- Pickhardt, W.; Grätz, S.; Borchardt, L. Direct mechanocatalysis: Using milling balls as catalysts. Chem. Eur. J. 2020, 26, 12903–12911. [Google Scholar] [CrossRef]
- Mlostoń, G.; Celeda, M.; Heimgartner, H.; Duda, D.; Obijalska, E.; Jasiński, M. Synthesis and selected transformations of 2-unsubstituted imidazole N-oxides using a ball-milling mechanochemical approach. Catalysts 2022, 12, 589. [Google Scholar] [CrossRef]
- Awadallah, A.M.; Ferwanah, A.-R.S.; El-Sawi, E.; Dalloul, H.M. Cyclocondensation reactions of nitrilimines: Synthesis of 1,2,4-triazin-6-ones and 1,2,4,5-tetrazines. Heterocycl. Commun. 2002, 8, 369–374. [Google Scholar] [CrossRef]
- Dalloul, H.M.M. Synthesis of spiroheterocycles containing thiadiazole thiadiazine and triazine moieties from nitrilimines. Phosphorus Sulfur Silicon Relat. Elem. 2011, 186, 1876–1884. [Google Scholar] [CrossRef]
- Omelańczuk, J.; Mikołajczyk, M. Chiral t-butylphenylphosphinothioic acid: A useful chiral solvating agent for direct determination of enantiomeric purity of alcohols, thiols, amines, diols, aminoalcohols and related compounds. Tetrahedron Asymmetry 1996, 7, 2687–2694. [Google Scholar] [CrossRef]
- Kowalczyk, A.; Jasiński, M. 4,5-Dihydro-1,2,4-triazin-6(1H)-ones. Chem. Heterocycl. Comp. 2022, 58, 585–587. [Google Scholar] [CrossRef]
- Camparini, A.; Celli, A.M.; Ponticelli, F. Synthesis and structure of dihydro-1,2,4-triazin-6(1H)ones. J. Heterocycl. Chem. 1978, 15, 1271–1276. [Google Scholar] [CrossRef]
- Taylor, E.C.; Macor, J.E. Efficient synthesis of 5-substituted-4,5-dihydro-1,2,4-triazin-6-ones and 5-substituted-1,2,4-triazin-6-ones. J. Heterocycl. Chem. 1985, 22, 409–411. [Google Scholar] [CrossRef]
- Boeglin, D.; Cantel, S.; Martinez, J.; Fehrentz, J.-A. Efficient solid-phase synthesis of 4,5-dihydro-1,2,4-triazin-6(1H)-ones. Tetrahedron Lett. 2003, 44, 459–462. [Google Scholar] [CrossRef]
- Kudelko, A.; Zieliński, W.; Jasiak, K. Synthesis of novel 1-[(1-ethoxymethylene)amino]imidazol-5(4H)-ones and 1,2,4-triazin-6(5H)-ones from optically active α-aminocarboxylic acid hydrazides. Tetrahedron Lett. 2013, 54, 4637–4640. [Google Scholar] [CrossRef]
- Amin, M.A.; Saad, H.A. Synthesis and biological activity of fused heteropolycyclic systems containing an indole moiety. Curr. Org. Synth. 2016, 13, 116–125. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).