
Accelerating First Principles Calculation of Multi-Component Alloy Steady-State Structure and Elastic Properties in Full Component Space



 ,
,
Abstract
:1. Introduction
2. Modeling and Dataset
2.1. First Principles Calculations
2.2. Feature Engineering
2.3. Machine Learning Design
3. Results and Discussion
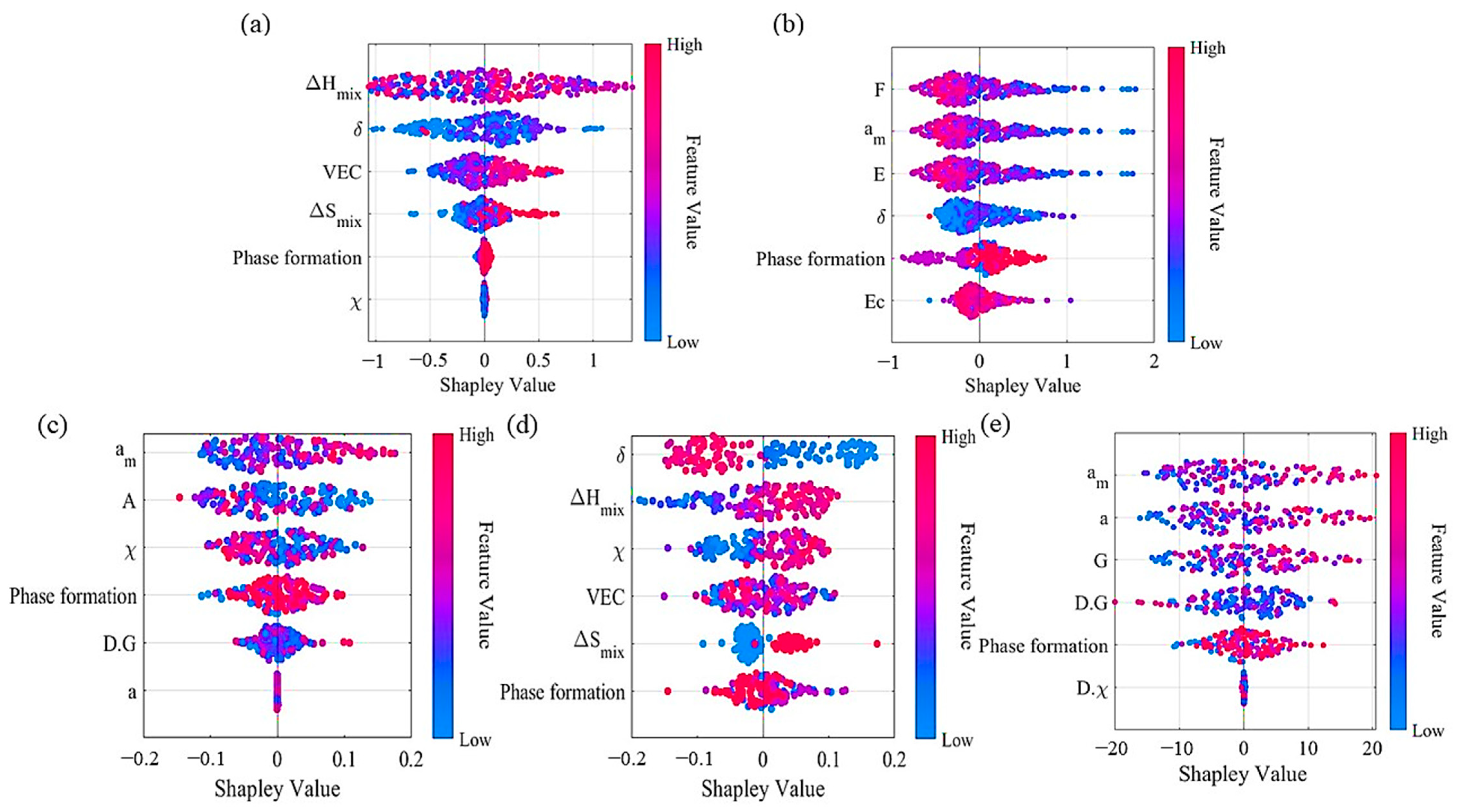
3.1. Feature Analysis
3.2. Model Evaluation and Interpretable Analysis
3.3. Multivariate System Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhang, W.; Liaw, P.K.; Zhang, Y. Science and technology in high-entropy alloys. Sci. China Mater. 2018, 61, 2–22. [Google Scholar] [CrossRef]
- Yeh, J.W.; Chen, S.K.; Lin, S.J.; Gan, J.Y.; Chin, T.S.; Shun, T.T.; Tsau, C.H.; Chang, S.Y. Nanostructured high-entropy alloys with multiple principal element: Novel alloy design concept and outcomes. Adv. Eng. Mater. 2004, 6, 299–363. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, X.; Liaw, P.K. Alloy design and properties optimization of high-entropy alloys. J. Miner. Met. Mater. Soc. 2012, 64, 830–838. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Q.; Han, B.; Song, L.; Li, J.; Yang, J. Investigation on microstructure and properties of AlxCoCrFeMnNi high entropy alloys by ultrasonic impact treatment. J. Alloys Compd. 2020, 816, 152–626. [Google Scholar] [CrossRef]
- Zhang, Y.; Zuo, T.T.; Tang, Z.; Gao, M.C.; Dahmen, K.A.; Liaw, P.K.; Lu, Z.P. Microstructures and properties of high-entropy alloys. Prog. Mater. Sci. 2014, 61, 1–93. [Google Scholar] [CrossRef]
- Lu, W.; Luo, X.; Yang, Y.; Le, W.; Huang, B.; Li, P. Co-free non-equilibrium medium-entropy alloy with outstanding tensile properties. J. Alloys Compd. 2020, 833, 155074. [Google Scholar] [CrossRef]
- Han, Y.; Li, H.; Feng, H.; Li, K.; Tian, Y.; Jiang, Z. Enhancing the strength and ductility of CoCrFeMnNi high-entropy alloy by nitrogen addition. Mater. Sci. Eng. 2020, 789, 139587. [Google Scholar] [CrossRef]
- Thiel, F.; Geissler, D.; Nielsch, K.; Kauffmann, A.; Seils, S.; Heilmaier, M.; Utt, D.; Albe, K.; Motylenko, M.; Rafaja, D.; et al. Origins of strength and plasticity in the precious metal based high-entropy alloy AuCuNiPdPt. Acta Mater. 2020, 185, 400–411. [Google Scholar] [CrossRef]
- Průša, F.; Cabibbo, M.; Šenková, A.; Kučera, V.; Veselka, Z.; Školáková, A.; Vojtěch, D.; Cibulková, J.; Čapek, J. High-strength ultrafine-grained CoCrFeNiNb high-entropy alloy prepared by mechanical alloying: Properties and strengthening mechanism. J. Alloys Compd. 2020, 835, 155308. [Google Scholar] [CrossRef]
- Tian, F.; Varga, L.K.; Chen, N.; Shen, J.; Vitos, L. Empirical design of single phase high-entropy alloys with high hardness. Intermetallics 2015, 58, 1–6. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Z.; Pi, J. Microstructure and wear behavior of the AlCrFeCoNi high-entropy alloy fabricated by additive manufacturing. Mater. Lett. 2020, 261, 127004. [Google Scholar] [CrossRef]
- Soni, V.; Gwalani, B.; Alam, T.; Dasari, S.; Zheng, Y.; Senkov, O.N.; Miracle, D.; Banerjee, R. Phase inversion in a two-phase, BCC+B2, refractory high entropy alloy. Acta Mater. 2020, 185, 89–97. [Google Scholar] [CrossRef]
- Zhao, Y.L.; Yang, T.; Li, Y.R.; Fan, L.; Han, B.; Jiao, Z.B.; Chen, D.; Liu, C.T.; Kai, J.J. Superior high-temperature properties and deformation-induced planar faults in a novel L12-strengthened high-entropy alloy. Acta Mater. 2020, 188, 517–527. [Google Scholar] [CrossRef]
- Ye, Y.F.; Wang, Q.; Lu, J.; Liu, C.T.; Yang, Y. High-entropy alloy: Challenges and prospects. Mater. Today 2016, 19, 349–362. [Google Scholar] [CrossRef]
- Qiao, L.; Zhu, J.; Teng, Y.; Bao, A.; Lai, Z.; Wang, Y. Dynamic solidification model of low-density FeCrNiAl multi-component alloy. Vacuum 2021, 184, 109873. [Google Scholar] [CrossRef]
- Yang, D.; Liu, Y.; Jiang, H.; Liao, M.; Qu, N.; Han, T.; Lai, Z.; Zhu, J. A novel FeCrNiAlTi-based high entropy alloy strengthened by refined grains. J. Alloys Compd. 2020, 823, 153729. [Google Scholar] [CrossRef]
- Jumaev, E.; Hong, S.H.; Kim, J.T.; Park, H.J.; Kim, Y.S.; Mun, S.C.; Park, J.-Y.; Song, G.; Lee, J.K.; Min, B.H.; et al. Chemical evolution-induced strengthening on AlCoCrNi dual-phase high-entropy alloy with high specific strength. J. Alloys Compd. 2019, 777, 828–834. [Google Scholar] [CrossRef]
- Kalidindi, S.R.; Brough, D.B.; Li, S.; Cecen, A.; Blekh, A.L.; Congo, F.Y.P.; Campbell, C. Role of materials data science and informatics in accelerated materials innovation. MRS Bull. 2016, 41, 596–602. [Google Scholar] [CrossRef]
- Brunton, S.L.; Kutz, J.N. Methods for data-driven multiscale model discovery for materials. J. Phys. Mater. 2019, 2, 044002. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Mooney, C.Z. Monte Carlo Simulation; Sage: Washington, DC, USA, 1997. [Google Scholar]
- Alder, B.J.; Wainwright, T.E. Studies in molecular dynamics. I. General method. J. Chem. Phys. 1959, 31, 459–466. [Google Scholar] [CrossRef]
- Boettinger, W.J.; Warren, J.A.; Beckermann, C.; Karma, A. Phase-field simulation of solidification. Annu. Rev. Mater. Res. 2002, 32, 163–194. [Google Scholar] [CrossRef]
- Strang, G.; Fix, G.J.; Griffin, D.S. An Analysis of the Finite-Element Method; Wellesley-Cambridge Press: Wellesley, MA, USA, 1974. [Google Scholar]
- Saal, J.E.; Berglund, I.S.; Sebastian, J.T.; Liaw, P.K.; Olson, G.B. Equilibrium high entropy alloy phase stability from experiments and thermodynamic modeling. Scr. Mater. 2018, 146, 5–8. [Google Scholar] [CrossRef]
- Ma, D.; Yao, M.; Pradeep, K.G.; Tasan, C.C.; Springer, H.; Raabe, D. Phase stability of non-equiatomic CoCrFeMnNi high entropy alloys. Acta. Mater. 2015, 98, 288–296. [Google Scholar] [CrossRef]
- Choi, W.-M.; Jo, Y.H.; Sohn, S.S.; Lee, S.; Lee, B.-J. Understanding the physical metallurgy of the CoCrFeMnNi high-entropy alloy: An atomistic simulation study. NPJ Comput. Mater. 2018, 4, 1. [Google Scholar] [CrossRef]
- Jarlöv, A.; Ji, W.; Zhu, Z.; Tian, Y.; Babicheva, R.; An, R.; Seet, H.L.; Nai, M.L.S.; Zhou, K. Molecular dynamics study on the strengthening mechanisms of Cr-Fe-Co-Ni high-entropy alloys based on the generalized stacking fault energy. J. Alloys Compd. 2022, 905, 164137. [Google Scholar] [CrossRef]
- Ma, D.; Grabowski, B.; Körmann, F.; Neugebauer, J.; Raabe, D. Ab initio thermodynamics of the CoCrFeMnNi high entropy alloy: Importance of entropy contributions beyond the configurational one. Acta. Mater. 2015, 100, 90–97. [Google Scholar] [CrossRef]
- Lederer, Y.; Toher, C.; Vecchio, K.S.; Curtarolo, S. The search for high entropy alloys: A high-throughput ab-initio approach. Acta Mater. 2018, 159, 364–383. [Google Scholar] [CrossRef]
- Jo, Y.H.; Choi, W.M.; Kim, D.G.; Zargaran, A.; Sohn, S.S.; Kim, H.S.; Lee, B.J.; Kim, N.J.; Lee, S. FCC to BCC transformation-induced plasticity based on thermodynamic phase stability in novel V10Cr10Fe45CoxNi35−x medium-entropy alloys. Sci. Rep. 2019, 9, 2948. [Google Scholar] [CrossRef]
- Schleder, G.R.; Padilha, A.C.M.; Acosta, C.M.; Costa, M.; Fazzio, A. From DFT to machine learning: Recent approaches to materials science-a review. J. Phys. Mater. 2019, 2, 032001. [Google Scholar] [CrossRef]
- Zhang, L.; Qian, K.; Huang, J.; Liu, M.; Shibuta, Y. Molecular dynamics simulation and machine learning of mechanical response in non-equiatomic FeCrNiCoMn high-entropy alloy. J. Mater. Res. Technol. 2021, 13, 2043–2054. [Google Scholar] [CrossRef]
- Dey, S.; Sultana, N.; Kaiser, M.S.; Dey, P.; Datta, S. Computational intelligence based design of age-hardenable aluminium alloys for different temperature regimes. Mater. Des. 2016, 92, 522–534. [Google Scholar] [CrossRef]
- Suh, J.S.; Suh, B.-C.; Lee, S.E.; Bae, J.H.; Moon, B.G. Quantitative analysis of mechanical properties associated with aging treatment and microstructure in Mg-Al-Zn alloys through machine learning. J. Mater. Sci. Technol. 2022, 107, 52–63. [Google Scholar] [CrossRef]
- Chen, Y.; Tian, Y.; Zhou, Y.; Fang, D.; Ding, X.; Sun, J.; Xue, D. Machine learning assisted multi-objective optimization for materials processing parameters: A case study in Mg alloy. J. Alloys Compd. 2020, 844, 156159. [Google Scholar] [CrossRef]
- Peng, J.; Yamamoto, Y.; Hawk, J.A.; Lara-Curzio, E.; Shin, D. Coupling physics in machine learning to predict properties of high-temperatures alloys. NPJ Comput. Mater. 2020, 6, 141. [Google Scholar] [CrossRef]
- Du, J.L.; Feng, Y.L.; Zhang, M. Construction of a machine-learning-based prediction model for mechanical properties of ultra-fine-grained Fe-C alloy. J. Mater. Res. Technol. 2021, 15, 4914–4930. [Google Scholar] [CrossRef]
- Guo, T.; Wu, L.; Li, T. Machine learning accelerated, high throughput, multi-objective optimization of multiprincipal element alloys. Small 2021, 17, 2102972. [Google Scholar] [CrossRef]
- Guo, T.; Li, J.; Wang, J.; Wang, W.Y.; Liu, Y.; Luo, X.; Kou, H.; Beaugnon, E. Microstructure and properties of bulk Al0.5CoCrFeNi high-entropy alloy by cold rolling and subsequent annealing. Mater. Sci. Eng. 2018, 729, 141–148. [Google Scholar] [CrossRef]
- Fu, Z.; Chen, W.; Xiao, H.; Zhou, L.; Zhu, D.; Yang, S. Fabrication and properties of nanocrystalline Co0.5FeNiCrTi0.5 high entropy alloy by MA-SPS technique. Mater. Des. 2013, 44, 535–539. [Google Scholar] [CrossRef]
- Nam, S.; Kim, M.J.; Hwang, J.Y.; Choi, H. Strengthening of Al0.15CoCrCuFeNiTi–C (x = 0, 1, 2) high-entropy alloys by grain refinement and using nanoscale carbides via powder metallurgical route. J. Alloys Comp. 2018, 762, 29–37. [Google Scholar] [CrossRef]
- Zhang, A.; Han, J.; Meng, J.; Su, B.; Li, P. Rapid preparation of AlCoCrFeNi high entropy alloy by spark plasma sintering from elemental powder mixture. Mater. Lett. 2016, 181, 82–85. [Google Scholar] [CrossRef]
- Roy, A.; Taufique, M.F.N.; Khakurel, H.; Devanathan, R.; Johnson, D.D.; Balasubramanian, G. Machine-learning-guided descriptor selection for predicting corrosion resistance in multi-principal element alloys. NPJ Mater. Degrad. 2022, 6, 9. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, H.; Tao, X.; Cai, H.; Liu, J.; Ouyang, Y.; Peng, Q.; Du, Y. Machine learning reveals the importance of the formation enthalpy and atom-size difference in forming phases of high entropy alloys. Mater. Des. 2020, 193, 108835. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, J.; Lin, P.; Chen, L.; Liaw, K. Solid-Solution Phase Formation Rules for Multi-component Alloys. Adv. Eng. Mater. 2008, 10, 534–538. [Google Scholar] [CrossRef]
- Li, X.; Wu, S.; Li, X.; Yuan, H.; Zhao, D. Particle Swarm Optimization-Support Vector Machine Model for Machinery Fault Diagnoses in High-Voltage Circuit Breakers. Chin. J. Mech. Eng. 2020, 33, 6. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, X.; Xiao, P.; Xu, X. An ensemble learning framework for convolutional neural network based on multiple classifiers. Soft Comput. 2020, 24, 3727–3735. [Google Scholar] [CrossRef]
- Deringer, V.L.; Bartók, A.P.; Bernstein, N.; Wilkins, D.M.; Ceriotti, M.; Csányi, G. Gaussian Process Regression for Materials and Molecules. Chem. Rev. 2021, 121, 10073–10141. [Google Scholar] [CrossRef]
- Choudhury, A. The Role of Machine Learning Algorithms in Materials Science: A State of Art Review on Industry 4.0. Arch. Comput. Methods Eng. 2021, 28, 3361–3381. [Google Scholar] [CrossRef]
- Hu, Y.J.; Zhao, G.; Zhang, B.; Yang, C.; Zhang, M.; Liu, Z.K.; Qian, X.; Qi, L. Local electronic descriptors for solute-defect interactions in bcc refractory metals. Nat. Commun. 2019, 10, 4484. [Google Scholar] [CrossRef]
- Koval, E.N.; Juaristi, J.I.; Muiño, R.D.; Alducin, M. Structure and properties of CoCrFeNiX multi-principal element alloys from ab initio calculations. J. Appl. Phys. 2020, 127, 145102. [Google Scholar] [CrossRef]
- Yang, T.; Xia, S.; Liu, S.; Wang, C.; Liu, S.; Zhang, Y.; Xue, J.; Yan, S.; Wang, Y. Effects of AL addition on microstructure and mechanical properties of Al CoCrFeNi High-entropy alloy. Mater. Sci. Eng. A 2015, 648, 15–22. [Google Scholar] [CrossRef]
- Yang, J.; Cao, J. Tree-based interpretable machine learning of the thermodynamic phases. Phys. Lett. A 2021, 412, 127589. [Google Scholar] [CrossRef]







 ’ represent the single-phase maximum modulus in these alloy systems.
’ represent the single-phase maximum modulus in these alloy systems.
’ represent the single-phase maximum modulus in these alloy systems.
’ represent the single-phase maximum modulus in these alloy systems.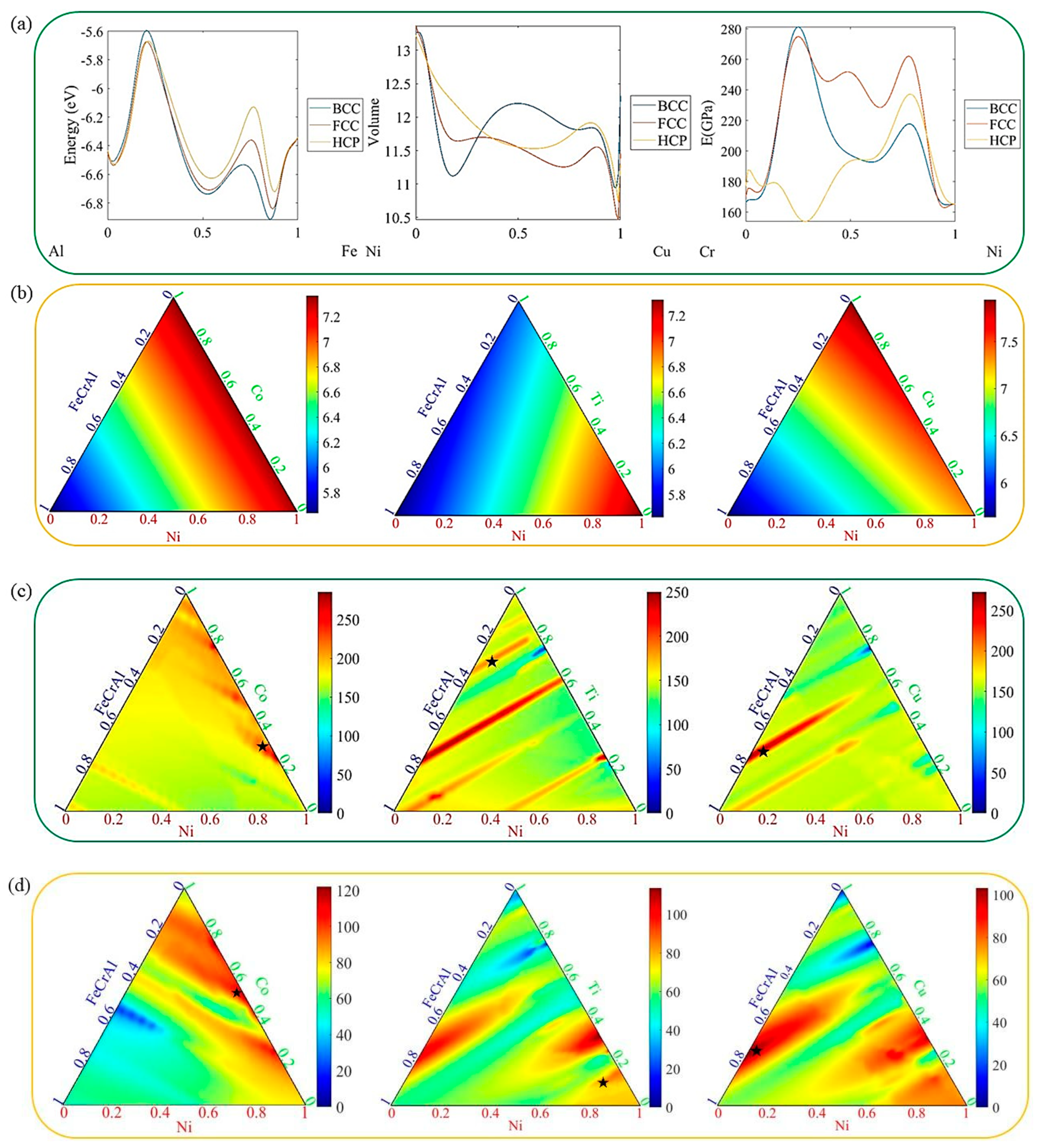

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Element Mass Fraction/wt | E/GPa | B/GPa | G/GPa | Literature | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fe | Ni | Cr | Al | Co | EV | PV | EV | PV | EV | PV | ||
| 1 | 0.25 | 0.25 | 0.25 | - | 0.25 | 214 | 209.86 | 147 | 140.87 | 86 | 90.55 | [52] |
| 2 | 0.20 | 0.20 | 0.20 | 0.20 | 0.20 | 230 | 233.11 | 154.13 | 149.69 | 94.56 | 93.32 | [52] |
| 3 | 0.25 | 0.25 | 0.23 | 0.02 | 0.25 | 203 | 199.78 | - | - | - | - | [53] |
| 4 | 0.29 | 0.11 | 0.26 | 0.07 | 0.27 | 235 | 232.76 | - | - | - | [53] | |
| 5 | 0.12 | 0.29 | 0.04 | 0.40 | 0.15 | 187 | 185.49 | - | - | - | - | [53] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, Z.; Zhang, Y.; Liu, Y.; Li, M.; Han, T.; Lai, Z.; Qu, N.; Zhu, J.; Yu, B. Accelerating First Principles Calculation of Multi-Component Alloy Steady-State Structure and Elastic Properties in Full Component Space. Materials 2023, 16, 6226. https://doi.org/10.3390/ma16186226
Yao Z, Zhang Y, Liu Y, Li M, Han T, Lai Z, Qu N, Zhu J, Yu B. Accelerating First Principles Calculation of Multi-Component Alloy Steady-State Structure and Elastic Properties in Full Component Space. Materials. 2023; 16(18):6226. https://doi.org/10.3390/ma16186226
Chicago/Turabian StyleYao, Zhixuan, Yan Zhang, Yong Liu, Mingwei Li, Tianyi Han, Zhonghong Lai, Nan Qu, Jingchuan Zhu, and Boyuan Yu. 2023. "Accelerating First Principles Calculation of Multi-Component Alloy Steady-State Structure and Elastic Properties in Full Component Space" Materials 16, no. 18: 6226. https://doi.org/10.3390/ma16186226
APA StyleYao, Z., Zhang, Y., Liu, Y., Li, M., Han, T., Lai, Z., Qu, N., Zhu, J., & Yu, B. (2023). Accelerating First Principles Calculation of Multi-Component Alloy Steady-State Structure and Elastic Properties in Full Component Space. Materials, 16(18), 6226. https://doi.org/10.3390/ma16186226