
Examining the Effect of Cu and Mn Dopants on the Structure of Zinc Blende ZnS Nanopowders



Abstract
:1. Introduction
2. Materials and Methods
2.1. Nanoparticle Synthesis and Characterization
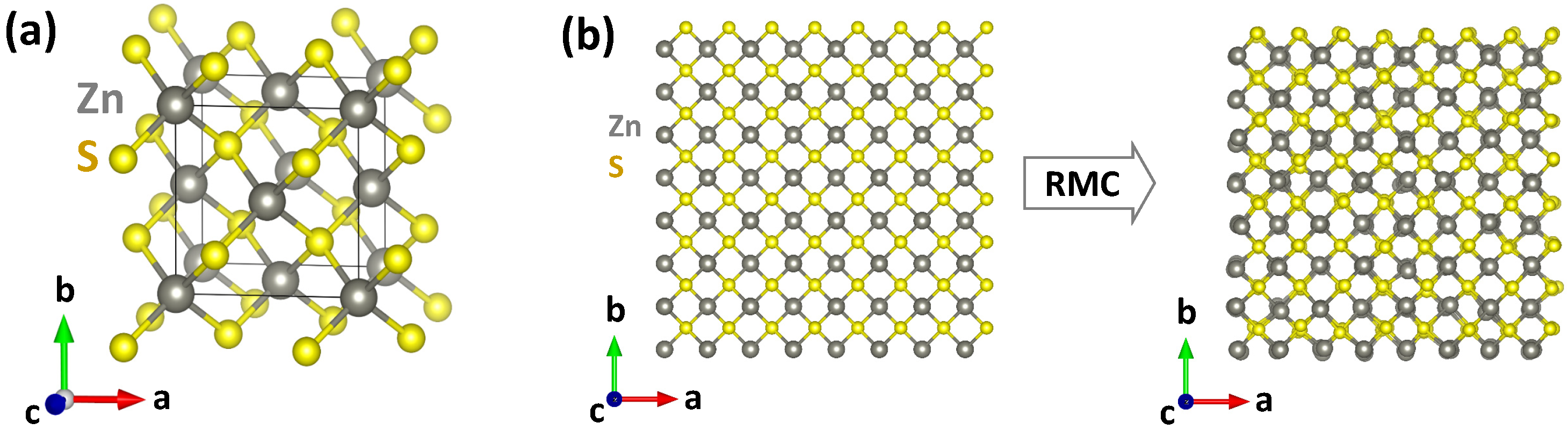
2.2. X-ray Absorption Spectroscopy and Data Analysis
2.3. First-Principles DFT Calculations
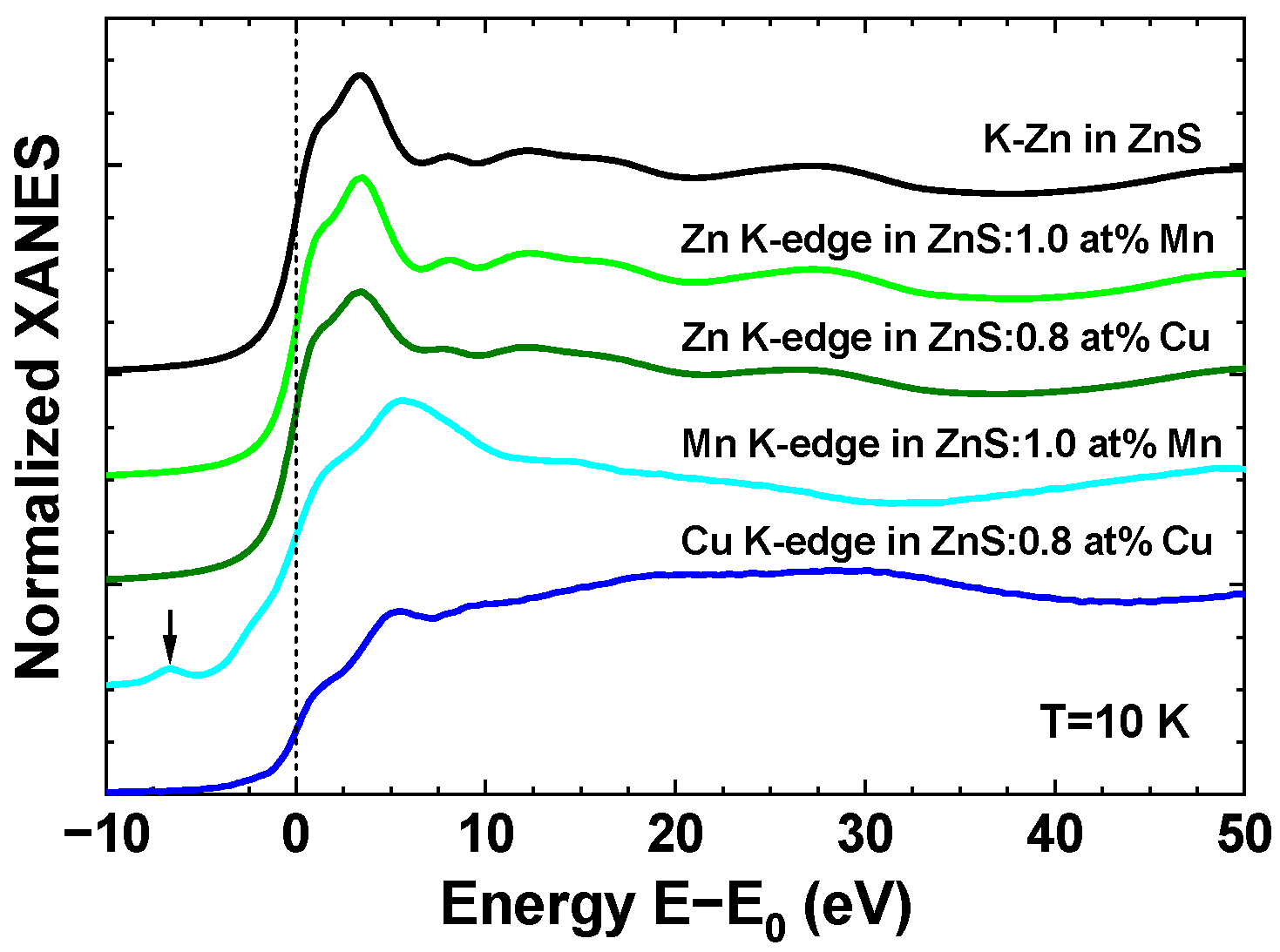
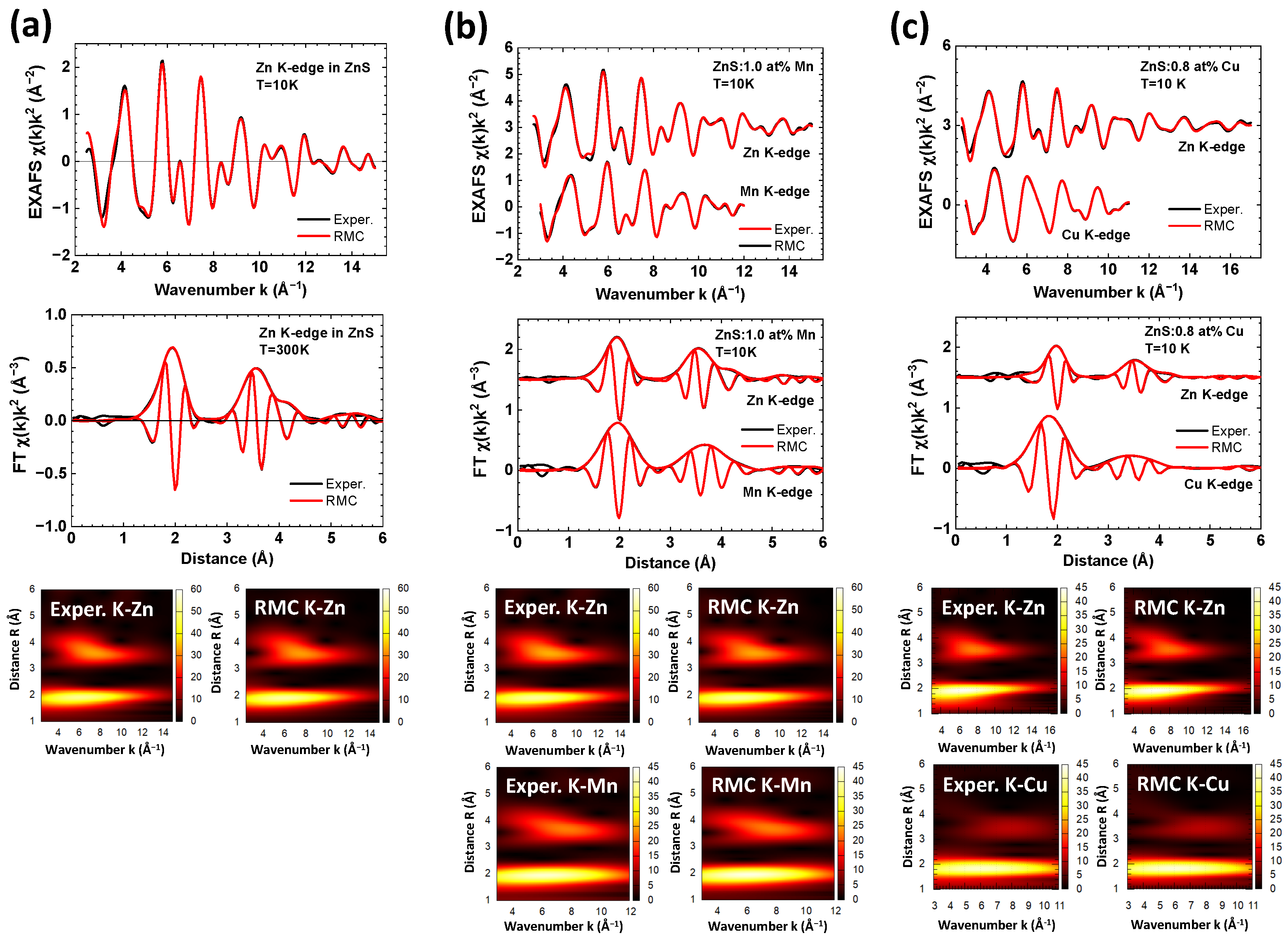
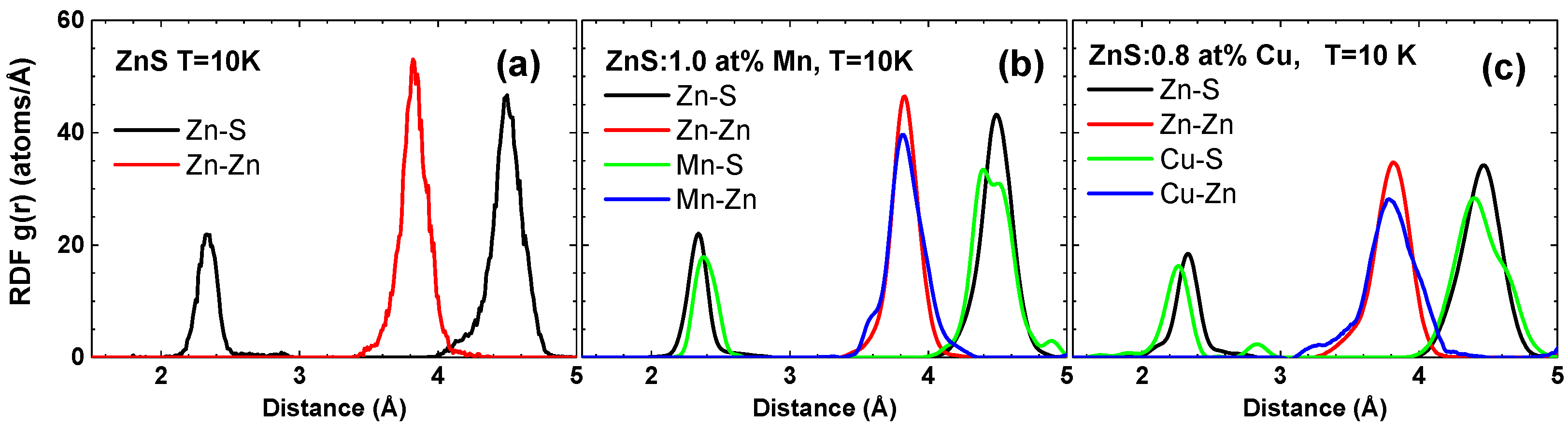
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | density functional theory |
| DI | deionized |
| EA | evolutionary algorithm |
| EDX | energy-dispersive X-ray |
| EXAFS | extended X-ray absorption fine structure |
| FT | Fourier transform |
| HS | high-spin |
| LCAO | linear combination of atomic orbitals |
| LS | low spin |
| MSRD | mean-squared relative displacements |
| MWST | microwave-assisted solvothermal |
| NP | nanoparticle |
| RDF | radial distribution function |
| RMC | reverse Monte Carlo |
| RT | room temperature |
| SCF | self-consistent field |
| SDS | sodium dodecyl sulfate |
| SEM | scanning electron microscope |
| TZV | triple-zeta valence |
| WT | wavelet transform |
| XANES | X-ray absorption near edge |
| XAS | X-ray absorption spectroscopy |
| XRD | X-ray diffraction |
References
- Fang, X.; Zhai, T.; Gautam, U.K.; Li, L.; Wu, L.; Bando, Y.; Golberg, D. ZnS nanostructures: From synthesis to applications. Prog. Mater. Sci. 2011, 56, 175–287. [Google Scholar] [CrossRef]
- Wang, X.; Huang, H.; Liang, B.; Liu, Z.; Chen, D.; Shen, G. ZnS nanostructures: Synthesis, properties, and applications. Crit. Rev. Solid State Mater. Sci. 2013, 38, 57–90. [Google Scholar] [CrossRef]
- Lee, G.J.; Wu, J.J. Recent developments in ZnS photocatalysts from synthesis to photocatalytic applications—A review. Powder Technol. 2017, 318, 8–22. [Google Scholar] [CrossRef]
- Hu, H.; Zhang, W. Synthesis and properties of transition metals and rare-earth metals doped ZnS nanoparticles. Opt. Mater. 2006, 28, 536–550. [Google Scholar] [CrossRef]
- Ummartyotin, S.; Infahsaeng, Y. A comprehensive review on ZnS: From synthesis to an approach on solar cell. Renew. Sustain. Energy Rev. 2016, 55, 17–24. [Google Scholar] [CrossRef]
- Xu, X.; Li, S.; Chen, J.; Cai, S.; Long, Z.; Fang, X. Design principles and material engineering of ZnS for optoelectronic devices and catalysis. Adv. Funct. Mater. 2018, 28, 1802029. [Google Scholar] [CrossRef]
- Yanagida, T.; Fujimoto, Y. Evaluations of pure zinc sulfide crystal scintillator. Jpn. J. Appl. Phys. 2014, 53, 032601. [Google Scholar] [CrossRef]
- Makowska, M.G.; Walfort, B.; Zeller, A.; Grunzweig, C.; Bucherl, T. Performance of the commercial PP/ZnS:Cu and PP/ZnS:Ag scintillation screens for fast neutron imaging. J. Imaging 2017, 3, 60. [Google Scholar] [CrossRef]
- Zboray, R.; Adams, R.; Kis, Z. Scintillator screen development for fast neutron radiography and tomography and its application at the beamline of the 10 MW BNC research reactor. Appl. Radiat. Isot. 2018, 140, 215–223. [Google Scholar] [CrossRef]
- Sun, H.; Wang, X.; Xia, Y.; Zhang, Z.; Ling, Y.; Zhou, Q. Surface-enhanced ZnS:Ag quantum dots scintillator. AIP Adv. 2019, 9, 105211. [Google Scholar] [CrossRef]
- Pino, F.; Delgado, J.; Carturan, S.; Mantovani, G.; Polo, M.; Fabris, D.; Maggioni, G.; Quaranta, A.; Moretto, S. Novel flexible and conformable composite neutron scintillator based on fully enriched lithium tetraborate. Sci. Rep. 2023, 13, 4799. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.R.F.; Leempoel, P.; Bard, A.J. Semiconductor Electrodes: LI. Efficient Electroluminescence at Electrode in Aqueous Electrolytes. J. Electrochem. Soc. 1983, 130, 1866–1875. [Google Scholar] [CrossRef]
- Djazovski, O.N.; Tanaka, S.; Kobayashi, H.; Semienov, N.N.; Pasynkov, V.V. Electro- and cathodoluminescent properties of ZnS:Cu, Mn and ZnS:Cu, Tb thin films. Jpn. J. Appl. Phys. 1995, 34, 4819. [Google Scholar] [CrossRef]
- Corrado, C.; Cooper, J.K.; Zhang, J.Z. Cu-doped ZnS nanocrystals: Synthesis, structure, and optical properties. Sci. Adv. Mater. 2012, 4, 254–265. [Google Scholar] [CrossRef]
- Saleh, M.; Lynn, K.G.; Jacobsohn, L.G.; McCloy, J.S. Luminescence of undoped commercial ZnS crystals: A critical review and new evidence on the role of impurities using photoluminescence and electrical transient spectroscopy. J. Appl. Phys. 2019, 125, 075702. [Google Scholar] [CrossRef]
- Mino, L.; Borfecchia, E.; Segura-Ruiz, J.; Giannini, C.; Martinez-Criado, G.; Lamberti, C. Materials characterization by synchrotron X-ray microprobes and nanoprobes. Rev. Mod. Phys. 2018, 90, 025007. [Google Scholar] [CrossRef]
- Giannini, C.; Holy, V.; De Caro, L.; Mino, L.; Lamberti, C. Watching nanomaterials with X-ray eyes: Probing different length scales by combining scattering with spectroscopy. Prog. Mater. Sci. 2020, 112, 100667. [Google Scholar] [CrossRef]
- von der Heyden, B.P. Shedding light on ore deposits: A review of synchrotron X-ray radiation use in ore geology research. Ore Geol. Rev 2020, 117, 103328. [Google Scholar] [CrossRef]
- Pudza, I.; Anspoks, A.; Aquilanti, G.; Kuzmin, A. Revealing the local structure of CuMo1−xWxO4 solid solutions by multi-edge X-ray absorption spectroscopy. Mater. Res. Bull. 2022, 153, 111910. [Google Scholar] [CrossRef]
- Bakradze, G.; Welter, E.; Kuzmin, A. Peculiarities of the local structure in new medium- and high-entropy, low-symmetry tungstates. J. Phys. Chem. Solids 2023, 172, 111052. [Google Scholar] [CrossRef]
- Bakradze, G.; Kalinko, A.; Kuzmin, A. Evidence of nickel ions dimerization in NiWO4 and NiWO4-ZnWO4 solid solutions probed by EXAFS spectroscopy and reverse Monte Carlo simulations. Acta Mater. 2021, 217, 117171. [Google Scholar] [CrossRef]
- Soo, Y.L.; Ming, Z.H.; Huang, S.W.; Kao, Y.H.; Bhargava, R.N.; Gallagher, D. Local structures around Mn luminescent centers in Mn-doped nanocrystals of ZnS. Phys. Rev. B 1994, 50, 7602–7607. [Google Scholar] [CrossRef]
- Dinsmore, A.D.; Hsu, D.S.; Qadri, S.B.; Cross, J.O.; Kennedy, T.A.; Gray, H.F.; Ratna, B.R. Structure and luminescence of annealed nanoparticles of ZnS:Mn. J. Appl. Phys. 2000, 88, 4985–4993. [Google Scholar] [CrossRef]
- Brieler, F.J.; Grundmann, P.; Froba, M.; Chen, L.; Klar, P.J.; Heimbrodt, W.; Krug von Nidda, H.A.; Kurz, T.; Loidl, A. Formation of Zn1−xMnxS nanowires within mesoporous silica of different pore sizes. J. Am. Chem. Soc. 2004, 126, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yang, J.; Zhang, Y.; Yang, L.; Wang, Y.; Wang, D.; Gao, M.; Liu, Y.; Liu, X.; Xie, Z. Multifunctional Zn0.99−xMn0.01CuxS nanowires: Structure, luminescence and magnetism. Mater. Res. Bull. 2010, 45, 705–709. [Google Scholar] [CrossRef]
- Cao, J.; Yang, J.; Zhang, Y.; Wang, Y.; Yang, L.; Wang, D.; Liu, Y.; Liu, X.; Xie, Z. XAFS analysis and luminescent properties of ZnS:Mn2+ nanoparticles and nanorods with cubic and hexagonal structure. Opt. Mater. 2010, 32, 643–647. [Google Scholar] [CrossRef]
- Curcio, A.L.; Bernardi, M.I.B.; Mesquita, A. Local structure and photoluminescence properties of nanostructured Zn1−xMnxS material. Phys. Status Solidi C 2015, 12, 1367–1371. [Google Scholar] [CrossRef]
- Trigub, A.L.; Trofimov, N.D.; Tagirov, B.R.; Nickolsky, M.S.; Kvashnina, K.O. Probing the local atomic structure of In and Cu in sphalerite by XAS spectroscopy enhanced by reverse Monte Carlo algorithm. Minerals 2020, 10, 841. [Google Scholar] [CrossRef]
- Kuzmin, A.; Dile, M.; Laganovska, K.; Zolotarjovs, A. Microwave-assisted synthesis and characterization of undoped and manganese doped zinc sulfide nanoparticles. Mater. Chem. Phys. 2022, 290, 126583. [Google Scholar] [CrossRef]
- Doebelin, N.; Kleeberg, R. Profex: A graphical user interface for the Rietveld refinement program BGMN. J. Appl. Crystallogr. 2015, 48, 1573–1580. [Google Scholar] [CrossRef]
- Welter, E.; Chernikov, R.; Herrmann, M.; Nemausat, R. A beamline for bulk sample x-ray absorption spectroscopy at the high brilliance storage ring PETRA III. AIP Conf. Proc. 2019, 2054, 1–6. [Google Scholar] [CrossRef]
- Kalinko, A. XAESA v0.07. 2023. Available online: https://gitlab.desy.de/aleksandr.kalinko/xaesa (accessed on 22 May 2023).
- Kuzmin, A.; Chaboy, J. EXAFS and XANES analysis of oxides at the nanoscale. IUCrJ 2014, 1, 571–589. [Google Scholar] [CrossRef] [PubMed]
- Timoshenko, J.; Kuzmin, A.; Purans, J. Reverse Monte Carlo modeling of thermal disorder in crystalline materials from EXAFS spectra. Comput. Phys. Commun. 2012, 183, 1237–1245. [Google Scholar] [CrossRef]
- Timoshenko, J.; Kuzmin, A.; Purans, J. EXAFS study of hydrogen intercalation into ReO3 using the evolutionary algorithm. J. Phys. Condens. Matter 2014, 26, 055401. [Google Scholar] [CrossRef] [PubMed]
- Timoshenko, J.; Kuzmin, A. Wavelet data analysis of EXAFS spectra. Comput. Phys. Commun. 2009, 180, 920–925. [Google Scholar] [CrossRef]
- Rehr, J.J.; Albers, R.C. Theoretical approaches to x-ray absorption fine structure. Rev. Mod. Phys. 2000, 72, 621–654. [Google Scholar] [CrossRef]
- Ankudinov, A.L.; Ravel, B.; Rehr, J.J.; Conradson, S.D. Real-space multiple-scattering calculation and interpretation of X-ray-absorption near-edge structure. Phys. Rev. B 1998, 58, 7565–7576. [Google Scholar] [CrossRef]
- Hedin, L.; Lundqvist, B.I. Explicit local exchange-correlation potentials. J. Phys. C Solid State Phys. 1971, 4, 2064. [Google Scholar] [CrossRef]
- Zabinsky, S.I.; Rehr, J.J.; Ankudinov, A.; Albers, R.C.; Eller, M.J. Multiple-scattering calculations of x-ray-absorption spectra. Phys. Rev. B 1995, 52, 2995–3009. [Google Scholar] [CrossRef]
- Ravel, B. A practical introduction to multiple scattering theory. J. Alloys Compd. 2005, 401, 118–126. [Google Scholar] [CrossRef]
- Rehr, J.J.; Kas, J.J.; Prange, M.P.; Sorini, A.P.; Takimoto, Y.; Vila, F. Ab initio theory and calculations of X-ray spectra. Comptes Rendus Phys. 2009, 10, 548–559. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. WIREs Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Oliveira, D.V.; Laun, J.; Peintinger, M.F.; Bredow, T. BSSE-correction scheme for consistent gaussian basis sets of double- and triple-zeta valence with polarization quality for solid-state calculations. J. Comput. Chem. 2019, 40, 2364–2376. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Account. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Skinner, B.J. Unit-cell edges of natural and synthetic sphalerites. Am. Mineral. 1961, 46, 1399–1411. [Google Scholar]
- Hotje, U.; Rose, C.; Binnewies, M. Lattice constants and molar volume in the system ZnS, ZnSe, CdS, CdSe. Solid State Sci. 2003, 5, 1259–1262. [Google Scholar] [CrossRef]
- Medling, S.; France, C.; Balaban, B.; Kozina, M.; Jiang, Y.; Bridges, F.; Carter, S.A. Understanding and improving electroluminescence in mill-ground ZnS: Cu, Cl phosphors. J. Phys. D Appl. Phys. 2011, 44, 205402. [Google Scholar] [CrossRef]
- Nietubyc, R.; Sobczak, E.; Attenkofer, K. X-ray absorption fine structure study of manganese compounds. J. Alloys Compd. 2001, 328, 126–131. [Google Scholar] [CrossRef]
- Modrow, H.; Bucher, S.; Rehr, J.J.; Ankudinov, A.L. Calculation and interpretation of K-shell x-ray absorption near-edge structure of transition metal oxides. Phys. Rev. B 2003, 67, 035123. [Google Scholar] [CrossRef]
- Henderson, G.S.; de Groot, F.M.; Moulton, B.J. X-ray Absorption Near-Edge Structure (XANES) Spectroscopy. Rev. Mineral. Geochem. 2014, 78, 75–138. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 1976, 32, 751–767. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| x (at%) | Lattice Parameter a (Å) | Crystallite Size d (nm) |
|---|---|---|
| 0 | 5.406 (2) | 5.8 (2) |
| 0.8 (Cu) | 5.400 (2) | 6.0 (2) |
| 1.0 (Mn) | 5.406 (2) | 5.8 (2) |
| MSRD (Å) | ||
|---|---|---|
| Atom Pair | ZnS & ZnS:Mn | ZnS:Cu |
| Zn–S1 | 0.005 ± 0.003 | 0.009 ± 0.003 |
| Zn–Zn2 | 0.010 ± 0.003 | 0.020 ± 0.003 |
| Zn–S3 | 0.016 ± 0.003 | 0.020 ± 0.003 |
| Mn–S1 | 0.006 ± 0.003 | |
| Mn–Zn2 | 0.018 ± 0.003 | |
| Mn–S3 | 0.023 ± 0.003 | |
| Cu–S1 | 0.010 ± 0.003 | |
| Cu–Zn2 | 0.039 ± 0.003 | |
| Cu–S3 | 0.028 ± 0.003 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuzmin, A.; Pudza, I.; Dile, M.; Laganovska, K.; Zolotarjovs, A. Examining the Effect of Cu and Mn Dopants on the Structure of Zinc Blende ZnS Nanopowders. Materials 2023, 16, 5825. https://doi.org/10.3390/ma16175825
Kuzmin A, Pudza I, Dile M, Laganovska K, Zolotarjovs A. Examining the Effect of Cu and Mn Dopants on the Structure of Zinc Blende ZnS Nanopowders. Materials. 2023; 16(17):5825. https://doi.org/10.3390/ma16175825
Chicago/Turabian StyleKuzmin, Alexei, Inga Pudza, Milena Dile, Katrina Laganovska, and Aleksejs Zolotarjovs. 2023. "Examining the Effect of Cu and Mn Dopants on the Structure of Zinc Blende ZnS Nanopowders" Materials 16, no. 17: 5825. https://doi.org/10.3390/ma16175825
APA StyleKuzmin, A., Pudza, I., Dile, M., Laganovska, K., & Zolotarjovs, A. (2023). Examining the Effect of Cu and Mn Dopants on the Structure of Zinc Blende ZnS Nanopowders. Materials, 16(17), 5825. https://doi.org/10.3390/ma16175825