

High Concentration Intrinsic Defects in MnSb2Te4


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Computational Details
3. Results and Discussion
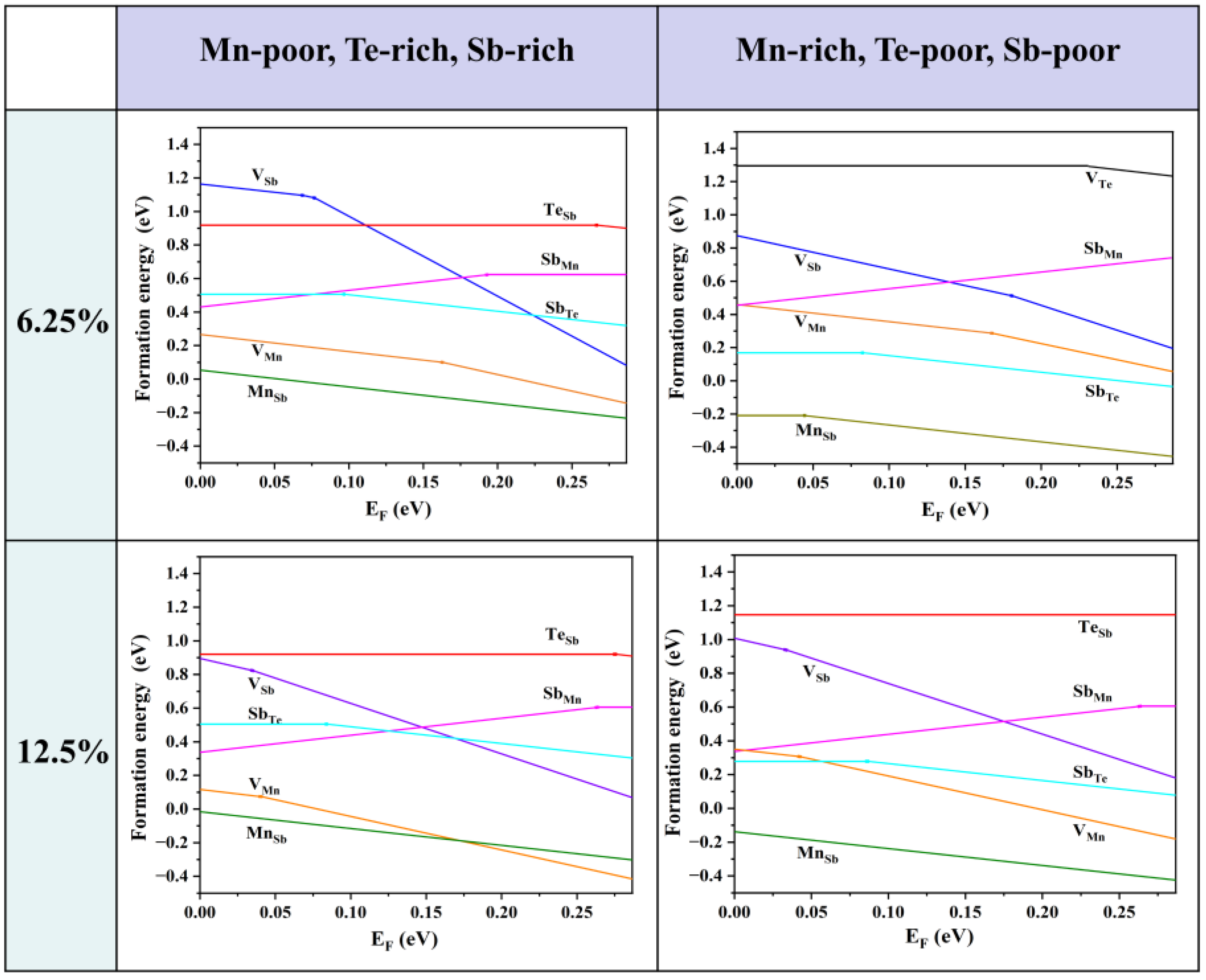
3.1. Intrinsic Defects of MnSb2Te4 at Low Concentrations
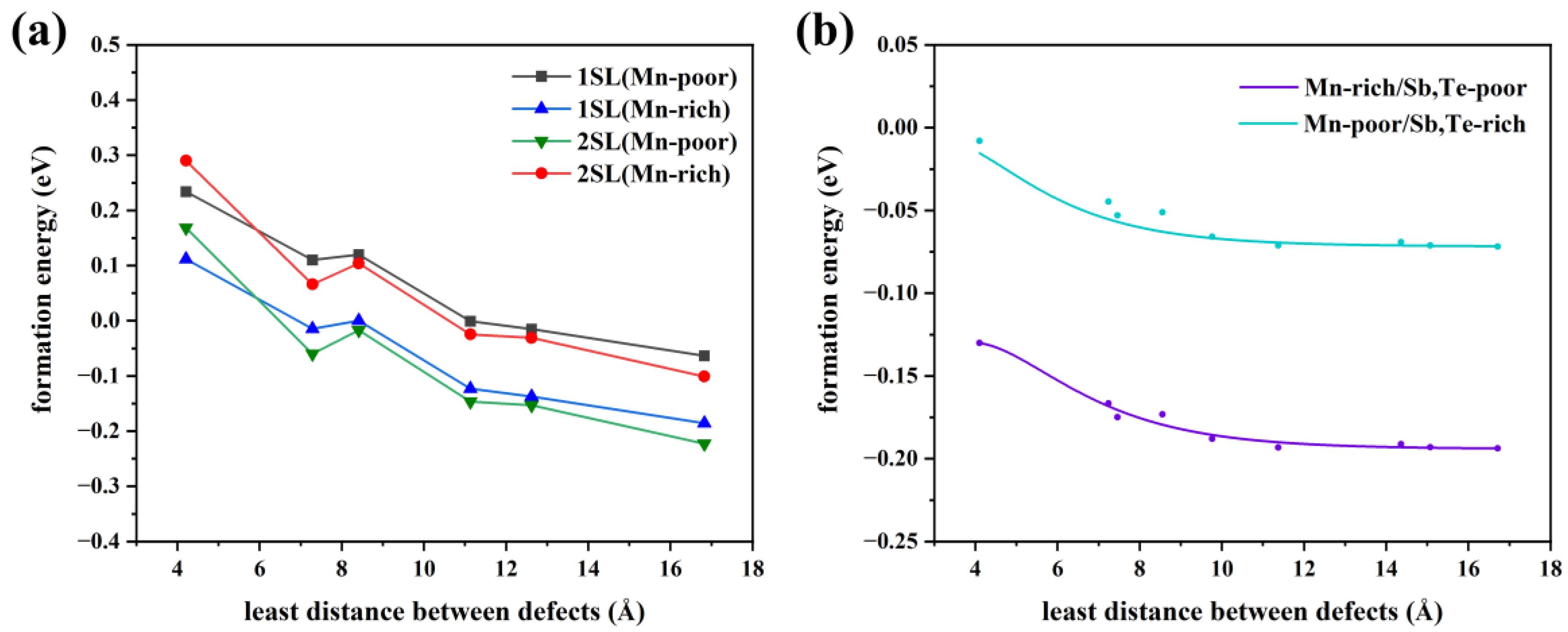
3.2. Defect Concentration of Mn Antisite Defect under a Thermal Equilibrium
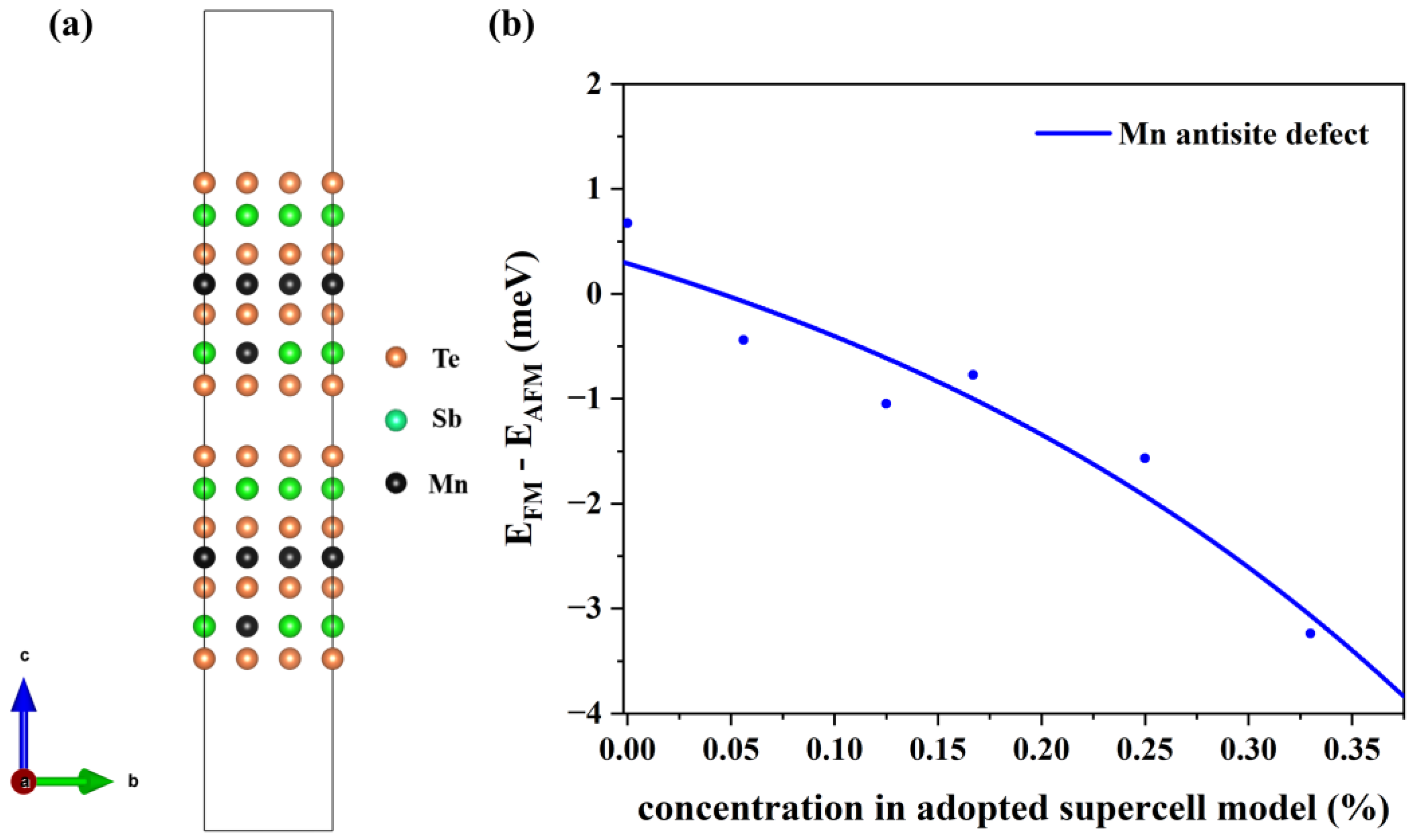
3.3. Magnetic Interlayer Coupling of MnSb2Te4 with Mn Antisite Defects
3.4. Magnetic Interlayer Coupling with Additional p-Type Doping
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gong, Y.; Guo, J.; Li, J.; Zhu, K.; Liao, M.; Liu, X.; Zhang, Q.; Gu, L.; Tang, L.; Feng, X. Experimental realization of an intrinsic magnetic topological insulator. Chin. Phys. Lett. 2019, 36, 076801. [Google Scholar] [CrossRef]
- Ovchinnikov, D.; Huang, X.; Lin, Z.; Fei, Z.; Cai, J.; Song, T.; He, M.; Jiang, Q.; Wang, C.; Li, H. Intertwined topological and magnetic orders in atomically thin Chern insulator MnBi2Te4. Nano Lett. 2021, 21, 2544–2550. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, Y.; Li, H.; Wu, Y.; Li, Y.; Li, J.; He, K.; Xu, Y.; Zhang, J.; Wang, Y. Robust axion insulator and Chern insulator phases in a two-dimensional antiferromagnetic topological insulator. Nat. Mater. 2020, 19, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Otrokov, M.M.; Klimovskikh, I.I.; Bentmann, H.; Estyunin, D.; Zeugner, A.; Aliev, Z.S.; Gaß, S.; Wolter, A.U.B.; Koroleva, A.V.; Shikin, A.M. Prediction and observation of an antiferromagnetic topological insulator. Nature 2019, 576, 416–422. [Google Scholar] [CrossRef]
- Deng, Y.; Yu, Y.; Shi, M.Z.; Guo, Z.; Xu, Z.; Wang, J.; Chen, X.H.; Zhang, Y. Quantum anomalous Hall effect in intrinsic magnetic topological insulator MnBi2Te4. Science 2020, 367, 895–900. [Google Scholar] [CrossRef]
- Zhang, D.; Shi, M.; Zhu, T.; Xing, D.; Zhang, H.; Wang, J. Topological axion states in the magnetic insulator MnBi2Te4 with the quantized magnetoelectric effect. Phys. Rev. Lett. 2019, 122, 206401. [Google Scholar] [CrossRef]
- Lian, B.; Liu, Z.; Zhang, Y.; Wang, J. Flat chern band from twisted bilayer MnBi2Te4. Phys. Rev. Lett. 2020, 124, 126402. [Google Scholar] [CrossRef]
- Li, J.; Li, Y.; Du, S.; Wang, Z.; Gu, B.-L.; Zhang, S.-C.; He, K.; Duan, W.; Xu, Y. Intrinsic magnetic topological insulators in van der Waals layered MnBi2Te4-family materials. Sci. Adv. 2019, 5, eaaw5685. [Google Scholar] [CrossRef]
- Zhou, L.; Tan, Z.; Yan, D.; Fang, Z.; Shi, Y.; Weng, H. Topological phase transition in the layered magnetic compound MnSb2Te4: Spin-orbit coupling and interlayer coupling dependence. Phys. Rev. B 2020, 102, 085114. [Google Scholar] [CrossRef]
- Eremeev, S.V.; Rusinov, I.P.; Koroteev, Y.M.; Vyazovskaya, A.Y.; Hoffmann, M.; Echenique, P.M.; Ernst, A.; Otrokov, M.M.; Chulkov, E.V. Topological magnetic materials of the (MnSb2Te4)·(Sb2Te3)n van der Waals compounds family. J. Phys. Chem. Lett. 2021, 12, 4268–4277. [Google Scholar] [CrossRef]
- Wimmer, S.; Sánchez-Barriga, J.; Küppers, P.; Ney, A.; Schierle, E.; Freyse, F.; Caha, O.; Michalička, J.; Liebmann, M.; Primetzhofer, D. Mn-Rich MnSb2Te4: A topological insulator with magnetic gap closing at high curie temperatures of 45–50 K. Adv. Mater. 2021, 33, 2102935. [Google Scholar] [CrossRef] [PubMed]
- Xi, M.; Chen, F.; Gong, C.; Tian, S.; Yin, Q.; Qian, T.; Lei, H. Relationship between antisite defects, magnetism, and band topology in MnSb2Te4 crystals with TC ≈ 40 K. J. Phys. Chem. Lett. 2022, 13, 10897–10904. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Du, M.-H.; Yan, J.; Wu, W. Native defects in antiferromagnetic topological insulator MnBi2Te4. Phys. Rev. Mater. 2020, 4, 121202. [Google Scholar] [CrossRef]
- Hou, F.; Yao, Q.; Zhou, C.-S.; Ma, X.-M.; Han, M.; Hao, Y.-J.; Wu, X.; Zhang, Y.; Sun, H.; Liu, C. Te-vacancy-induced surface collapse and reconstruction in antiferromagnetic topological insulator MnBi2Te4. ACS Nano 2020, 14, 11262–11272. [Google Scholar] [CrossRef]
- Du, M.H.; Yan, J.; Cooper, V.R.; Eisenbach, M. Tuning fermi levels in intrinsic antiferromagnetic topological insulators MnBi2Te4 and MnBi4Te7 by defect engineering and chemical doping. Adv. Funct. Mater. 2021, 31, 2006516. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.-L.; Zheng, Q.; Huang, Z.; Wang, X.; Chi, M.; Wu, Y.; Chakoumakos, B.C.; McGuire, M.A.; Sales, B.C. Site mixing for engineering magnetic topological insulators. Phys. Rev. X 2021, 11, 021033. [Google Scholar] [CrossRef]
- Li, H.; Li, Y.; Lian, Y.; Xie, W.; Chen, L.; Zhang, J.; Wu, Y.; Fan, S. Glassy magnetic ground state in layered compound MnSb2Te4. Sci. China Mater. 2022, 65, 477–485. [Google Scholar] [CrossRef]
- Jiang, Y.; Sun, Y.Y.; Chen, M.; Wang, Y.; Li, Z.; Song, C.; He, K.; Wang, L.; Chen, X.; Xue, Q.-K. Fermi-level tuning of epitaxial Sb2Te3 thin films on graphene by regulating intrinsic defects and substrate transfer doping. Phys. Rev. Lett. 2012, 108, 066809. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Janesko, B.G. Replacing hybrid density functional theory: Motivation and recent advances. Chem. Soc. Rev. 2021, 50, 8470–8495. [Google Scholar] [CrossRef] [PubMed]
- Shinde, R.; Yamijala, S.S.; Wong, B.M. Improved band gaps and structural properties from Wannier–Fermi–Löwdin self-interaction corrections for periodic systems. J. Phys. Condens. Matter 2020, 33, 115501. [Google Scholar] [CrossRef]
- Petersilka, M.; Gossmann, U.J.; Gross, E.K.U. Excitation energies from time-dependent density-functional theory. Phys. Rev. Lett. 1996, 76, 1212. [Google Scholar] [CrossRef]
- Sham, L.J.; Schlüter, M. Density-functional theory of the band gap. Phys. Rev. B 1985, 32, 3883. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Anisimov, V.I.; Zaanen, J.; Andersen, O.K. Band theory and Mott insulators: Hubbard U instead of Stoner I. Phys. Rev. B 1991, 44, 943. [Google Scholar] [CrossRef]
- Yin, Y.; Ma, X.; Yan, D.; Yi, C.; Yue, B.; Dai, J.; Zhao, L.; Yu, X.; Shi, Y.; Wang, J.-T. Pressure-driven electronic and structural phase transition in intrinsic magnetic topological insulator MnSb2Te4. Phys. Rev. B 2021, 104, 174114. [Google Scholar] [CrossRef]
- Zhang, S.B.; Wei, S.-H.; Zunger, A. Stabilization of ternary compounds via ordered arrays of defect pairs. Phys. Rev. Lett. 1997, 78, 4059. [Google Scholar] [CrossRef]
- Makov, G.; Payne, M.C. Periodic boundary conditions in ab initio calculations. Phys. Rev. B 1995, 51, 4014. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Nambu, Y.; Koretsune, T.; Xiangyu, G.; Yamamoto, T.; Brown, C.M.; Kageyama, H. Realization of interlayer ferromagnetic interaction in MnSb2Te4 toward the magnetic Weyl semimetal state. Phys. Rev. B 2019, 100, 195103. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kanai, S.; Matsukura, F.; Ohno, H. Magnetic and transport properties of Sb2Te3 doped with high concentration of Cr. Appl. Phys. Express 2017, 10, 103001. [Google Scholar] [CrossRef]
- Han, Y.; Sun, S.; Qi, S.; Xu, X.; Qiao, Z. Interlayer ferromagnetism and high-temperature quantum anomalous Hall effect in p-doped MnBi2Te4 multilayers. Phys. Rev. B 2021, 103, 245403. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, J.; Peng, Y.-H.; Lin, J.-Y.; Cen, Y.-J.; Yang, X.-B.; Zhao, Y.-J. High Concentration Intrinsic Defects in MnSb2Te4. Materials 2023, 16, 5496. https://doi.org/10.3390/ma16155496
Xiong J, Peng Y-H, Lin J-Y, Cen Y-J, Yang X-B, Zhao Y-J. High Concentration Intrinsic Defects in MnSb2Te4. Materials. 2023; 16(15):5496. https://doi.org/10.3390/ma16155496
Chicago/Turabian StyleXiong, Jie, Yin-Hui Peng, Jia-Yi Lin, Yu-Jie Cen, Xiao-Bao Yang, and Yu-Jun Zhao. 2023. "High Concentration Intrinsic Defects in MnSb2Te4" Materials 16, no. 15: 5496. https://doi.org/10.3390/ma16155496
APA StyleXiong, J., Peng, Y.-H., Lin, J.-Y., Cen, Y.-J., Yang, X.-B., & Zhao, Y.-J. (2023). High Concentration Intrinsic Defects in MnSb2Te4. Materials, 16(15), 5496. https://doi.org/10.3390/ma16155496