

Overview of the Structural, Electronic and Optical Properties of the Cubic and Tetragonal Phases of PbTiO3 by Applying Hubbard Potential Correction

 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Computational Model
3. Results and Discussion
3.1. Geometry Optimization
3.1.1. Appropriate Pseudopotential Methods
3.1.2. Appropriate k-Points and Cut-Off Energy
3.1.3. Hubbard Potential Correction
3.2. Chemical Bonds
3.2.1. Bond Lengths, Mulliken Charges and Effective Valence Charges
3.2.2. Electron Charge Density
3.3. Electronic Properties
Band Structure and Density of States
3.4. Optical Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jain, A.; Wang, Y.G.; Shi, L.N. Recent Developments in BaTiO3 Based Lead-Free Materials for Energy Storage Applications. J. Alloys Compd. 2022, 928, 167066. [Google Scholar] [CrossRef]
- Shi, X.-L.; Wu, H.; Liu, Q.; Zhou, W.; Lu, S.; Shao, Z.; Dargusch, M.; Chen, Z.-G. SrTiO3-Based Thermoelectrics: Progress and Challenges. Nano Energy 2020, 78, 105195. [Google Scholar] [CrossRef]
- Baasandorj, L.; Chen, Z. Recent Developments on Relaxor-PbTiO3 Ferroelectric Crystals. Crystals 2021, 12, 56. [Google Scholar] [CrossRef]
- Baudry, L.; Lukyanchuk, I.; Vinokur, V.M. Ferroelectric Symmetry-Protected Multibit Memory Cell. Sci. Rep. 2017, 7, 42196. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.W.; Son, J.Y. Nonvolatile Ferroelectric Memory Based on PbTiO3 Gated Single-Layer MoS2 Field-Effect Transistor. Electron. Mater. Lett. 2018, 14, 59–63. [Google Scholar] [CrossRef]
- Moret, M.P.; Devillers, M.A.C.; Wörhoff, K.; Larsen, P.K. Optical Properties of PbTiO3, PbZrxTi1−xO3, and PbZrO3 Films Deposited by Metalorganic Chemical Vapor on SrTiO3. J. Appl. Phys. 2002, 92, 468–474. [Google Scholar] [CrossRef]
- Yoon, Y.S.; Kang, W.N.; Yom, S.S.; Kim, T.W.; Jung, M.; Kim, H.J.; Park, T.H.; Na, H.K. Electrical and Optical Properties of PbTiO3 Thin Films on p -Si Grown by Metalorganic Chemical Vapor Deposition at Low Temperature. Appl. Phys. Lett. 1993, 63, 1104–1106. [Google Scholar] [CrossRef]
- Kim, Y.T.; Lee, C.W. Dielectric Properties of PbTiO3 Thin Film Capacitors Deposited on Tungsten Nitride/Tungsten Bilayers. Ferroelectrics 1995, 166, 159–163. [Google Scholar] [CrossRef]
- Zhang, S.; Li, F.; Luo, J.; Sahul, R.; Shrout, T.R. Relaxor-PbTiO3 Single Crystals for Various Applications. IEEE Trans. Ultrason. Ferroelect. Freq. Contr. 2013, 60, 1572–1580. [Google Scholar] [CrossRef]
- Kanda, K.; Inoue, J.; Saito, T.; Fujita, T.; Higuchi, K.; Maenaka, K. Fabrication and Characterization of Double-Layer Pb(Zr,Ti)O3 Thin Films for Micro-Electromechanical Systems. Jpn. J. Appl. Phys. 2012, 51, 09LD12. [Google Scholar] [CrossRef]
- Okuyama, M.; Hamakawa, Y. PbTiO3 Ferroelectric Thin Films and Their Pyroelectric Application. Ferroelectrics 1991, 118, 261–278. [Google Scholar] [CrossRef]
- Yoshiasa, A.; Nakatani, T.; Nakatsuka, A.; Okube, M.; Sugiyama, K.; Mashimo, T. High-Temperature Single-Crystal X-Ray Diffraction Study of Tetragonal and Cubic Perovskite-Type PbTiO3 Phases. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Shirane, G.; Pepinsky, R.; Frazer, B.C. X-Ray and Neutron Diffraction Study of Ferroelectric PbTiO3. Acta Cryst. 1956, 9, 131–140. [Google Scholar] [CrossRef]
- Robertson, J. Band Offsets of Wide-Band-Gap Oxides and Implications for Future Electronic Devices. J. Vac. Sci. Technol. B 2000, 18, 1785. [Google Scholar] [CrossRef]
- Peng, C.H.; Chang, J.-F.; Desu, S.B. Optical Properties of PZT, PLZT, and PNZT Thin Films. MRS Proc. 1991, 243, 21. [Google Scholar] [CrossRef]
- Mabud, S.A.; Glazer, A.M. Lattice Parameters and Birefringence in PbTiO3 Single Crystals. J. Appl. Crystallogr. 1979, 12, 49–53. [Google Scholar] [CrossRef]
- Cohen, R.E.; Krakauer, H. Electronic Structure Studies of the Differences in Ferroelectric Behavior of BaTiO3 and PbTiO3. Ferroelectrics 1992, 136, 65–83. [Google Scholar] [CrossRef]
- Shirane, G.; Hoshino, S. On the Phase Transition in Lead Titanate. J. Phys. Soc. Jpn. 1951, 6, 265–270. [Google Scholar] [CrossRef]
- Nelmes, R.J.; Kuhs, W.F. The Crystal Structure of Tetragonal PbTiO3 at Room Temperature and at 700 K. Solid State Commun. 1985, 54, 721–723. [Google Scholar] [CrossRef]
- Ramakanth, S.; Hamad, S.; Venugopal Rao, S.; James Raju, K.C. Magnetic and Nonlinear Optical Properties of BaTiO3 Nanoparticles. AIP Adv. 2015, 5, 057139. [Google Scholar] [CrossRef]
- Achehboune, M.; Khenfouch, M.; Boukhoubza, I.; Derkaoui, I.; Mothudi, B.M.; Zorkani, I.; Jorio, A. Effect of Yb Concentration on the Structural, Magnetic and Optoelectronic Properties of Yb Doped ZnO: First Principles Calculation. Opt. Quant. Electron. 2021, 53, 709. [Google Scholar] [CrossRef]
- Achehboune, M.; Khenfouch, M.; Boukhoubza, I.; Derkaoui, I.; Mothudi, B.M.; Zorkani, I.; Jorio, A. A DFT Study on the Electronic Structure, Magnetic and Optical Properties of Er Doped ZnO: Effect of Er Concentration and Native Defects. Comput. Condens. Matter 2022, 31, e00627. [Google Scholar] [CrossRef]
- Derkaoui, I.; Achehboune, M.; Boukhoubza, I.; El Adnani, Z.; Rezzouk, A. Improved First-Principles Electronic Band Structure for Cubic (Pmm) and Tetragonal (P4mm, P4/mmm) Phases of BaTiO3 Using the Hubbard U Correction. Comput. Mater. Sci. 2023, 217, 111913. [Google Scholar] [CrossRef]
- Teng, Z.; Jiang, J.; Chen, G.; Ma, C.; Zhang, F. The Electronic Structures and Optical Properties of B, C or N Doped BaTiO3. AIP Advances 2018, 8, 095216. [Google Scholar] [CrossRef]
- Piskunov, S.; Heifets, E.; Eglitis, R.I.; Borstel, G. Bulk Properties and Electronic Structure of SrTiO3, BaTiO3, PbTiO3 Perovskites: An Ab Initio HF/DFT Study. Comput. Mater. Sci. 2004, 29, 165–178. [Google Scholar] [CrossRef]
- Lv, H.; Gao, H.; Yang, Y.; Liu, L. Density Functional Theory (DFT) Investigation on the Structure and Electronic Properties of the Cubic Perovskite PbTiO3. Appl. Catal. A Gen. 2011, 404, 54–58. [Google Scholar] [CrossRef]
- Meyer, B.; Padilla, J.; Vanderbilt, D. Theory of PbTiO3, BaTiO3, and SrTiO3 Surfaces. Faraday Disc. 1999, 114, 395–405. [Google Scholar] [CrossRef]
- Brehm, J.A.; Takenaka, H.; Lee, C.-W.; Grinberg, I.; Bennett, J.W.; Schoenberg, M.R.; Rappe, A.M. Density Functional Theory Study of Hypothetical PbTiO3-Based Oxysulfides. Phys. Rev. B 2013, 89, 195202. [Google Scholar] [CrossRef]
- Anisimov, V.I.; Zaanen, J.; Andersen, O.K. Band Theory and Mott Insulators: Hubbard U Instead of Stoner I. Phys. Rev. B 1991, 44, 943–954. [Google Scholar] [CrossRef]
- Arroyo-de Dompablo, M.E.; Morales-García, A.; Taravillo, M. DFT+ U Calculations of Crystal Lattice, Electronic Structure, and Phase Stability under Pressure of TiO2 Polymorphs. J. Chem. Phys. 2011, 135, 054503. [Google Scholar] [CrossRef]
- Parhizgar, S.S.; Beheshtian, J. Effect of Nitrogen Doping on Electronic and Optical Properties of ZnO Sheet: DFT+U Study. Comput. Condens. Matter 2018, 15, 1–6. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First Principles Methods Using CASTEP. Z. fur Krist. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft Self-Consistent Pseudopotentials in a Generalized Eigenvalue Formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Sinha, T.P.; Mookerjee, A. Electronic Structure, Chemical Bonding, and Optical Properties of Paraelectric BaTiO3. Phys. Rev. B 2000, 62, 8828–8834. [Google Scholar] [CrossRef]
- Glazer, A.M.; Mabud, S.A. Powder Profile Refinement of Lead Zirconate Titanate at Several Temperatures. II. Pure PbTiO3. Acta Crystallogr. B Struct. Sci. 1978, 34, 1065–1070. [Google Scholar] [CrossRef]
- Megaw, H.D. Crystal Structure of Double Oxides of the Perovskite Type. Proc. Phys. Soc. 1946, 58, 133–152. [Google Scholar] [CrossRef]
- Ghosez, P.; Cockayne, E.; Waghmare, U.V.; Rabe, K.M. Lattice Dynamics of BaTiO3, PbTiO3, and PbZrO3: A Comparative First-Principles Study. Phys. Rev. B 1999, 60, 836–843. [Google Scholar] [CrossRef]
- Zhao, Y.; Yan, J. Effects of N Concentration on Electronic and Optical Properties of N-Doped PbTiO3. J. Semicond. 2015, 36, 093005. [Google Scholar] [CrossRef]
- Bilc, D.I.; Orlando, R.; Shaltaf, R.; Rignanese, G.-M.; Íñiguez, J.; Ghosez, P. Hybrid Exchange-Correlation Functional for Accurate Prediction of the Electronic and Structural Properties of Ferroelectric Oxides. Phys. Rev. B 2008, 77, 165107. [Google Scholar] [CrossRef]
- Kang, T.D.; Lee, H.; Xing, G.; Izumskaya, N.; Avrutin, V.; Xiao, B.; Morkoç, H. Dielectric Functions and Critical Points of PbTiO3, PbZrO3, and PbZr0.57Ti0.43O3 Grown on SrTiO3 Substrate. Appl. Phys. Lett. 2007, 91, 022918. [Google Scholar] [CrossRef]
- Železný, V.; Chvostová, D.; Šimek, D.; Máca, F.; Mašek, J.; Setter, N.; Hong Huang, Y. The Variation of PbTiO3 Bandgap at Ferroelectric Phase Transition. J. Phys. Condens. Matter 2016, 28, 025501. [Google Scholar] [CrossRef] [PubMed]
- Bäuerle, D.; Braun, W.; Saile, V.; Sprüssel, G.; Koch, E.E. Vacuum Ultraviolet Reflectivity and Band Structure of SrTiO3 and BaTiO3. Z Physik B 1978, 29, 179–184. [Google Scholar] [CrossRef]
- Cardona, M. Optical Properties and Band Structure of SrTiO3 and BaTiO3. Phys. Rev. 1965, 140, A651–A655. [Google Scholar] [CrossRef]
- Cai, M.-Q.; Yin, Z.; Zhang, M.-S. First-Principles Study of Optical Properties of Barium Titanate. Appl. Phys. Lett. 2003, 83, 2805–2807. [Google Scholar] [CrossRef]
- Issam, D.; Achehboune, M.; Boukhoubza, I.; Hatel, R.; El Adnani, Z.; Rezzouk, A. Investigation of the Crystal Structure, Electronic and Optical Properties of Cr-Doped BaTiO3 on the Ti Site Using First Principles Calculations. J. Phys. Chem. Solids 2023, 175, 111209. [Google Scholar] [CrossRef]
- Foster, C.M.; Bai, G.-R.; Csencsits, R.; Vetrone, J.; Jammy, R.; Wills, L.A.; Carr, E.; Amano, J. Single-Crystal Pb(ZrxTi1−x)O3 Thin Films Prepared by Metal-Organic Chemical Vapor Deposition: Systematic Compositional Variation of Electronic and Optical Properties. J. Appl. Phys. 1997, 81, 2349–2357. [Google Scholar] [CrossRef]
- Foster, C.M.; Bai, G.-R.; Li, Z.; Jammy, R.; Wills, L.A.; Hiskes, R. Properties Variation with Composition of Single-Crystal Pb(Zrx Ti1−x,)O3 Thin Films Prepared by MOCVD. MRS Proc. 1995, 401, 139. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
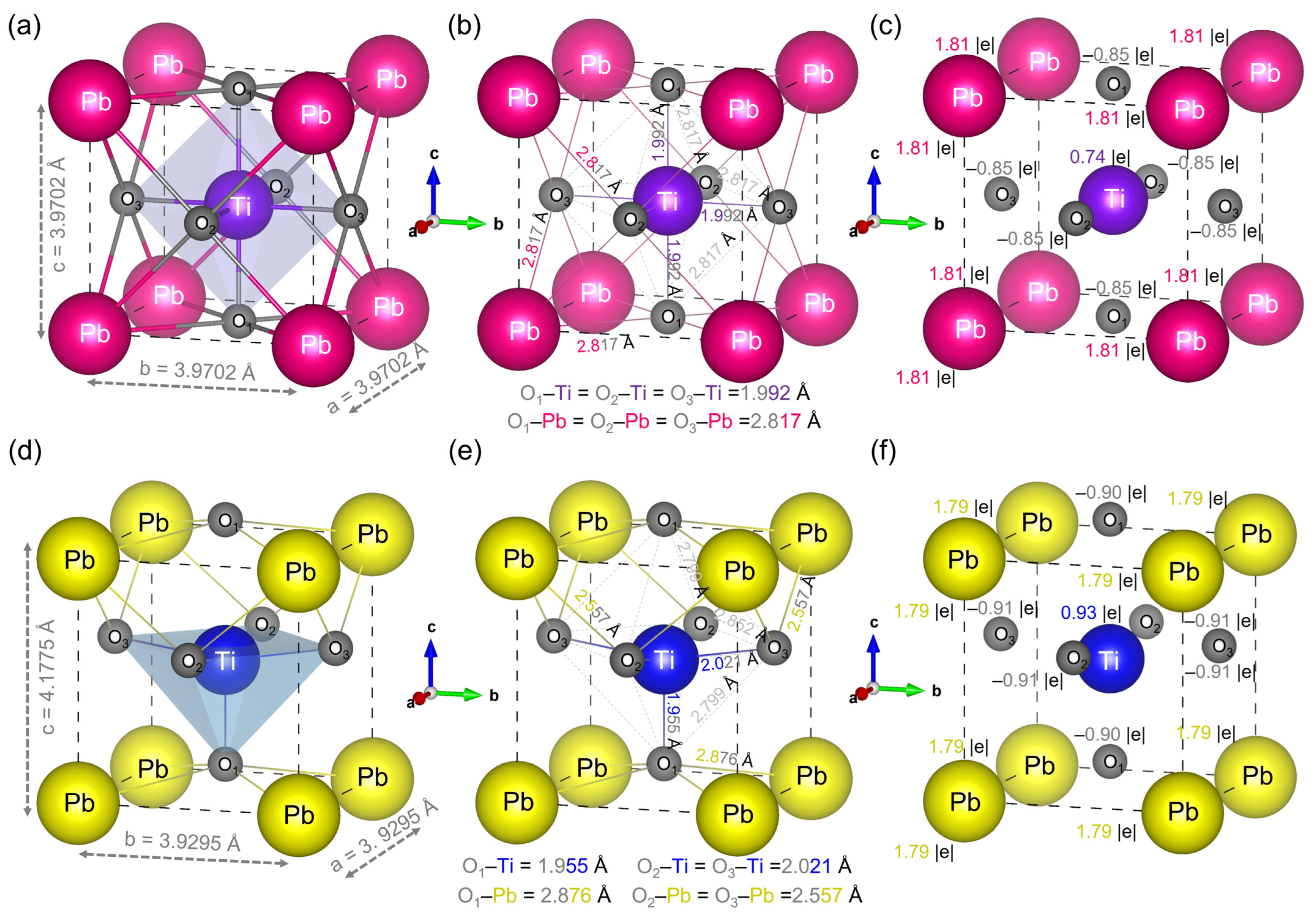{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PbTiO3: Cubic Phase (Pmm) (Pseudo-Potential Method: Norm-Conserving) | |||||||||
| k-points | Cut-off (eV) | aexp = bexp = cexp (Å) | aopt = bopt = copt (Å) | D (%) | Vexp (Å3) | Vopt (Å3) | D (%) | ||
|---|---|---|---|---|---|---|---|---|---|
| 2 × 2 × 2 | 560 | 3.9702 [a] | 3.9700 | 0.0050 | 62.58 [a] | 62.573 | 0.0111 | ||
| PbTiO3: Tetragonal phase (P4mm) (Pseudo-potential method: Norm-conserving) | |||||||||
| k-points | Cut-off (eV) | Exp. lattice parameters (Å) | Opt. lattice parameters (Å) | D (%) | |||||
| aexp = bexp | cexp | cexp/aexp | aopt = bopt | copt | copt/aopt | a, b | c | ||
| 2 × 2 × 2 | 480 | 3.904 [b] | 4.152 [b] | 1.063 [b] | 3.9295 | 4.1775 | 1.063 | 0.6489 | 0.6104 |
| Phases; Local Symmetry | This Calculation | Experimental Data | Previous Theoretical Results (DFT Functional) |
|---|---|---|---|
| 3.93 (LDA), 3.96 (PWGGA), 3.96 (PBE), 4.02 (BLYP), 3.93 (P3PW), 3.96 (P3LYP), 3.924 (HF) [25], 3.980 (GGA/PBE), 4.010 (GGA/RPBE), 3.977 (GGA/PW91), 3.899 (LDA/CA-PZ) [26], 3.970 (LDA) [38], and 3.987 (GGA/PBE) [39]. | |||
| Cubic | (GGA/PBE) | 3.9702 [12] | |
| (Pmm); | a = b = c = 3.9700 | 3.97 [13] | |
| 3.970 [36] | |||
| Tetragonal (P4mm); | (GGA/PBE) | a = 3.9043, c = 4.1407, c/a = 1.060 [12]. | a = 3.87, c = 4.07, c/a = 1.05 (LDA) [28], a = 3.872, and c/a = 1.041 (LDA); a = 3.834 and c/a = 1.221 (GGA/PBE) [40]. |
| a = b = 3.9295 | a = 3.904, c = 4.152, c/a = 1.063 [13]. | ||
| c = 4.1775 | a = 3.905, c = 4.156, c/a = 1.064 [36]. | ||
| c/a = 1.063 | a = 3.896, c = 4.144, c/a = 1.063 [37]. |
| Method | Up(O) | Ud(Ti) | Ud(Pb) | Eg (eV) | D (%) | |
|---|---|---|---|---|---|---|
| This Cal. | Exp Data. | |||||
| GGA/PBE | PbTiO3: Cubic phase (Pmm) (Pseudo-Potential Method: Norm-Conserving) | |||||
| 0 | 0 | 0 | 2.207 | - | - | |
| 3.5 | 6 | 0 | 3.400 | No exp. data for cubic phase | - | |
| 3.5 | 6.5 | 0 | 3.395 | - | ||
| 3.5 | 7 | 0 | 3.382 | - | ||
| 4 | 5.5 | 0 | 3.431 | - | ||
| 4.5 | 5 | 0 | 3.388 | - | ||
| 5.5 | 4.5 | 0 | 3.395 | - | ||
| 6.5 | 4 | 0 | 3.395 | - | ||
| 7.5 | 3.5 | 0 | 3.387 | - | ||
| 8 | 3.5 | 0 | 3.421 | - | ||
| 8.5 | 3 | 0 | 3.396 | - | ||
| 9 | 3 | 0 | 3.426 | - | ||
| 10 | 2.5 | 0 | 3.390 | - | ||
| PbTiO3: Tetragonal phase (P4mm) (Pseudo-potential method: Norm-conserving) | ||||||
| 0 | 0 | 0 | 2.213 | - | - | |
| 5 | 5.5 | 0 | 3.383 | 3.40 [14,15] | −0.502 | |
| 5 | 6 | 0 | 3.414 | 0.410 | ||
| 5 | 7 | 0 | 3.400 | 0 | ||
| 5 | 8 | 0 | 3.386 | −0.413 | ||
| 5.5 | 9 | 0 | 3.421 | 0.613 | ||
| 5.5 | 9.5 | 0 | 3.390 | −0.294 | ||
| 6.5 | 10 | 0 | 3.416 | 0.468 | ||
| 9.5 | 3.5 | 0 | 3.377 | −0.681 | ||
| 10 | 3.5 | 0 | 3.402 | 0.058 | ||
| Species | Mulliken Charges (e) | Effective Valence Charges (e) |
|---|---|---|
| PbTiO3: Cubic phase (Pmm) | ||
| Pb | 1.81 | 10.0 |
| Ti | 0.74 | 1.81 |
| O | −0.85 | 0.00 |
| PbTiO3: Tetragonal phase (P4mm) | ||
| Pb | 1.79 | 10 |
| Ti | 0.93 | 1.57 |
| O1 | −0.90 | 0.00 |
| O2 | −0.91 | 0.00 |
| O3 | −0.91 | 0.00 |
| Synthesis Method | Technique-DFT/Functional | Refractive Index | Ref. |
|---|---|---|---|
| Sol–gel PbTiO3 | Transmittance | 2.58 | [15] |
| MOCVD PbTiO3 on SiTiO3 | Ellipsometry | 2.66 | [6] |
| MOCVD PbTiO3 on SiTiO3 | Prism coupling | 2.675 | [47] |
| MOCVD PbTiO3 on SiTiO3 | Prism coupling | 2.67 | [48] |
| Primitive cell of PbTiO3 (cubic) | GGA/PBE | 3.27 | This Calc. |
| Primitive cell of PbTiO3 (cubic) | GGA/PBE + U | 2.65 | This Calc. |
| Primitive cell of PbTiO3 (tetragonal) | GGA/PBE | 2.91 | This Calc. |
| Primitive cell of PbTiO3 (tetragonal) | GGA/PBE + U | 2.41 | This Calc. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Derkaoui, I.; Achehboune, M.; Eglitis, R.I.; Popov, A.I.; Rezzouk, A. Overview of the Structural, Electronic and Optical Properties of the Cubic and Tetragonal Phases of PbTiO3 by Applying Hubbard Potential Correction. Materials 2023, 16, 4302. https://doi.org/10.3390/ma16124302
Derkaoui I, Achehboune M, Eglitis RI, Popov AI, Rezzouk A. Overview of the Structural, Electronic and Optical Properties of the Cubic and Tetragonal Phases of PbTiO3 by Applying Hubbard Potential Correction. Materials. 2023; 16(12):4302. https://doi.org/10.3390/ma16124302
Chicago/Turabian StyleDerkaoui, Issam, Mohamed Achehboune, Roberts I. Eglitis, Anatoli I. Popov, and Abdellah Rezzouk. 2023. "Overview of the Structural, Electronic and Optical Properties of the Cubic and Tetragonal Phases of PbTiO3 by Applying Hubbard Potential Correction" Materials 16, no. 12: 4302. https://doi.org/10.3390/ma16124302
APA StyleDerkaoui, I., Achehboune, M., Eglitis, R. I., Popov, A. I., & Rezzouk, A. (2023). Overview of the Structural, Electronic and Optical Properties of the Cubic and Tetragonal Phases of PbTiO3 by Applying Hubbard Potential Correction. Materials, 16(12), 4302. https://doi.org/10.3390/ma16124302