Abstract
Using density functional theory and semiclassical Boltzmann transport equation, the lattice thermal conductivity and electronic transport performance of monolayer SnI2 were systematically investigated. The results show that its room temperature lattice thermal conductivities along the zigzag and armchair directions are as low as 0.33 and 0.19 W/mK, respectively. This is attributed to the strong anharmonicity, softened acoustic modes, and weak bonding interactions. Such values of the lattice thermal conductivity are lower than those of other famous two-dimensional thermoelectric materials such as MoO3, SnSe, and KAgSe. The two quasi-degenerate band valleys for the valence band maximum make it a p-type thermoelectric material. Due to its ultralow lattice thermal conductivities, coupled with an ultrahigh Seebeck coefficient, monolayer SnI2 possesses an ultrahigh figure of merits at 800 K, approaching 4.01 and 3.34 along the armchair and zigzag directions, respectively. The results indicate that monolayer SnI2 is a promising low-dimensional thermoelectric system, and would stimulate further theoretical and experimental investigations of metal halides as thermoelectric materials.
1. Introduction
With more than 60% of energy in the world lost in the form of waste heat, the thermoelectric system has attracted widespread attention, since it directly converts the waste heat to electric energy through the Seebeck effect. It has the advantages of small size, high reliability, no pollutants, and a feasibility in a wide temperature range. Such a system is widely used in aerospace exploration and industrial production, such as in space probes, thermoelectric generators, and precise temperature controls [1,2]. The converting efficiency of TE materials is ruled by the dimensionless figure of merit (ZT); ZT = S2σT/(κL+ κe), where S, σ, T, κL, and κe are the Seebeck coefficient, electrical conductivity, absolute temperature, lattice thermal conductivity, and electronic thermal conductivity, respectively. The electronic transport properties S, σ, and κe, have a complex coupling relationship, and are difficult to decouple, even though they can be modified via carrier concentration [3,4,5]. Historically, two aspects were established to enhance ZT: one aspect is to optimize carrier concentration by the band structure, engineered to enhance the power factor (σS2) [6,7,8,9], and the other aspect is to reduce the lattice thermal conductivity, via alloying and nanostructuring [10]. Alternatively, it is more attractive to seek materials with an intrinsic low lattice thermal conductivity (generally associated with complex crystal structures [11]), strong anharmonicity [12,13], lone pair electrons [14,15], and liquid-like behavior [16,17,18], etc.
The group IVA metal dihalides are candidates for semiconductor optical devices and perovskite solar cells, due to their excellent properties, such as the visible-range band gap and the thickness-dependent band structure [19,20,21,22]. However, the application of the Pb-based materials, such as the layered 2H-PbI2, has been greatly limited by their toxicity and environmental unfriendliness [23,24,25]. In addition, the surface of the bulk SnI2 is generally very rough and accompanied by many defects, which strongly scatters carriers [26]. Fortunately, its vdW monolayer has been experimentally realized, via molecular beam epitaxy [27]. The low dimensionality provides an effective conductive channel for carriers, and also suppresses phonon thermal transport [28]. Thus, monolayer SnI2 is a preferable option over bulk SnI2 and monolayer PbI2 in use as a TE material, and deserves to be carefully explored in a systematic study.
In this work, combining first-principles calculations and the semiclassical Boltzmann transport equation, the TE properties of monolayer SnI2 were systematically explored. Results show that the intrinsic ultralow lattice thermal conductivity originates from the strong anharmonicity, weak bonding, and softened acoustic modes. The Grüneisen parameter, phonons scattering phase space, and phonon relaxation time were calculated to understand the micro-mechanism of the phonon transports. The two quasi-degenerate band valleys for the valence band maximum (VBM) in its electronic band structure led to a p-type TE material. The maximum ZT value along the armchair and zigzag directions at 800 K reach 4.01 and 3.34, respectively, using optimal p-type doping. These results indicate that monolayer SnI2 exhibits an extraordinary TE response, and is an ideal material for TE applications.
2. Computational Methods
The first-principles calculations were implemented in the Vienna ab initio simulation package (VASP, VASP.5.3, Wien, Austria) [29]. The generalized gradient approximation (GGA) [30,31] in the Perdew–Burke–Ernzerhof (PBE) [32] form was employed to deal with the exchange–correlation functional, with a cutoff of 300 eV on a 9 × 9 × 1 Monkhorst–Pack k-mesh. To screen the interactions between adjacent images, the length of the unit cell of 20 Å was used along the z direction. The geometry structure was fully relaxed, with a criterion of convergence for residual forces of 0.001 eV/Å, and the total energy difference converged to within 10−8 eV/Å. To obtain an accurate band gap and electronic transport performance, the Heyd–Suseria–Ernzerhof (HSE06) [33] method was employed.
The electronic transport properties of the monolayer SnI2 were calculated by solving the semiclassical Boltzmann transport equation, utilizing the BoltzTraP code (Georg K. H. Madsen, Århus, Denmark) [34] with a dense 35 × 35 × 1 k-mesh. The constant relaxation time approach (CRTA) was used, since the relaxation time is not strongly dependent on the energy scale of kBT, and has accurately predicted TE properties of multitudinous materials. In this work, the electrons relaxation time was calculated using the deformation potential (DP) theory [35], which considers the primarily acoustic phonon scatterings, but ignores effects of the optical phonons in the single parabolic band (SPB) model. Based on the rigid band approximation (RBA) [36] and CRTA, the transport coefficients S, σ, and κe can be obtained by:
where kB, f0, and Ξ(ε) are the Boltzmann constant, the Fermi–Dirac distribution function, and the transport distribution function, respectively. The calculation of the relaxation time τ was extremely difficult, due to the various complex scattering mechanism in the crystals. The relaxation time was calculated based on the DP theory in the SPB model, which considers the predominant scatterings between carriers and acoustic phonons in the low-energy region. In fact, the scattering matrix element () of the acoustic phonons in the long-wavelength can be approximated as kBTE12/Cii, where Cii and E1 are the elastic and DP constants, respectively. Thus, for the 2D material, the carrier mobility and electrons relaxation time can be approached as [37,38]
where h, m*, and md are the Planck constant, the effective mass along the transport direction, and the averaged effective mass, respectively.
The harmonic second-order interaction force constants (2nd IFCs) and phonon spectrum were calculated using the Phonopy package [39], using a 2 × 2 × 1 supercell with a 5 × 5 × 1 k-mesh using the finite-difference method [40]. The anharmonic third-order interaction force constants (3rd IFCs), which consider the interactions between the sixth-nearest-neighbor atoms, were obtained using the ShengBTE package (ShengBTE version 1.0.2, Wu Li, Grenoble, France; Phonopy version 2.11.0, Atsushi Togo, Sakyo, Japan) [41] with the same supercell. The lattice thermal conductivity and phonon transport properties were obtained using the self-consistent iterative solution of the Boltzmann transport equation, with a dense 36 × 36 × 1 mesh, which had a good convergence. Based on the Boltzmann transport equation, with the Fourier’s low of heat conduction, the matrix elements of the phonon thermal conductivity can be expressed by [42,43]
where α and β are the Cartesian indices, Nr is the total number of q-points sampled in the first Brillouin zone, and cλ, vλ, and τλ are the mode-specific heat capacities, phonon group velocity, and the relaxation time, respectively. Here, the phonon thermal properties were calculated based on the 3rd IFCs, ignoring the fourth- and higher-order terms. This strategy described the phonon behavior of the most anharmonic materials [44,45]. To define the effective thickness of two-dimensional (2D) materials, the summation of interlayer distance and the vdW radii of the outermost surface atoms was adopted [44].
3. Results and Discussion
3.1. Geometry and Electronic Structure
Monolayer SnI2 crystallizes in a hexagonal lattice with space group P-3m1 (164), as shown in Figure 1. The optimized lattice parameter is 4.57 Å, which is in good agreement with the previous experimental data of 4.48 Å [27]. The structure is analogous to H-MoS2 [46], consisting of three layers, with Sn atoms as the middle layer, and I atoms as the upper and lower atomic layers. The electron localization function (ELF) provides a deeper insight to characterize the nature of, and strengthen, the chemical band. Figure 1c shows the calculated three-dimensional (3D) ELF map (isosurface level of 0.97). The ELF around I is in the shape of a “mushroom”, suggesting the existence of the lone pair electrons. Moreover, the electron sharing is better visualized by the 2D ELF map in Figure 1d. The interstitial electrons between Sn and I are close to I atoms, and the value of the area between them is 0.5, which shows the characteristics of a free electron gas and indicates a weak bonding between Sn and I atoms. The electronic repulsion between the lone pair electrons and the Sn–I bonding electrons results in strong anharmonicity, such as in CuSbS2 [14]. The complex structure and lone pair electrons are also beneficial to its low lattice thermal conductivity.
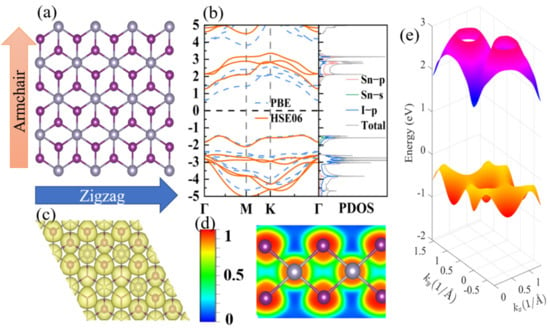
Figure 1.
(a) Top view of the atomic structure of SnI2 monolayer. (b) The electronic band structures calculated with PBE and HSE06 hybrid functional potentials. (c,d) the 3D and 2D ELF maps. (e) The 3D electronic band structure calculated with PBE.
The electronic structure plays a crucial role in characterizing the electronic transport properties. As shown in Figure 1b, the electronic band structures obtained from the PBE and HSE06 hybrid function potentials are presented. They exhibit analogous band structures, except for a more accurate bandgap (2.71 eV) that approaches the experiment value (2.9 eV) [27] for the latter structure. The conduction band mainly originates from the p orbitals of the Sn atom and the p orbitals of the I atom, whereas the valence band consists of the s orbital of the Sn atom and the p orbitals of the I atom. Below the VBM, there are two quasi-degenerate band valleys along the Γ–M and Γ–K directions with an energy difference of ~0.03 eV, which is far less than those in SnTe (~0.35eV) [47] and PbTe (~0.15 eV) [1]. The multi-energy valley enhances the TE performance and is verified in many materials [48,49]. However, the behavior of band valleys degenerate do not exist for the conduction band minimum (CBM), which is located at the Γ point. Hence, it is expected that the TE performance of the p-type could be superior to that of the n-type.
3.2. Electronic Transport Properties
All the parameters for electronic transport, calculated according to the DP theory, are tabulated in Table 1. Based on the reasonable relaxation time, all the electronic transport coefficients, Seebeck coefficient S, electrical conductivity σ, and power factor (PF), as a function of temperature at the corresponding optimal carrier concentration for p-type SnI2, range from 1 × 1013 to 1 × 1014 cm−2, and are presented in Figure 2a–c. For comparison, those for n-type SnI2 at the same condition are also plotted in Figure 2d–f. Here, the tiny anisotropy characteristics are ignored.

Table 1.
The calculated DP constant E1, elastic constant, effective mass, carrier mobility, and relaxation time along the zigzag and armchair directions in monolayer SnI2 at 300 K.
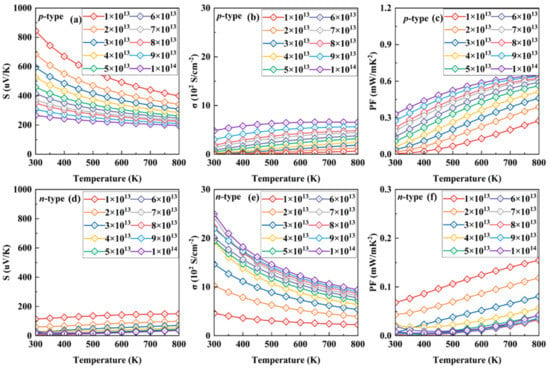
Figure 2.
Seebeck coefficient, electronic conductivity, and PF as a function of temperature under various concentrations of p- (a–c) and n-type (d–f) doping.
Increasing the carrier concentration, the chemical potential enters in the deeper energy levels for both p- and n-types of SnI2, resulting in a decreased Seebeck coefficient at the same temperature. It is clear that the values under p-type doping are always far larger than those under n-type at the same condition, which can be attributed to the multi-band character near the VBM, as shown in Figure 1b. For example, the absolute values of the S under p- and n-types of doping at 700 K are 439 and 146 uV/K, respectively, for a doping concentration of 1 × 1013 cm−2. In addition, the trend of S under p-type doping is opposite to that under n-type. The former decreases while the latter increases upon heating.
Compared with S, the electronic conductivity σ exhibits a different behavior, due to the complete relationship between them, as Equations (1) and (2) show. Interestingly, in all cases, the electrical conductivities for p-type SnI2 are always far less than those for n-type. For example, the values under p- and n-type doping are 0.46 and 2.43 S/cm−2, respectively, at 700 K for a doping concentration of 1 × 1013 cm−2. At 800 K, the electrical conductivity under p-type (n-type) increases from ~0.67 S/cm−2 (~2.24 S/cm−2) for 1 × 1013 cm−2 to 6.35 S/cm−2 (9.43 S/cm−2) for 1 × 1014 cm−2.
Ultimately, the PF decouples the complete relationship between the Seebeck coefficient and electrical conductivity. It is clearly seen that the p-type SnI2 possesses significantly higher PF values than those of the n-type system. This is in good agreement with the previous analysis for the electronic structure. At 800 K, the PF for p-type increases from ~0.27 mW/mK2 for 1 × 1013 cm−2 to 0.62 mW/mK2 for 1 × 1014 cm−2. For the n-type system, however, the PF decreases from ~0.15 mW/mK2 for 1 × 1013 cm−2 to 0.04 mW/mK2 for 1 × 1014 cm−2. Thus, it is expected that the p-type SnI2 possesses a more excellent performance than that of the n-type. According to the Wiedemann–Franz law, κe = LσT, where L is the Lorenz number, the classical value L = (πkB)2/3e2 ≈ 2.44 × 10−8 WΩK−2 is adopted. This value meets the result of κ0-σS2T [50]. Thus, the electronic conductivity is proportional to the electronic thermal conductivity. The results in the present work also obey this rule.
3.3. Phonon Transport Properties
As shown in Figure 3a, the calculated total κL is very low in the wide temperature of 300–800 K. Remarkably, the low room temperature phonon thermal conductivities of 0.33 and 0.19 W/mK along the armchair and zigzag directions, respectively, are fundamentally lower than those previously reported values for 2D MO3 (1.57 W/mk) [51], SnSe (2.77 W/mK) [13], and CaP3 (0.65 W/mK) [52]. Through analyzing the contributions of the acoustic phonon branches along the in-plane (TA and LA) or out-of-plane (ZA), as well as the optical phonon modes to the total κL, results show that the main contribution to the κL is from the ZA mode. In the following, we reveal the origins of such ultralow phonon conductivity, and present a comprehensive analysis to support this result.
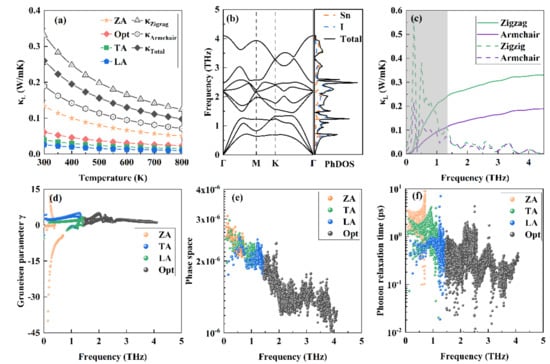
Figure 3.
(a) Calculated lattice thermal conductivity. The contributions from the ZA, TA, LA, and optical modes to the total κL, as well as the lattice thermal conductivities along the armchair and the zigzag directions, are shown. (b) Phonon dispersion curves and corresponding PhDOS. (c) Cumulative thermal conductivity and the derivatives (dashed line) with respect to frequency. (d–f) Grüneisen parameter γ, phonon scattering phase space, and phonon relaxation time of the ZA, TA, LA, and optical modes.
The phonon dispersion curves are presented in Figure 3b. With three atoms in its primitive cell, there are three acoustic and six optical phonon modes for monolayer SnI2. It is dynamically stable, since no imaginary frequency is observed. Near the long wave limit, the TA and LA branches are in linear trend, whereas the ZA branch exhibits a quadratic trend. These features are typical for 2D materials, and can be explained with the elastic theory of thin plate [53]. It is clearly seen that a narrow phonon gap of about 0.1 THz separates the phonon modes into the acoustic phonon part (0~1.35 THz) and the optical phonon part (1.45~4.10 THz). The cutoff acoustic phonon frequency, which is as low as 1.35 THz, is lower than those for SnSe (1.6 THz) and SnS (1.9 THz) [13]. The low-lying acoustic modes, as well as the soft mode for TA near the M point, imply the weak bonding between Sn and I atoms, consistent with the previous analysis. From the corresponding phonon density of states (PhDOS), the contributions from the I atomic vibrations are apparently larger than those from Sn, within the region of 2.8 to 4.10 THz. Both Sn and I evidently contribute in a wide energy range, implying the nature of covalent bonding [12]. In addition, it is recalled that the decoupling of the in-plane and out-of-plane phonon modes results in ultrahigh phonon thermal conductivity for graphene, due to its one atom plane nature [53,54]. However, such full decoupling behavior should not be observed for a finite thickness 2D system, such as the case of the present studied monolayer SnI2.
To explain the anomalous thermal transport behavior of monolayer SnI2, the cumulative lattice conductivity, and their derivatives, with respect to frequency at 300 K are calculated and presented in Figure 3c. Clearly, the κL is mainly caused by phonons of the cutoff acoustic part (the shadow region < 1.35 THz). Specifically, the contributions from ZA, TA, LA, and the optical branches are 51.21%, 15.14%, 10%, and 23.54%, respectively. For graphene, the low frequency (0–5 THz) also dominates the κL and the ZA mode, and contributes 75% to κL [54]. Comparing with graphene, the structure of the monolayer SnI2 lacks mirror symmetry. Thus, the mirror symmetry does not reflect whether the ZA mode dominates the thermal transport [55].
Generally, there are two factors that dominate the κL: (i) anharmonic interaction matrix elements, and (ii) the inverse of phonon phase space volume. The Grüneisen parameter γ is generally employed to quantify the strength of anharmonicity [41]. Figure 3d shows the calculated γ of the ZA, TA, LA, and optical modes of the SnI2 monolayer. As a large |γ| implies strong anharmonicity, it can be seen that the ZA mode exhibits giant anharmonicity. The large |γ| of this out-of-plane mode means that the anharmonicity of the bonding between Sn and I atoms in the vertical direction of the monolayer plate is strong. The phonon scattering phase space reveals all available scattering processes, which are ruled by the energy and (quasi)momentum conservation. As shown in Figure 3e, the total scatting phase space of the ZA, TA, and LA modes are 1.55 × 10−3, 1.41 × 10−3, and 1.35 × 10−3, respectively, which confirms more abundant scattering channels of the ZA mode, than those of the TA and LA modes. The larger the phonon phase space, the greater the contribution to the κL, thus, validating the decreasing contributions to κL from the ZA, TA, and then LA modes. The phonon relaxation time provides a deeper microcosmic insight to understand the ultralow κL of SnI2. Compared with the monolayer SnP3 [38], monolayer SnI2 possesses a shorter phonon relaxation time, implying an ultralow κL.
3.4. Thermoelectric Figure of Merit
By combining the phonon and electron transport coefficients, the ZT of SnI2 under p- and n-types of doping as functions of temperature and carrier concentration are presented in Figure 4. Owing to the calculated thermal conductivity, along the zigzag direction is slightly larger than the armchair direction, and it is expected that the ZT along the armchair direction is higher than that along the zigzag direction. In addition, the p-type SnI2 is obviously superior to the n-type, since the two quasi-degenerate band valleys in VBM leads to a larger Seebeck coefficient. Results show that monolayer SnI2 is thermally stable up to 800 K, by performing ab initio molecular dynamic (AIMD) simulations (see Figure S1). The values of ZT are as high as 4.01 and 3.34 along the armchair and zigzag directions, respectively, around concentrations of 1.0 × 1013 and 1.2 × 1013 cm−2, at 800 K. Such large values are better than the experimental value of 2.6 for the well-known TE material SnSe along the specific axis at 925 K [3]. In addition, the doping carrier concentrations at such levels have been realized experimentally in monolayer MoS2 [56]. Therefore, such a high TE value for monolayer SnI2 is possible, and indicates its excellent potential TE performance.
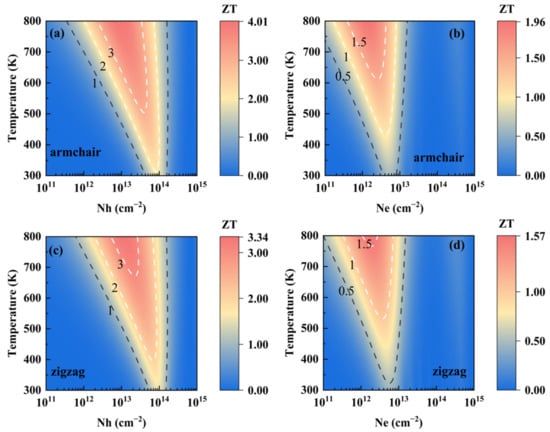
Figure 4.
Contour maps of ZT as functions of both temperature and carrier concentration for monolayer SnI2: (a) p-type and (b) n-type along the armchair direction, (c) p-type and (d) n-type along the zigzag direction.
To clearly compare the TE performances of the monolayer SnI2 with some famous TE materials, the κL and the max ZT are listed in Table 2. According to these results, the monolayer SnI2 is an emerging candidate for TE devices, due to its ultrahigh ZT. As seen, the κL of monolayer SnI2 is very low, almost a twentieth of the monolayer SnP3 [38], and is greatly lower than those of tin selenide and tin sulfide [13,48]. Such low value of the κL for monolayer SnI2 make it a typical 2D TE system. After all, the electronic transport properties can be easily modulated in experiments, while the lattice thermal conductivity is very difficult to change. In addition, the κL values at the level of 0.1-0.5 W/mK in experiments are very few. To the best of our knowledge, the complex systems of CsAg5Te3 (0.18 W/mK) [57] and CsCu5Se3 (0.4~0.8 W/mK) [58], as well as the bulk superlattice material Bi4O4SeCl2 (0.1 W/mK) [59], are typical low κL TE materials. Overall, the low thermal conductivity, as well as the high ZT, make monolayer SnI2 a good TE material for low-dimensional devices.
Table 2.
κL at 300 K and the max ZT at corresponding maximum thermodynamic temperature for monolayer SnI2, as well as some typical TE materials.
4. Conclusions
In summary, the TE properties of monolayer SnI2 were studied using DFT and the semi-classical Boltzmann transport equation. The results indicate that this 2D material possesses intrinsically ultralow lattice thermal conductivity. Its strong anharmonicity, weak bonding, and softened acoustic branches greatly suppress the phonon transport, and result in an ultralow κL of 0.33 and 0.18 W/mK at 300 K along the zigzag and armchair directions, respectively. The p-type SnI2 possesses a superior electric transport performance than the n-type one, due to the two quasi-degenerate band valleys in its VBM. The ZT at 800 K under p-type doping is as high as 4.01 along the armchair direction. Collectively, these results indicate the great advantages of monolayer SnI2 for converting heat energy with high efficiency at high temperatures. Generally, when the ZT value of a material exceeds 1.0, it is considered as an ideal TE material. Therefore, the high ZT of monolayer SnI2 demonstrates it as an emerging candidate for TE applications.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ma15093147/s1, Figure S1: Free energy fluctuations with respect to time and equilibrium structures of SnI2 monolayer by AIMD simulations at 800 K.
Author Contributions
Conceptualization, Q.-Y.X., K.-W.Z. and B.-T.W.; methodology, Q.-Y.X., P.-F.L., J.-J.M. and B.-T.W.; software, B.-T.W.; validation, Q.-Y.X., K.-W.Z. and B.-T.W.; formal analysis, Q.-Y.X.; investigation, Q.-Y.X., P.-F.L. and F.-G.K.; resources, B.-T.W.; data curation, Q.-Y.X. and B.-T.W.; writing—original draft preparation, Q.-Y.X.; writing—review and editing, K.-W.Z. and B.-T.W.; supervision, K.-W.Z. and B.-T.W.; project administration, K.-W.Z. and B.-T.W.; funding acquisition, B.-T.W. All authors have read and agreed to the published version of the manuscript.
Funding
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (Grants No. 12074381, No. 12005230, and No. 12104458), the Guangdong Basic and Applied Basic Research Foundation (Grant No. 2021A1515110587), and the Youth Science Foundation of Jiangxi Province (Grants 20171BAB211009).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding authors. The data are not publicly available due to ongoing research in the project.
Conflicts of Interest
There are no conflicts to declare.
References
- Pei, Y.; Shi, X.; LaLonde, A.; Wang, H.; Chen, L.; Snyder, G.J. Convergence of electronic bands for high performance bulk thermoelectrics. Nature 2011, 473, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Zhao, L.D.; Kanatzidis, M.G. Rationally Designing High-Performance Bulk Thermoelectric Materials. Chem. Rev. 2016, 116, 12123–12149. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.D.; Tan, G.; Hao, S.; He, J.; Pei, Y.; Chi, H.; Wang, H.; Gong, S.; Xu, H.; Dravid, V.P.; et al. Ultrahigh power factor and thermoelectric performance in hole-doped single-crystal SnSe. Science 2016, 351, 141–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhao, L.-D. Thermoelectric materials: Energy conversion between heat and electricity. J. Mater. 2015, 1, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.D.; Chang, C.; Tan, G.J.; Kanatzidis, M.G. SnSe: A remarkable new thermoelectric material. Energy Environ. Sci. 2016, 9, 3044–3060. [Google Scholar] [CrossRef]
- Zhao, L.D.; He, J.; Wu, C.I.; Hogan, T.P.; Zhou, X.; Uher, C.; Dravid, V.P.; Kanatzidis, M.G. Thermoelectrics with earth abundant elements: High performance p-type PbS nanostructured with SrS and CaS. J. Am. Chem. Soc. 2012, 134, 7902–7912. [Google Scholar] [CrossRef]
- Zhao, L.D.; Wu, H.J.; Hao, S.Q.; Wu, C.I.; Zhou, X.Y.; Biswas, K.; He, J.Q.; Hogan, T.P.; Uher, C.; Wolverton, C.; et al. All-scale hierarchical thermoelectrics: MgTe in PbTe facilitates valence band convergence and suppresses bipolar thermal transport for high performance. Energy Environ. Sci. 2013, 6, 3346–3355. [Google Scholar] [CrossRef]
- Pei, Y.Z.; May, A.F.; Snyder, G.J. Self-Tuning the Carrier Concentration of PbTe/Ag2Te Composites with Excess Ag for High Thermoelectric Performance. Adv. Energy Mater. 2011, 1, 291–296. [Google Scholar] [CrossRef]
- Zebarjadi, M.; Joshi, G.; Zhu, G.; Yu, B.; Minnich, A.; Lan, Y.; Wang, X.; Dresselhaus, M.; Ren, Z.; Chen, G. Power factor enhancement by modulation doping in bulk nanocomposites. Nano Lett. 2011, 11, 2225–2230. [Google Scholar] [CrossRef]
- Shi, X.; Yang, J.; Salvador, J.R.; Chi, M.; Cho, J.Y.; Wang, H.; Bai, S.; Yang, J.; Zhang, W.; Chen, L. Multiple-filled skutterudites: High thermoelectric figure of merit through separately optimizing electrical and thermal transports. J. Am. Chem. Soc. 2011, 133, 7837–7846. [Google Scholar] [CrossRef]
- Zhang, Y.; Skoug, E.; Cain, J.; Ozoliņš, V.; Morelli, D.; Wolverton, C. First-principles description of anomalously low lattice thermal conductivity in thermoelectric Cu-Sb-Se ternary semiconductors. Phys. Rev. B 2012, 85, 054306. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.-F.; Bo, T.; Xu, J.; Yin, W.; Zhang, J.; Wang, F.; Eriksson, O.; Wang, B.-T. First-principles calculations of the ultralow thermal conductivity in two-dimensional group-IV selenides. Phys. Rev. B 2018, 98, 235426. [Google Scholar] [CrossRef]
- Guo, R.Q.; Wang, X.J.; Kuang, Y.D.; Huang, B.L. First-principles study of anisotropic thermoelectric transport properties of IV-VI semiconductor compounds SnSe and SnS. Phys. Rev. B 2015, 92, 115202. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.Z.; Jia, T.T.; Zhang, J.H.; Wang, Y.X.; Zhang, Y.S. Dual effects of lone-pair electrons and rattling atoms in CuBiS2 on its ultralow thermal conductivity. Phys. Rev. B 2017, 96, 235205. [Google Scholar] [CrossRef]
- Nielsen, M.D.; Ozolins, V.; Heremans, J.P. Lone pair electrons minimize lattice thermal conductivity. Energy Environ. Sci. 2013, 6, 570–578. [Google Scholar] [CrossRef]
- Liu, H.; Yuan, X.; Lu, P.; Shi, X.; Xu, F.; He, Y.; Tang, Y.; Bai, S.; Zhang, W.; Chen, L.; et al. Ultrahigh thermoelectric performance by electron and phonon critical scattering in Cu2 Se1-x Ix. Adv. Mater. 2013, 25, 6607–6612. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Zhou, Y.; Hu, M. Two-Channel Thermal Transport in Ordered-Disordered Superionic Ag2Te and Its Traditionally Contradictory Enhancement by Nanotwin Boundary. J. Phys. Chem. Lett. 2018, 9, 5704–5709. [Google Scholar] [CrossRef]
- Luo, Y.; Yang, X.; Feng, T.; Wang, J.; Ruan, X. Vibrational hierarchy leads to dual-phonon transport in low thermal conductivity crystals. Nat. Commun. 2020, 11, 2554. [Google Scholar] [CrossRef]
- Ravindran, P.; Delin, A.; Ahuja, R.; Johansson, B.; Auluck, S.; Wills, J.M.; Eriksson, O. Optical properties of monoclinic SnI2from relativistic first-principles theory. Phys. Rev. B 1997, 56, 6851–6861. [Google Scholar] [CrossRef] [Green Version]
- Zhong, M.; Zhang, S.; Huang, L.; You, J.; Wei, Z.; Liu, X.; Li, J. Large-scale 2D PbI2 monolayers: Experimental realization and their indirect band-gap related properties. Nanoscale 2017, 9, 3736–3741. [Google Scholar] [CrossRef]
- Yagmurcukardes, M.; Peeters, F.M.; Sahin, H. Electronic and vibrational properties of PbI2: From bulk to monolayer. Phys. Rev. B 2018, 98, 085431. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.M.; Hou, B.; Wu, Y.; Chen, Y.; Xia, Y.-J.; Mei, H.; Kong, M.R.; Peng, L.; Shao, H.Z.; Cao, J.; et al. Towards high-temperature electron-hole condensate phase in monolayer tetrels metal halides: Ultra-long excitonic lifetimes, phase diagram and exciton dynamics. Mater. Today Phys. 2022, 22, 100604. [Google Scholar] [CrossRef]
- Sandoval, S.; Kepic, D.; Perez Del Pino, A.; Gyorgy, E.; Gomez, A.; Pfannmoeller, M.; Tendeloo, G.V.; Ballesteros, B.; Tobias, G. Selective Laser-Assisted Synthesis of Tubular van der Waals Heterostructures of Single-Layered PbI2 within Carbon Nanotubes Exhibiting Carrier Photogeneration. ACS Nano 2018, 12, 6648–6656. [Google Scholar] [CrossRef] [Green Version]
- Peng, B.; Mei, H.D.; Zhang, H.; Shao, H.Z.; Xu, K.; Ni, G.; Jin, Q.Y.; Soukoulis, C.M.; Zhu, H.Y. High thermoelectric efficiency in monolayer PbI2 from 300 K to 900 K. Inorg. Chem. Front. 2019, 6, 920–928. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.; Huang, J.W.; Dong, N.N.; Hong, S.; Yan, C.; Liu, Y.C.; Qiu, J.S.; Wang, J.; Sun, Z.Y. Liquid Exfoliation of Two-Dimensional PbI2 Nanosheets for Ultrafast Photonics. ACS Photonics 2019, 6, 1051–1057. [Google Scholar] [CrossRef]
- Howie, R.A.; Moser, W.; Trevena, I.C. The crystal structure of tin(II) iodide. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 1972, 28, 2965–2971. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Q.Q.; Zheng, F.; Shi, Z.Q.; Li, Q.Y.; Lv, Y.Y.; Chen, Y.; Zhang, P.; Li, S.C. Direct Growth of van der Waals Tin Diiodide Monolayers. Adv. Sci. 2021, 8, 2100009. [Google Scholar] [CrossRef]
- Arab, A.; Li, Q. Anisotropic thermoelectric behavior in armchair and zigzag mono- and fewlayer MoS2 in thermoelectric generator applications. Sci. Rep. 2015, 5, 13706. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter. 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B Condens. Matter 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Blochl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B Condens. Matter. 1994, 49, 16223–16233. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E. Assessment and validation of a screened Coulomb hybrid density functional. J. Chem. Phys. 2004, 120, 7274–7280. [Google Scholar] [CrossRef] [PubMed]
- Madsen, G.K.H.; Singh, D.J. BoltzTraP. A code for calculating band-structure dependent quantities. Comput. Phys. Commun. 2006, 175, 67–71. [Google Scholar] [CrossRef] [Green Version]
- Bardeen, J.; Shockley, W. Deformation Potentials and Mobilities in Non-Polar Crystals. Phys. Rev. 1950, 80, 72–80. [Google Scholar] [CrossRef]
- Lee, M.S.; Mahanti, S.D. Validity of the rigid band approximation in the study of the thermopower of narrow band gap semiconductors. Phys. Rev. B 2012, 85, 165149. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Zhang, G.; Zhang, Y.W. Polarity-reversed robust carrier mobility in monolayer MoS2 nanoribbons. J. Am. Chem. Soc. 2014, 136, 6269–6275. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.L.; Liu, P.F.; Zhang, J.; Zhang, P.; Zhou, W.X.; Xie, G.; Wang, B.T. Monolayer SnP3: An excellent p-type thermoelectric material. Nanoscale 2019, 11, 19923–19932. [Google Scholar] [CrossRef]
- Baroni, S.; de Gironcoli, S.; Dal Corso, A.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef] [Green Version]
- Togo, A.; Oba, F.; Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type andCaCl2-typeSiO2at high pressures. Phys. Rev. B 2008, 78, 134106. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Carrete, J.; Katcho, N.A.; Mingo, N. ShengBTE: A solver of the Boltzmann transport equation for phonons. Comput. Phys. Commun. 2014, 185, 1747–1758. [Google Scholar] [CrossRef]
- Tong, Z.; Li, S.; Ruan, X.; Bao, H. Comprehensive first-principles analysis of phonon thermal conductivity and electron-phonon coupling in different metals. Phys. Rev. B 2019, 100, 144306. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, A.; Hu, Y.; Gu, X.; Tong, Z.; Bao, H. Anomalous thermal transport in metallic transition-metal nitrides originated from strong electron–phonon interactions. Mater. Today Phys. 2020, 15, 100256. [Google Scholar] [CrossRef]
- Wang, Y.X.; Xu, N.; Li, D.Y.; Zhu, J. Thermal Properties of Two Dimensional Layered Materials. Adv. Funct. Mater. 2017, 27, 1604134. [Google Scholar] [CrossRef]
- Peng, B.; Zhang, H.; Shao, H.; Xu, Y.; Zhang, R.; Lu, H.; Zhang, D.W.; Zhu, H. First-Principles Prediction of Ultralow Lattice Thermal Conductivity of Dumbbell Silicene: A Comparison with Low-Buckled Silicene. ACS Appl. Mater. Interfaces 2016, 8, 20977–20985. [Google Scholar] [CrossRef]
- Onodera, T.; Morita, Y.; Nagumo, R.; Miura, R.; Suzuki, A.; Tsuboi, H.; Hatakeyama, N.; Endou, A.; Takaba, H.; Dassenoy, F.; et al. A computational chemistry study on friction of h-MoS(2). Part II. Friction anisotropy. J. Phys. Chem. B 2010, 114, 15832–15838. [Google Scholar] [CrossRef]
- Zhao, L.D.; Zhang, X.; Wu, H.; Tan, G.; Pei, Y.; Xiao, Y.; Chang, C.; Wu, D.; Chi, H.; Zheng, L.; et al. Enhanced Thermoelectric Properties in the Counter-Doped SnTe System with Strained Endotaxial SrTe. J. Am. Chem. Soc. 2016, 138, 2366–2373. [Google Scholar] [CrossRef]
- Wang, D.Y.; He, W.K.; Chang, C.; Wang, G.T.; Wang, J.F.; Zhao, L.D. Thermoelectric transport properties of rock-salt SnSe: First-principles investigation. J. Mater. Chem. C 2018, 6, 12016–12022. [Google Scholar] [CrossRef]
- Huang, S.; Wang, Z.Y.; Xiong, R.; Yu, H.Y.; Shi, J. Significant enhancement in thermoelectric performance of Mg3Sb2 from bulk to two-dimensional mono layer. Nano Energy 2019, 62, 212–219. [Google Scholar] [CrossRef]
- Wang, N.; Li, M.; Xiao, H.; Zu, X.; Qiao, L. Layered LaCuOSe: A Promising Anisotropic Thermoelectric Material. Phys. Rev. Appl. 2020, 13, 024038. [Google Scholar] [CrossRef]
- Tong, Z.; Dumitrica, T.; Frauenheim, T. Ultralow Thermal Conductivity in Two-Dimensional MoO3. Nano Lett. 2021, 21, 4351–4356. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-L.; Liu, P.-F.; Wu, Y.-Y.; Zhang, P.; Xie, G.; Wang, B.-T. Significant enhancement of the thermoelectric properties of CaP3 through reducing the dimensionality. Adv. Mater. 2020, 1, 3322–3332. [Google Scholar] [CrossRef]
- Liu, D.; Every, A.G.; Tománek, D. Continuum approach for long-wavelength acoustic phonons in quasi-two-dimensional structures. Phys. Rev. B 2016, 94, 165432. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, L.; Li, W.; Carrete, J.; Mingo, N.; Broido, D.A.; Reinecke, T.L. Phonon thermal transport in strained and unstrained graphene from first principles. Phys. Rev. B 2014, 89, 155426. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, E.; Duan, F.; Wei, D.; Zheng, X.; Tang, C.; Ouyang, T.; Yao, Y.; Qin, G.; Zhong, J. Unique Arrangement of Atoms Leads to Low Thermal Conductivity: A Comparative Study of Monolayer Mg2C. J. Phys. Chem. Lett. 2021, 12, 10353–10358. [Google Scholar] [CrossRef]
- Hippalgaonkar, K.; Wang, Y.; Ye, Y.; Qiu, D.Y.; Zhu, H.; Wang, Y.; Moore, J.; Louie, S.G.; Zhang, X. High thermoelectric power factor in two-dimensional crystals of MoS2. Phys. Rev. B 2017, 95, 115407. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Tan, G.; Shen, J.N.; Hao, S.; Wu, L.M.; Calta, N.; Malliakas, C.; Wang, S.; Uher, C.; Wolverton, C.; et al. Concerted Rattling in CsAg5 Te3 Leading to Ultralow Thermal Conductivity and High Thermoelectric Performance. Angew. Chem. Int. Ed. Engl. 2016, 55, 11431–11436. [Google Scholar] [CrossRef]
- Ma, N.; Li, Y.Y.; Chen, L.; Wu, L.M. alpha-CsCu5Se3: Discovery of a Low-Cost Bulk Selenide with High Thermoelectric Per formance. J. Am. Chem. Soc. 2020, 142, 5293–5303. [Google Scholar] [CrossRef]
- Gibson, Q.D.; Zhao, T.; Daniels, L.M.; Walker, H.C.; Daou, R.; Hebert, S.; Zanella, M.; Dyer, M.S.; Claridge, J.B.; Slater, B.; et al. Low thermal conductivity in a modular inorganic material with bonding anisotropy and mismatch. Science 2021, 373, 1017–1022. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).