
Lithium-Rich Rock Salt Type Sulfides-Selenides (Li2TiSexS3−x): High Energy Cathode Materials for Lithium-Ion Batteries




Abstract
1. Introduction
2. Materials and Methods
2.1. Wet Mechanochemical Synthesis of Li2TiSexS3−x
2.2. Structural Characterizations
2.3. Electrochemical Characterizations
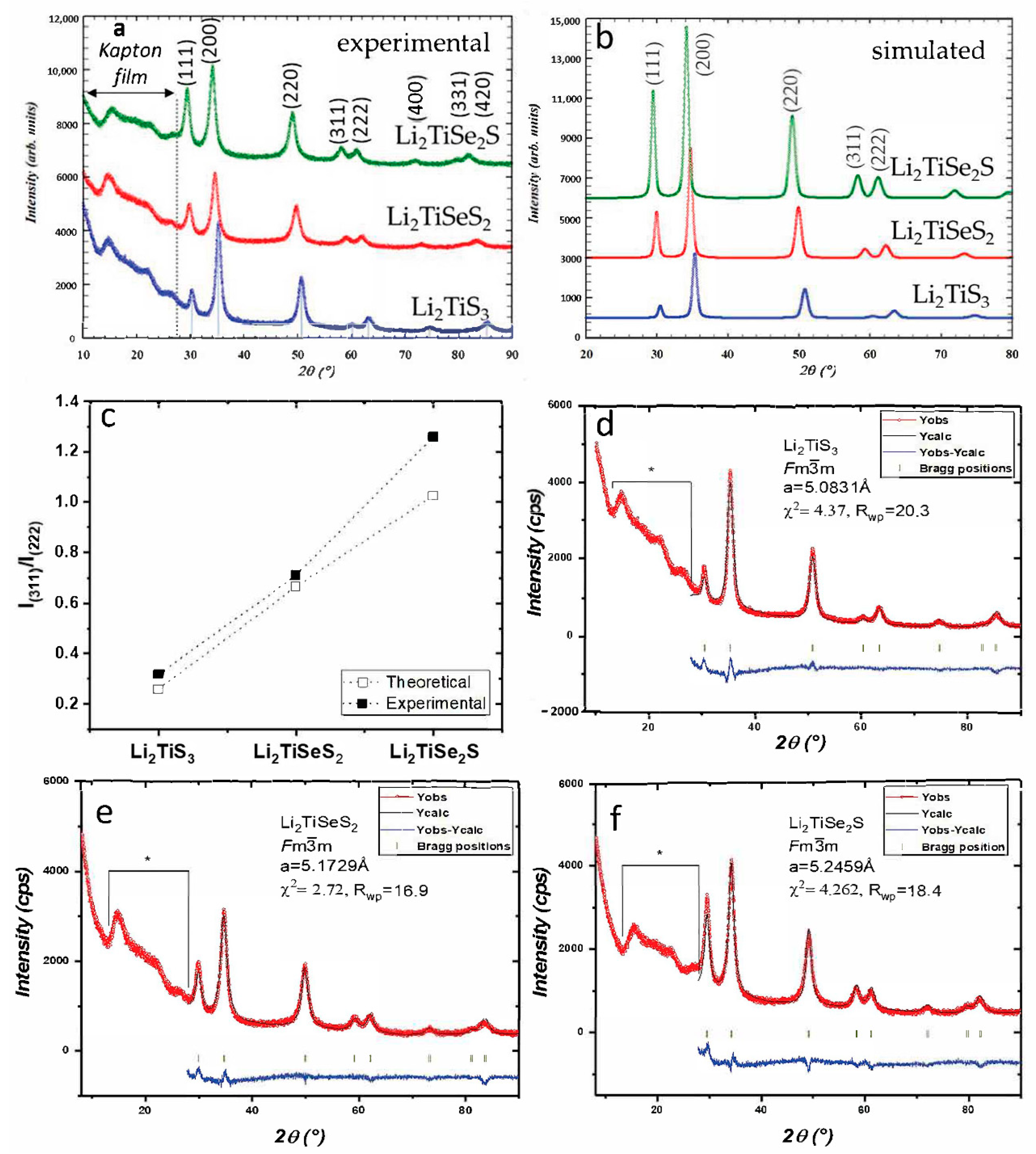
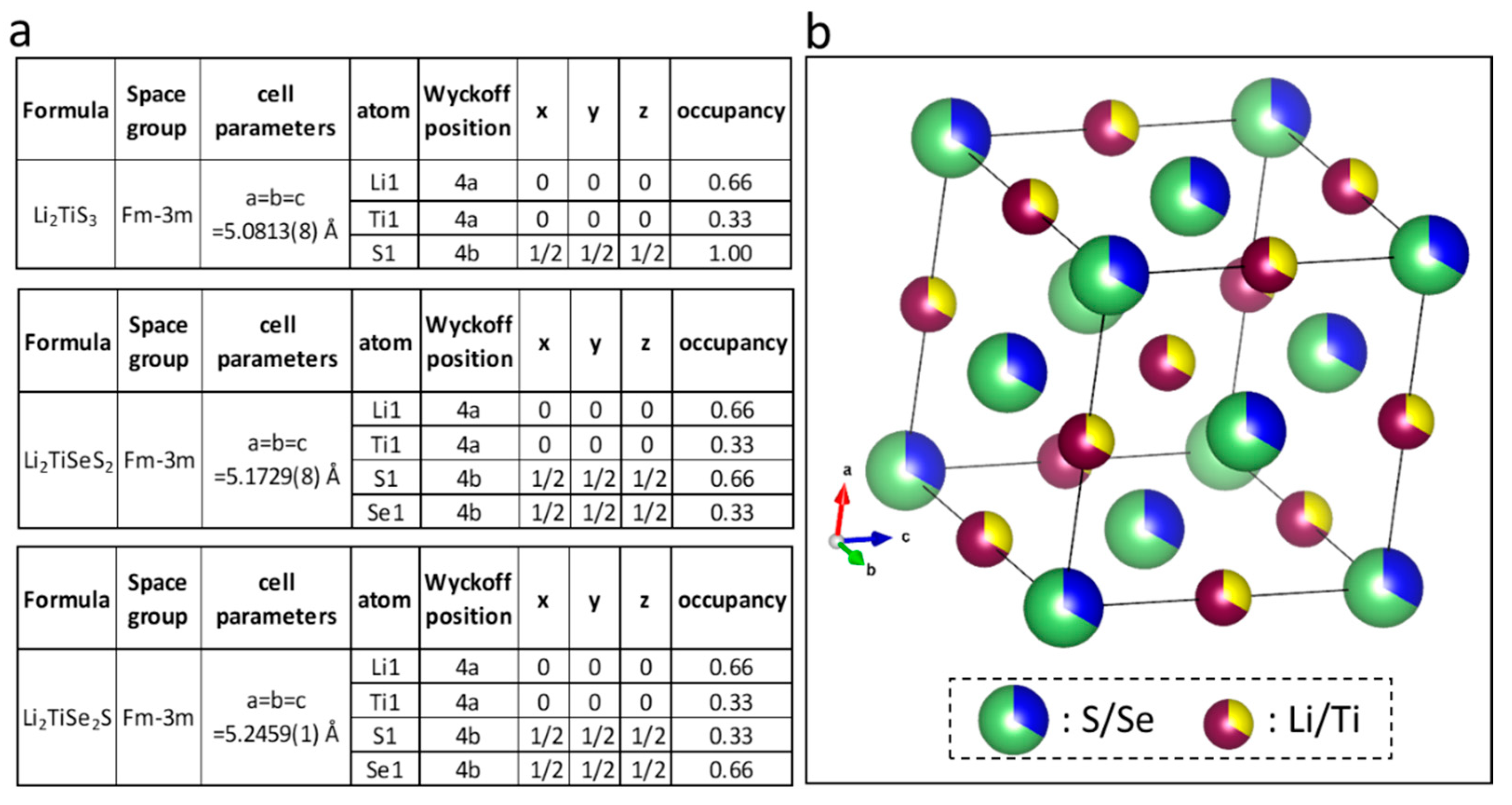
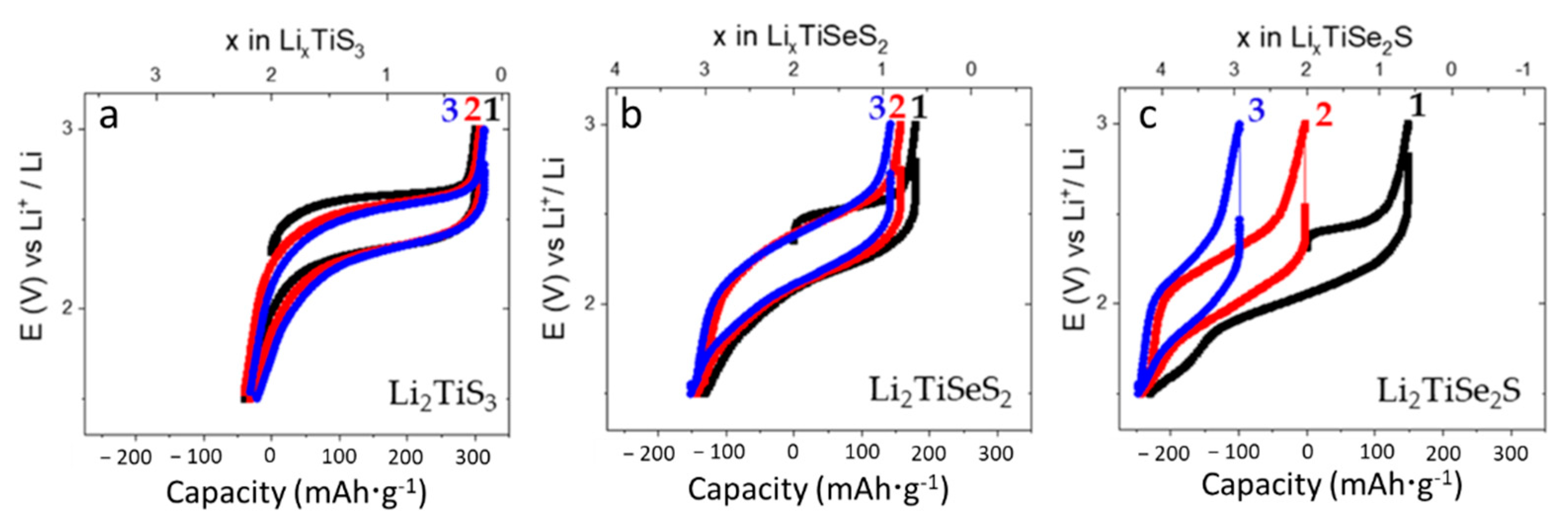
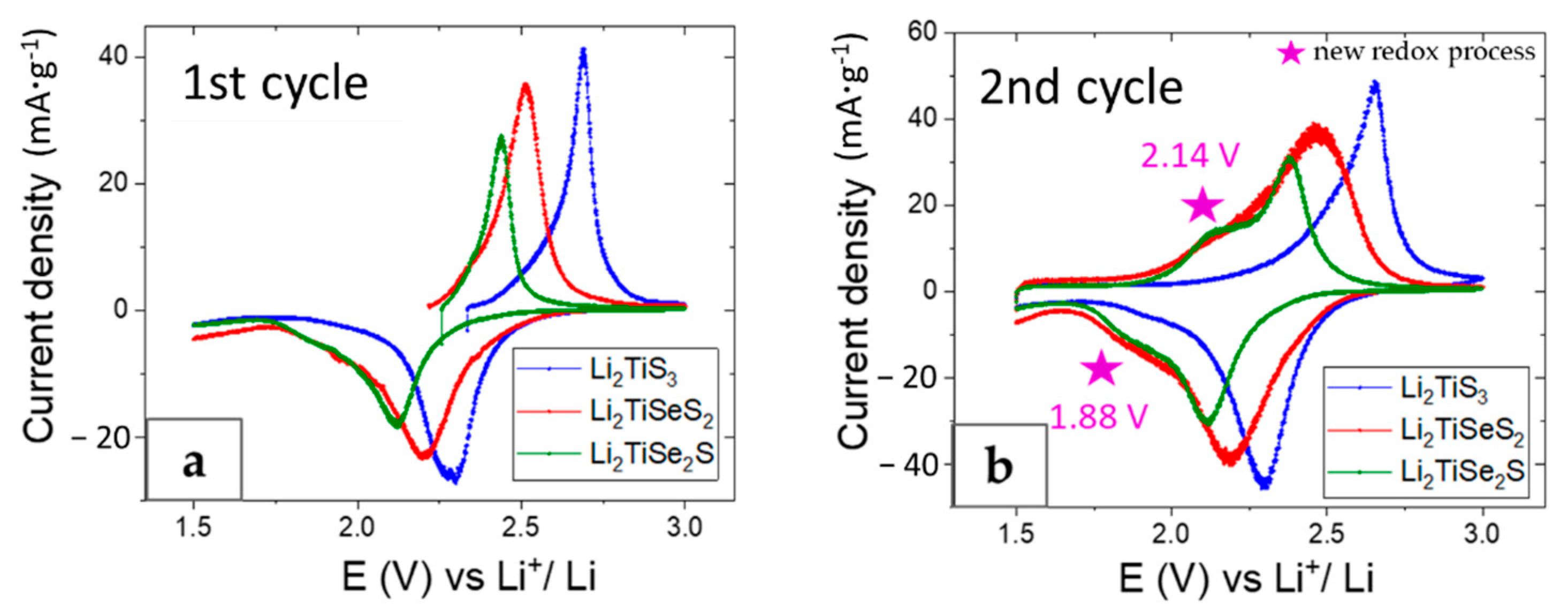
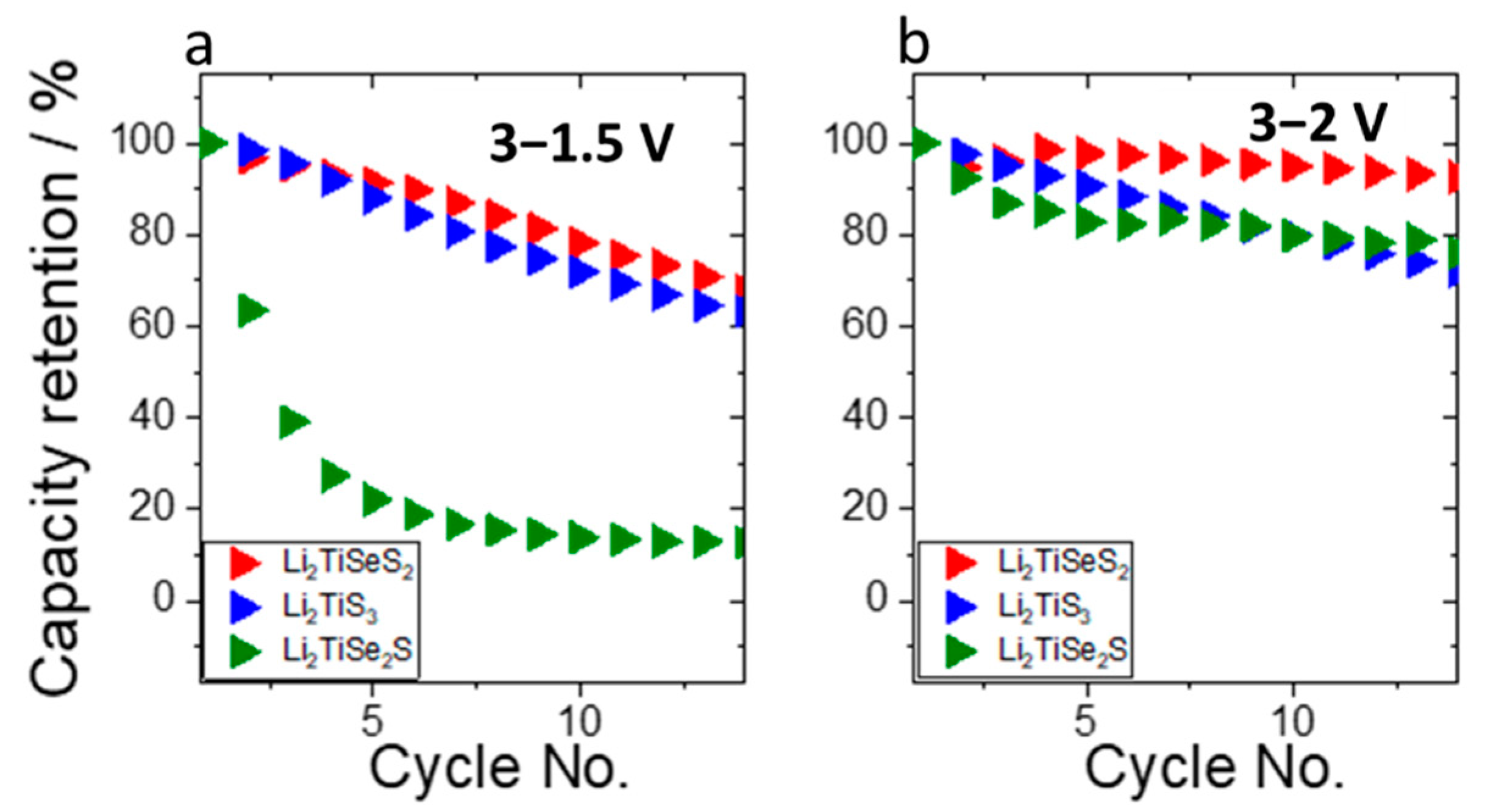
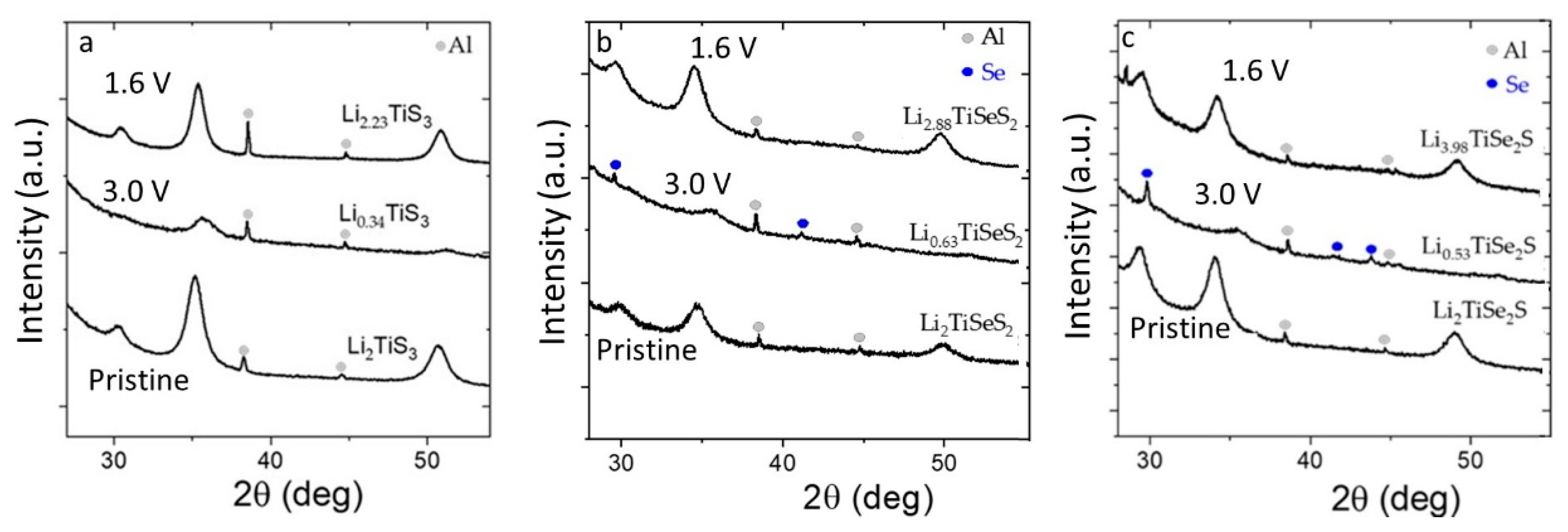
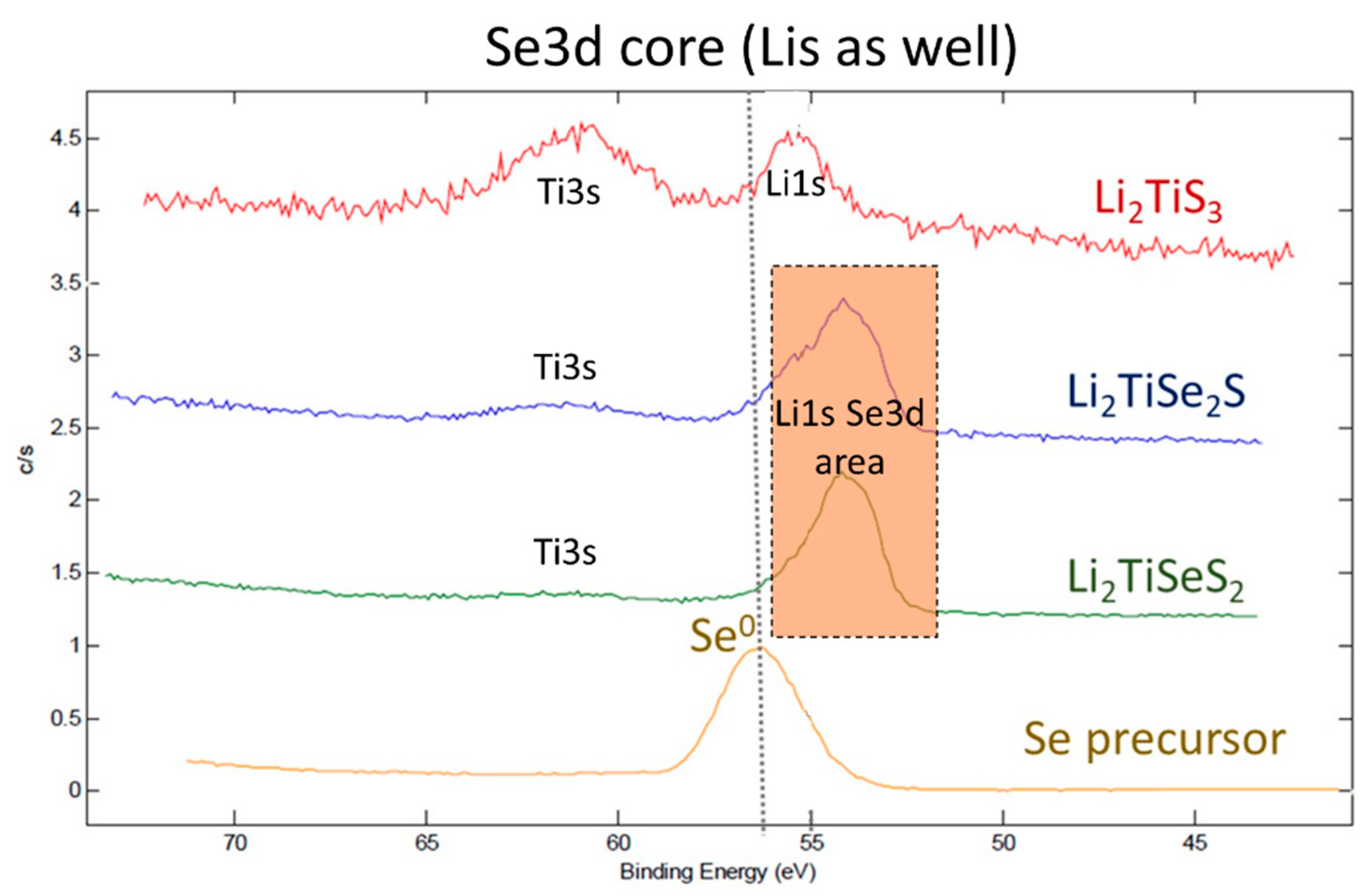
3. Results
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Larcher, D.; Tarascon, J.-M. Towards greener and more sustainable batteries for electrical energy storage. Nat. Chem. 2015, 7, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Rozier, P.; Tarascon, J.M. Review—Li-rich layered oxide cathodes for next-generation li-ion batteries: Chances and challenges. J. Electrochem. Soc. 2015, 162, A2490–A2499. [Google Scholar] [CrossRef]
- Schmuch, R.; Wagner, R.; Hörpel, G.; Placke, T.; Winter, M. Performance and cost of materials for lithium-based rechargeable automotive batteries. Nat. Energy 2018, 3, 267–278. [Google Scholar] [CrossRef]
- Wang, J.; Liu, C.; Xu, G.; Miao, C.; Wen, M.; Xu, M.; Wang, C.; Xiao, W. Strengthened the structural stability of in-situ F− doping Ni-rich LiNi0.8Co0.15Al0.05O2 cathode materials for lithium-ion batteries. Chem. Eng. J. 2022, 438, 135537. [Google Scholar] [CrossRef]
- Yabuuchi, N. Material design concept of lithium-excess electrode materials with rocksalt-related structures for rechargeable non-aqueous batteries. Chem. Rec. 2018, 19, 690–707. [Google Scholar] [CrossRef]
- Li, W.; Lee, S.; Manthiram, A. High-nickel NMA: A cobalt-free alternative to NMC and NCA cathodes for lithium-ion batteries. Adv. Mater. 2020, 32, 2002718. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, B.; Ning, L.; Feng, W. Hierarchically porous micro-rod lithium-rich cathode material Li1.2Ni0.13Mn0.54Co0.13O2 for high performance lithium-ion batteries. Electrochim. Acta 2014, 118, 67–74. [Google Scholar] [CrossRef]
- He, X.; Wang, J.; Kloepsch, R.; Krueger, S.; Jia, H.; Liu, H.; Vortmann, B.; Li, J. Enhanced electrochemical performance in lithium ion batteries of a hollow spherical lithium-rich cathode material synthesized by a molten salt method. Nano Res. 2014, 7, 110–118. [Google Scholar] [CrossRef]
- Song, B.; Liu, Z.; On Lai, M.; Lu, L. Structural evolution and the capacity fade mechanism upon long-term cycling in li-rich cathode material. Phys. Chem. Chem. Phys. 2012, 14, 12875–12883. [Google Scholar] [CrossRef]
- Muralidharan, N.; Essehli, R.; Hermann, R.P.; Parejiya, A.; Amin, R.; Bai, Y.; Du, Z.; Belharouak, I. LiNixFeyAlzO2, a new cobalt-free layered cathode material for advanced Li-ion batteries. J. Power Sour. 2020, 471, 228389. [Google Scholar] [CrossRef]
- Xiao, J.; Chen, X.; Sushko, P.V.; Sushko, M.L.; Kovarik, L.; Feng, J.; Deng, Z.; Zheng, J.; Graff, G.L.; Nie, Z.; et al. High-Performance LiNi0.5Mn1.5O4 spinel controlled by Mn3+ concentration and site disorder. Adv. Mater. 2012, 24, 2109–2116. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Myung, S.-T.; Yoon, C.S.; Kang, S.G.; Sun, Y.-K. Comparative study of LiNi0.5Mn1.5O4−δ and LiNi0.5Mn1.5O4 cathodes having two crystallographic structures: Fd3-m and P4332. Chem. Mater. 2004, 16, 906–914. [Google Scholar] [CrossRef]
- Pang, Q.; Liang, X.; Kwok, C.Y.; Nazar, L.F. Advances in lithium–sulfur batteries based on multifunctional cathodes and electrolytes. Nat. Energy 2016, 1, 16132. [Google Scholar] [CrossRef]
- Zhuang, Z.; Kang, Q.; Wang, D.; Li, Y. Single-atom catalysis enables long-life, high-energy lithium-sulfur batteries. Nano Res. 2020, 13, 1856–1866. [Google Scholar] [CrossRef]
- Xu, Z.-L.; Kim, S.J.; Chang, D.; Park, K.-Y.; Dae, K.S.; Dao, K.P.; Yuk, J.M.; Kang, K. Visualization of regulated nucleation and growth of lithium sulfides for high energy lithium sulfur batteries. Energy Environ. Sci. 2019, 12, 3144–3155. [Google Scholar] [CrossRef]
- Lee, J.; Urban, A.; Li, X.; Su, D.; Hautier, G.; Ceder, G. Unlocking the potential of cation-disordered oxides for rechargeable lithium batteries. Science 2014, 343, 519–522. [Google Scholar] [CrossRef]
- Yabuuchi, N.; Takeuchi, M.; Nakayama, M.; Shiiba, H.; Ogawa, M.; Nakayama, K.; Ohta, T.; Endo, D.; Ozaki, T.; Inamasu, T.; et al. High-capacity electrode materials for rechargeable lithium batteries: Li3NbO4-based system with cation-disordered rocksalt structure. Proc. Natl. Acad. Sci. USA 2015, 112, 7650–7655. [Google Scholar] [CrossRef]
- Wang, R.; Li, X.; Liu, L.; Lee, J.; Seo, D.-H.; Bo, S.-H.; Urban, A.; Ceder, G. A disordered rock-salt Li-excess cathode material with high capacity and substantial oxygen redox activity: Li1.25Nb0.25Mn0.5O2. Electrochem. Commun. 2015, 60, 70–73. [Google Scholar] [CrossRef]
- Sakuda, A.; Takeuchi, T.; Okamura, K.; Kobayashi, H.; Sakaebe, H.; Tatsumi, K.; Ogumi, Z. Rock-Salt-Type Lithium Metal Sulphides as Novel Positive-Electrode Materials. Sci. Rep. 2015, 4, 4883. [Google Scholar] [CrossRef]
- Sakuda, A.; Takeuchi, T.; Shikano, M.; Ohara, K.; Fukuda, K.; Uchimoto, Y.; Ogumi, Z.; Kobayashi, H.; Sakaebe, H. Development of Li2TiS3–Li3NbS4 by a mechanochemical process. J. Ceram. Soc. Jpn. 2017, 125, 268–271. [Google Scholar] [CrossRef][Green Version]
- Grimaud, A.; Hong, W.T.; Shao-Horn, Y.; Tarascon, J.-M. Anionic redox processes for electrochemical devices. Nat. Mater. 2016, 15, 121. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Xia, D. Anionic redox in rechargeable lithium batteries. Adv. Mater. 2017, 29, 1701054. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Lim, H.S. Factors that affect cycle-life and possible degradation mechanisms of a Li-Ion cell based on LiCoO2. J. Power Sour. 2002, 111, 130–136. [Google Scholar] [CrossRef]
- Sakuda, A.; Takeuchi, T.; Kobayashi, H.; Sakaebe, H.; Tatsumi, K.; Ogumi, Z. Preparation of novel electrode materials based on Lithium Niobium Sulfides. Electrochemistry 2014, 82, 880–883. [Google Scholar] [CrossRef]
- Sakuda, A.; Kuratani, K.; Takeuchi, T.; Kiuchi, H.; Kawaguchi, T.; Shikano, M.; Sakaebe, H.; Kobayashi, H. Cubic Rocksalt Li2SnS3 and a solid solution with Li3NbS4 prepared by mechanochemical synthesis. Electrochemistry 2017, 85, 580–584. [Google Scholar] [CrossRef][Green Version]
- Whittingham, M.S. Lithium incorporation in crystalline and amorphous chalcogenides: Thermodynamics, mechanism and structure. J. Electroanal. Chem. Interfacial Electrochem. 1981, 118, 229–239. [Google Scholar] [CrossRef]
- Chianelli, R.R.; Dines, M.B. Reaction of butyllithium with transition metal trichalcogenides. Inorg. Chem. 1975, 14, 2417–2421. [Google Scholar] [CrossRef]
- Eftekhari, A. The Rise of Lithium–Selenium Batteries. Sustain. Energy Fuels 2017, 1, 14–29. [Google Scholar] [CrossRef]
- Hu, J.; Zhong, H.; Yan, X.; Zhang, L. Confining selenium disulfide in 3D sulfur-doped mesoporous carbon for rechargeable lithium batteries. Appl. Surf. Sci. 2018, 457, 705–711. [Google Scholar] [CrossRef]
- Celasun, Y.; Colin, J.-F.; Martinet, S.; Peralta, D. Synthesis Method. EP 3626681, 19 March 2020. [Google Scholar]
- Rodriguez-Carvajal, J. Recent Developments of the Program FULLPROF, Commission on Powder Diffraction. IUCr Newsl. 2001, 26, 12–19. [Google Scholar]
- Sakuda, A.; Ohara, K.; Kawaguchi, T.; Fukuda, K.; Nakanishi, K.; Arai, H.; Uchimoto, Y.; Ohta, T.; Matsubara, E.; Ogumi, Z.; et al. A reversible rocksalt to amorphous phase transition involving anion redox. Sci. Rep. 2018, 8, 15086. [Google Scholar] [CrossRef] [PubMed]
- Huo, S.H.; Qian, M.; Schaffer, G.B.; Crossin, E. 21—Aluminium powder metallurgy. In Fundamentals of Aluminium Metallurgy; Lumley, R., Ed.; Woodhead Publishing Series in Metals and Surface Engineering; Woodhead Publishing: Cambridge, UK, 2011; pp. 655–701. [Google Scholar] [CrossRef]
- Cui, Y.; Abouimrane, A.; Lu, J.; Bolin, T.; Ren, Y.; Weng, W.; Sun, C.; Maroni, V.A.; Heald, S.M.; Amine, K. (De)Lithiation mechanism of Li/SeSx (x = 0–7) batteries determined by in situ synchrotron X-ray diffraction and X-ray absorption spectroscopy. J. Am. Chem. Soc. 2013, 135, 8047–8056. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition | Lattice Parameter a (Å) | 1st Charge Capacity (mAh·g−1) | 1st Discharge Capacity (mAh·g−1) | Average Charge Potential (V vs. Li+/Li) | Average Discharge Potential (V vs. Li+/Li) |
|---|---|---|---|---|---|
| Li2TiS3 | 5.0831 | 300 | 339 | 2.46 | 2.23 |
| Li2TiSeS2 | 5.1729 | 179 | 310 | 2.34 | 2.06 |
| Li2TiSe2S | 5.2459 | 149 | 379 | 2.24 | 1.98 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Celasun, Y.; Colin, J.-F.; Martinet, S.; Benayad, A.; Peralta, D. Lithium-Rich Rock Salt Type Sulfides-Selenides (Li2TiSexS3−x): High Energy Cathode Materials for Lithium-Ion Batteries. Materials 2022, 15, 3037. https://doi.org/10.3390/ma15093037
Celasun Y, Colin J-F, Martinet S, Benayad A, Peralta D. Lithium-Rich Rock Salt Type Sulfides-Selenides (Li2TiSexS3−x): High Energy Cathode Materials for Lithium-Ion Batteries. Materials. 2022; 15(9):3037. https://doi.org/10.3390/ma15093037
Chicago/Turabian StyleCelasun, Yagmur, Jean-François Colin, Sébastien Martinet, Anass Benayad, and David Peralta. 2022. "Lithium-Rich Rock Salt Type Sulfides-Selenides (Li2TiSexS3−x): High Energy Cathode Materials for Lithium-Ion Batteries" Materials 15, no. 9: 3037. https://doi.org/10.3390/ma15093037
APA StyleCelasun, Y., Colin, J.-F., Martinet, S., Benayad, A., & Peralta, D. (2022). Lithium-Rich Rock Salt Type Sulfides-Selenides (Li2TiSexS3−x): High Energy Cathode Materials for Lithium-Ion Batteries. Materials, 15(9), 3037. https://doi.org/10.3390/ma15093037