
Dissolution of β-C2S Cement Clinker: Part 1 Molecular Dynamics (MD) Approach for Different Crystal Facets

 ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Methods and Modeling Approach
Model Construction
3. Results and Discussion
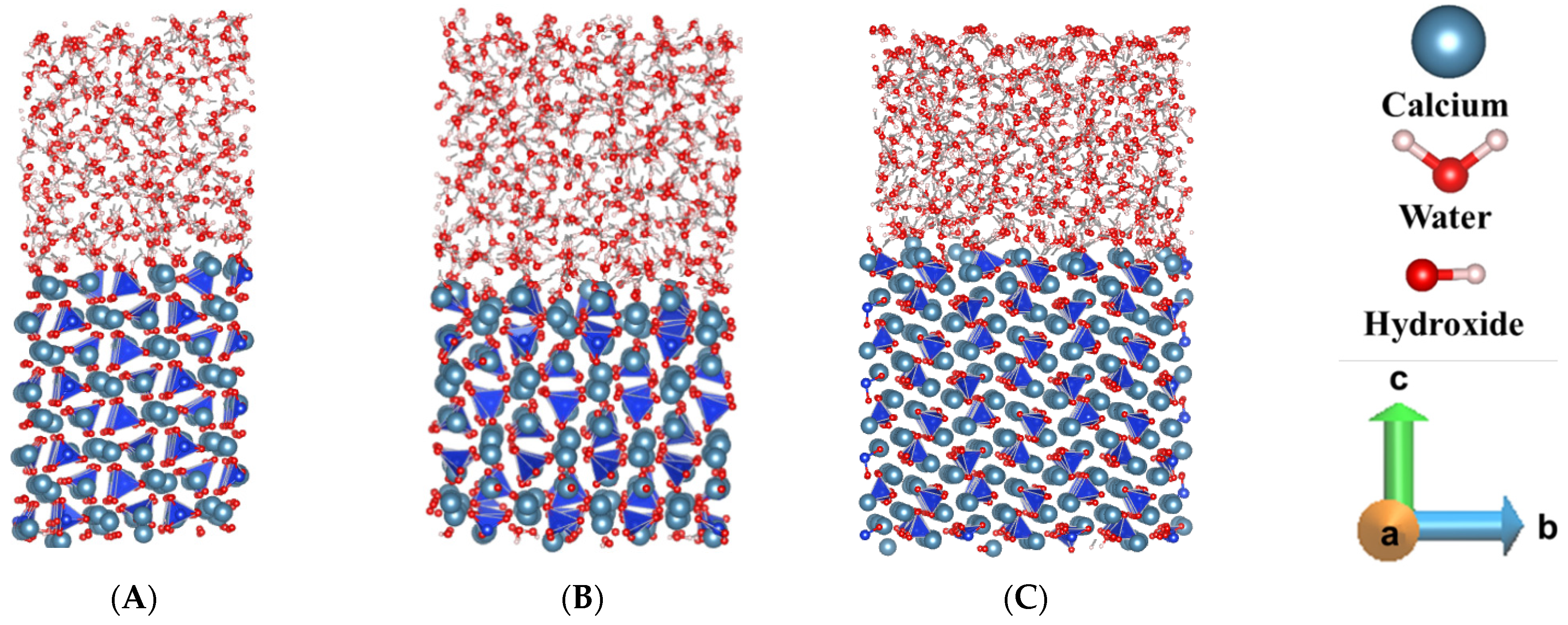
3.1. Pre-Hydration of β-C2S
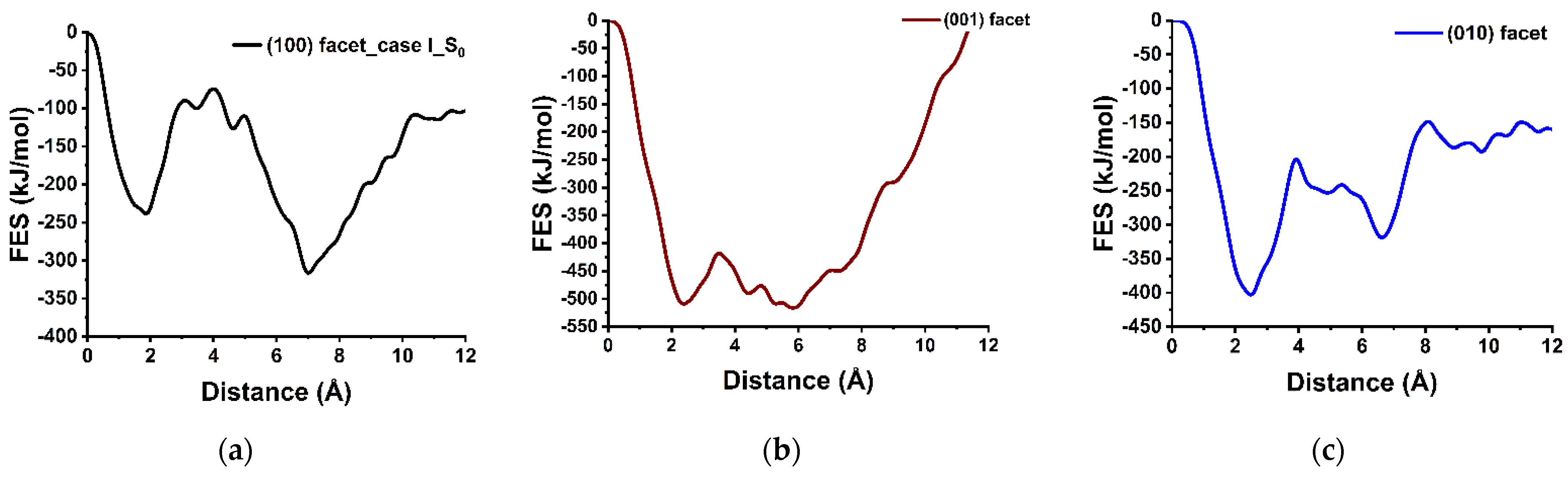
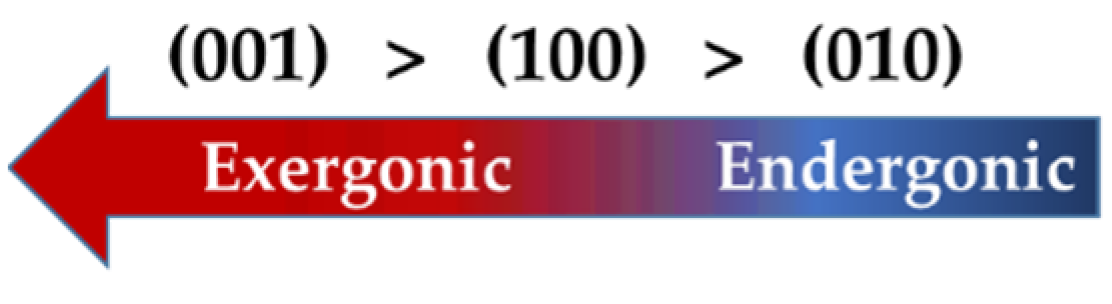
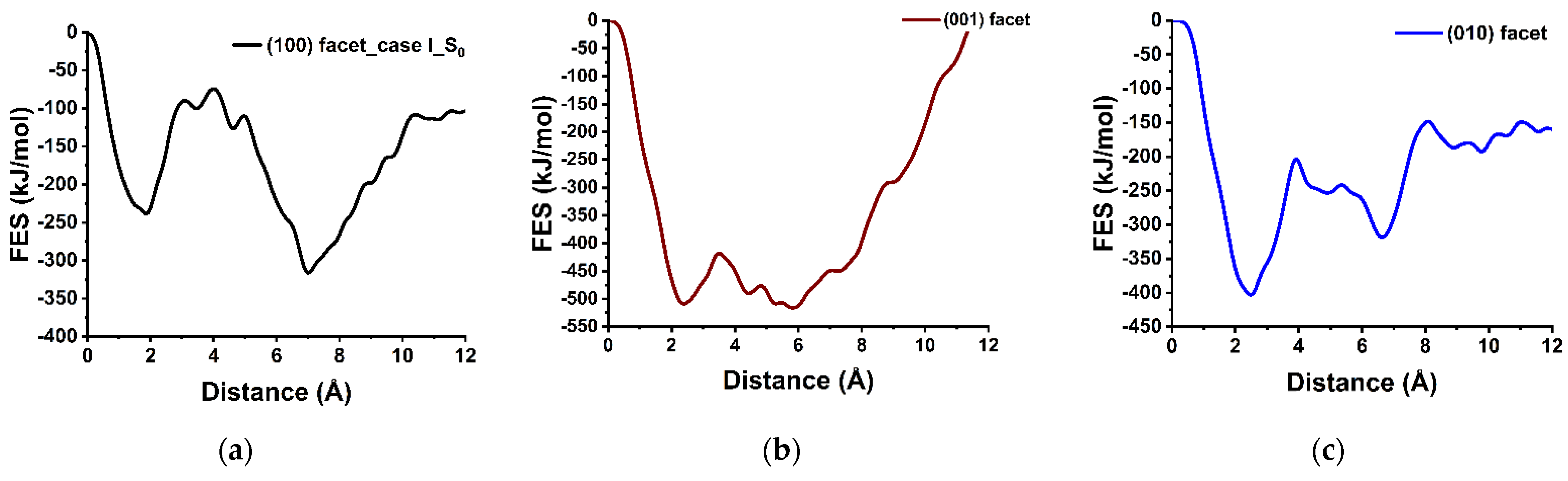
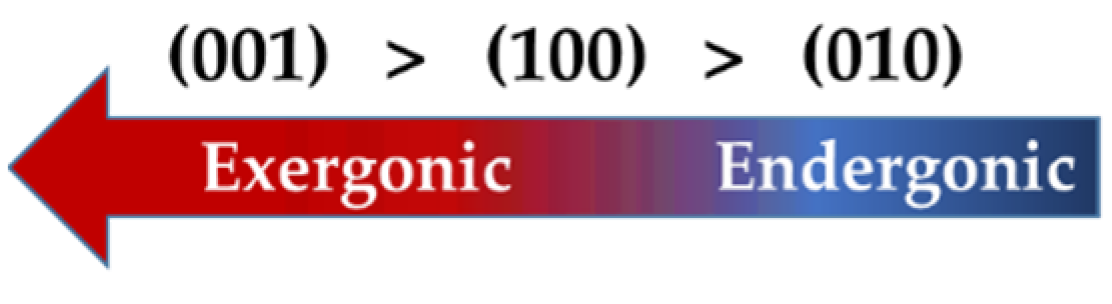
3.2. Dissolution of Calcium from (100), (001), and (010) Facets of β-C2S
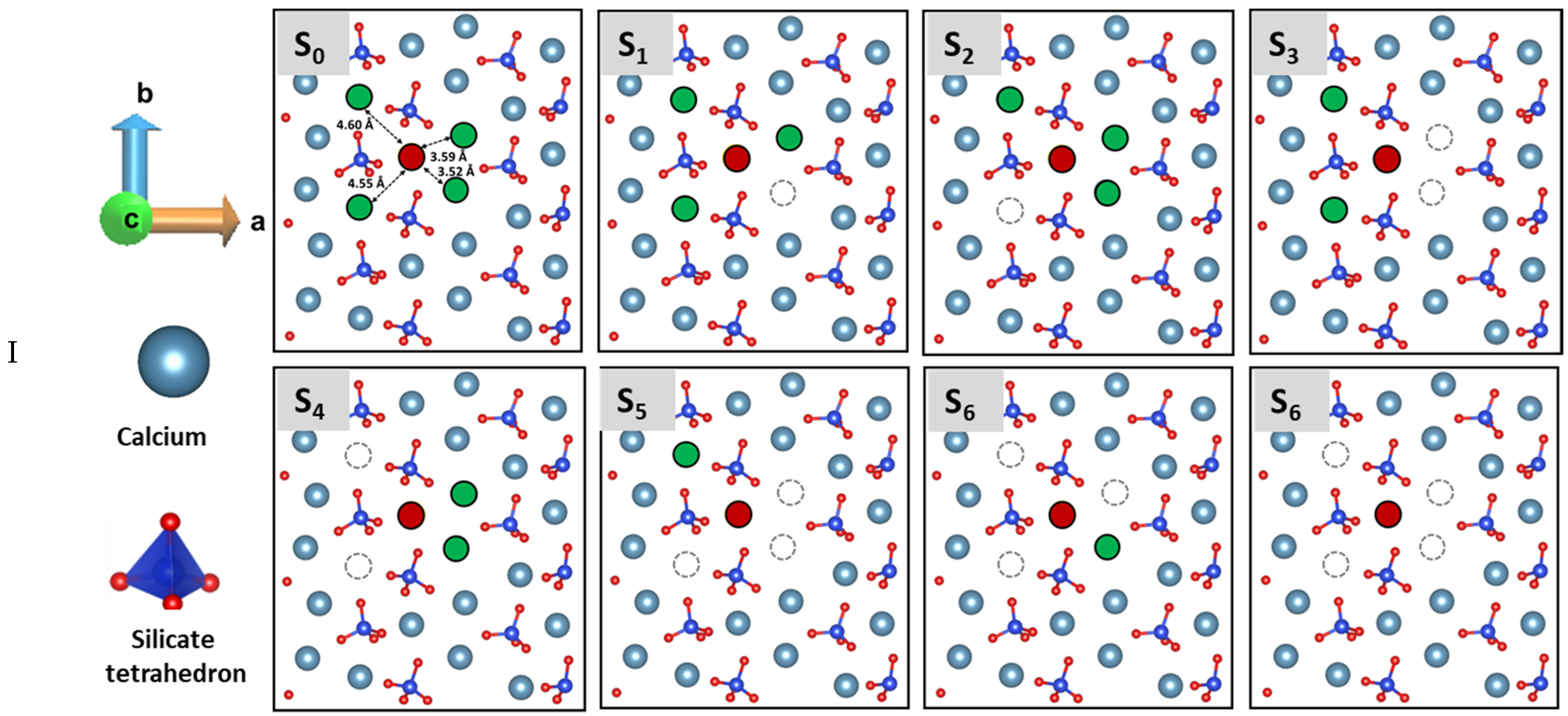
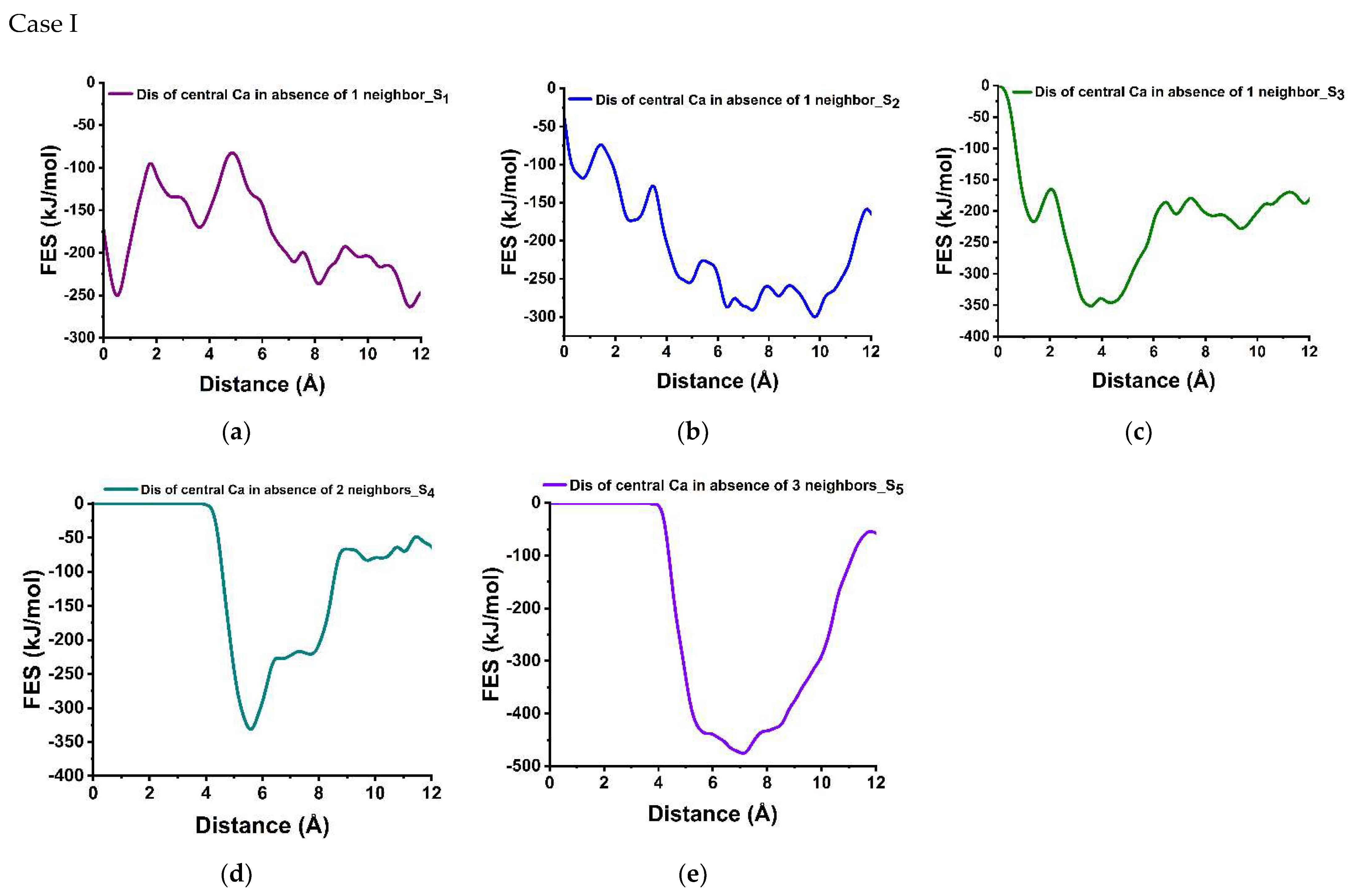
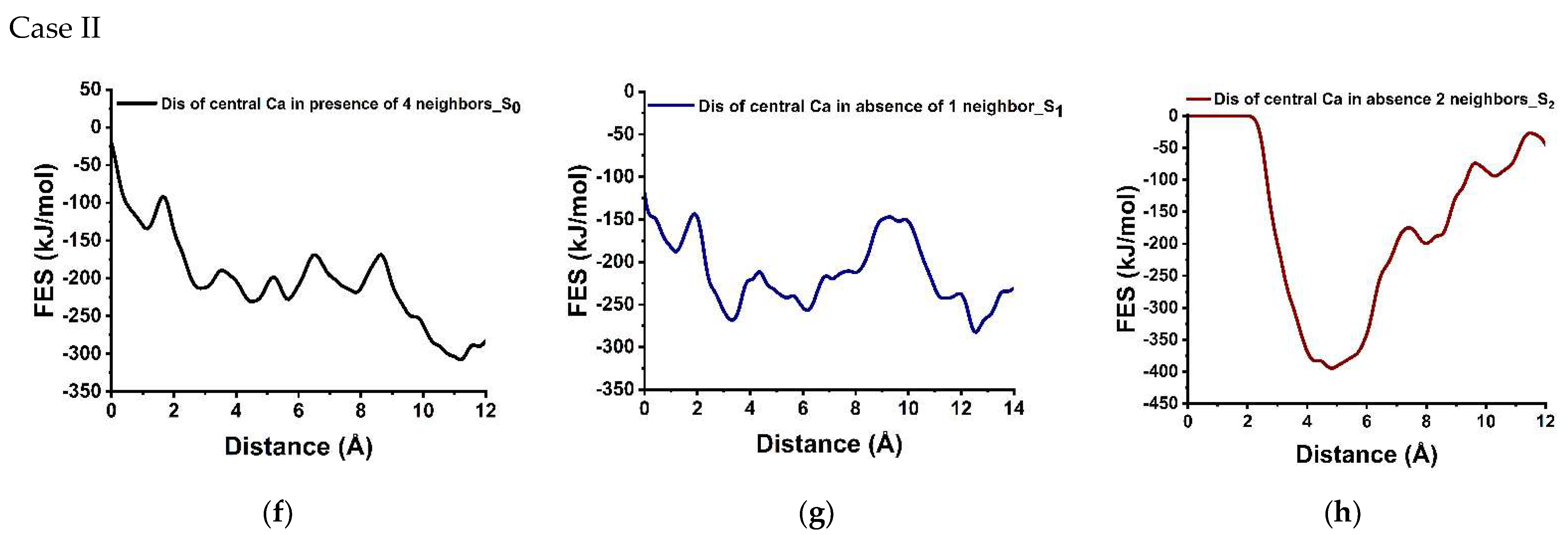
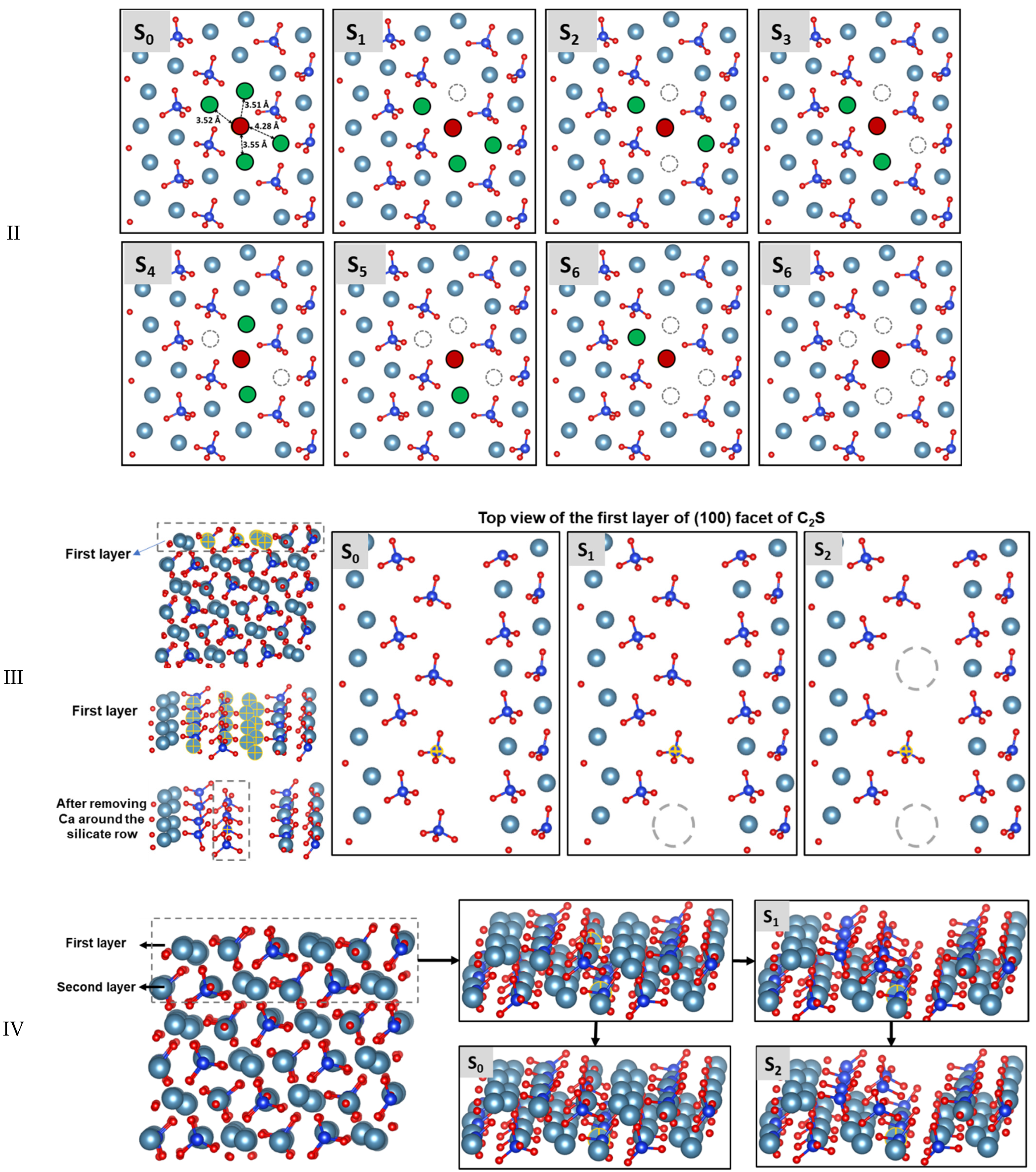
3.3. Dissolution Scenarios of Calcium from the First Layer of (100) Facet of β-C2S (Case I & Case II)
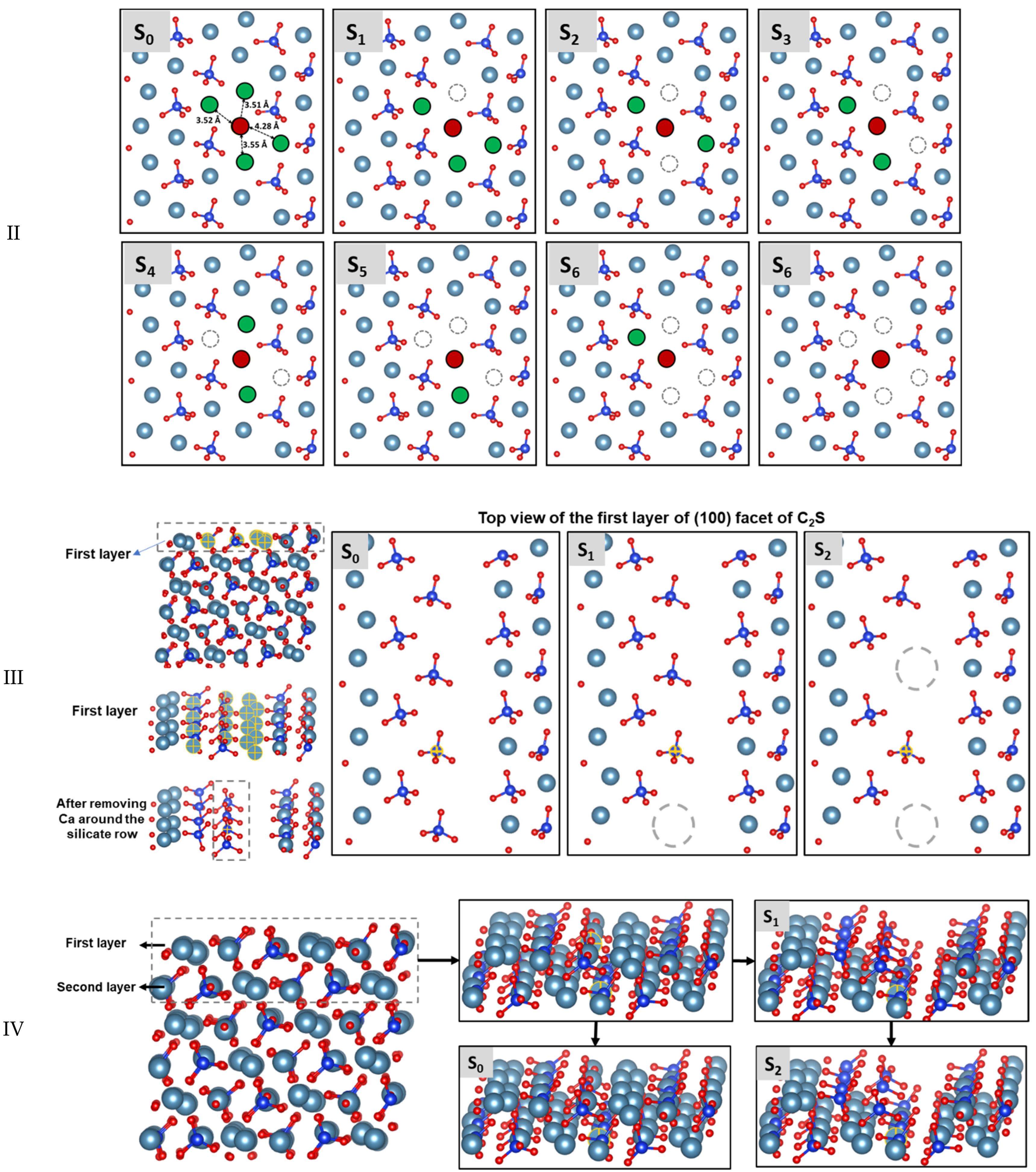
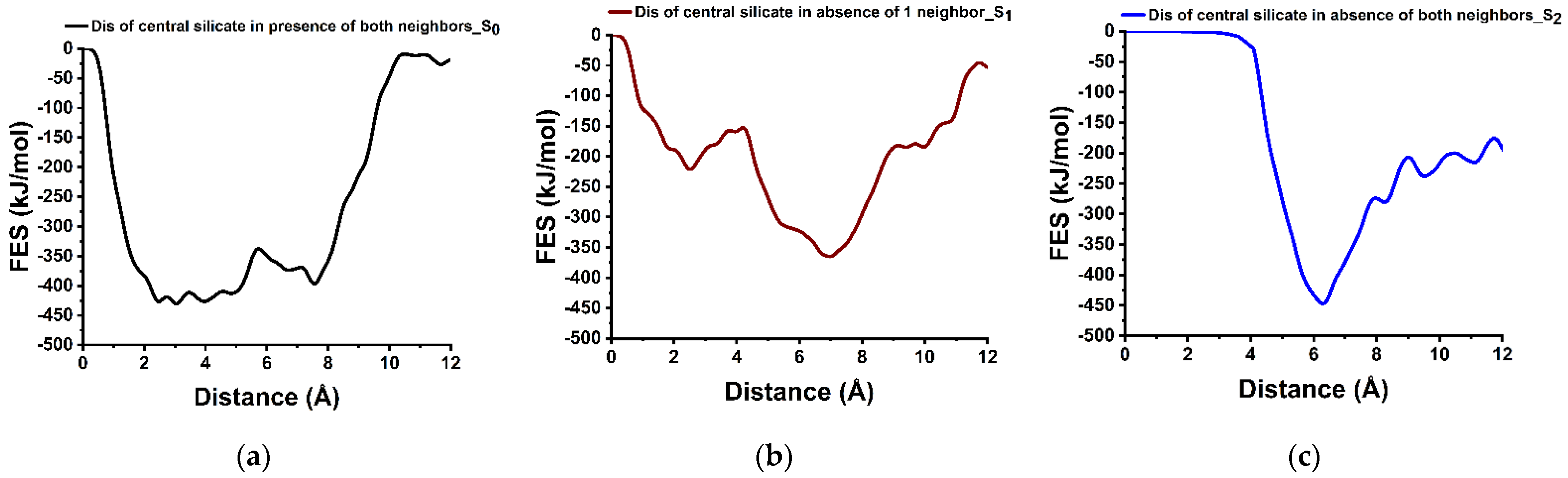
3.4. Dissolution Scenarios of Silicate from the First Layer of (100) Facet of β-C2S (Case III)
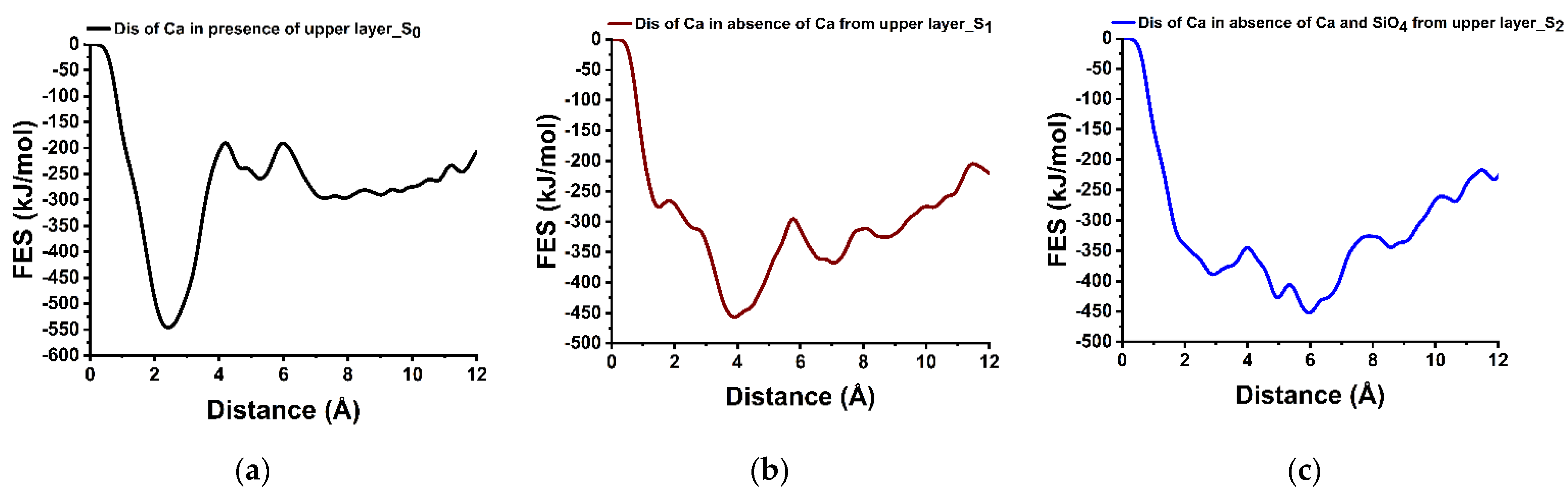
3.5. Dissolution Scenarios of Calcium from the Second Layer of (100) Facet of β-C2S (Case VI)
3.6. Upscaling the Dissolution Rate for (100) Facets of β-C2S
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MD | Molecular Dynamics |
| C2S | Belite, Dicalcium Silicate, 2CaO SiO2 |
| C3S | Alite, Tricalcium Silicate, 3CaO SiO2 |
| KMC | Kinetic Monte Carlo |
| ReaxFF | Reactive Force Field |
| LAMMPS | Large-scale Atomic/Molecular Massively Parallel Simulator |
| PLUMED | PLUgin for MolEcular Dynamics |
| MetaD | Metadynamics |
| CVs | Collective Variables |
| TS | Transition State |
| VNL | Virtual Nano Lab |
| FES | Free Energy Surfaces (kJ/mol) |
| HFTN | Hessian-free truncated Newton algorithm |
| NVT | Nose-Hoover thermostat |
| NPT | Nose-Hoover pressure barostat |
| ΔG* | Free Energy of Activation Barrier (kJ/mol) |
| ΔG | Free Energy Change (kJ/mol) |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Facet of β-C2S | Orthogonal Cell Dimension (Å3/10−30 m3) | No. of Atoms in the Simulation Cell |
|---|---|---|
| (100) | 23.51, 27.34, 48.99 | 1899 |
| (001) | 23.56, 27.38, 40.73 | 2141 |
| (010) | 27.75, 33.97, 47.74 | 3848 |
References
- Lea, F.M. Lea's Chemistry of Cement and Concrete, 4th ed.; Elsevier Butterworth-Heinemann: Oxford, UK, 2004; ISBN 0750662565. [Google Scholar]
- Taylor, H.F.W. Cement Chemistry; Thomas Telford: London, UK, 1997. [Google Scholar]
- Barcelo, L.; Kline, J.; Walenta, G.; Gartner, E. Cement and carbon emissions. Mater Struct 2014, 47, 1055–1065. [Google Scholar] [CrossRef]
- Kurdowski, W.; Duszak, S.; Trybalska, B. Belite produced by means of low-temperature synthesis. Cem. Concr. Res. 1997, 27, 51–62. [Google Scholar] [CrossRef]
- Scrivener, K.L.; John, V.M.; Gartner, E.M. Eco-efficient cements: Potential economically viable solutions for a low-CO2 cement-based materials industry. Cem. Concr. Res. 2018, 114, 2–26. [Google Scholar] [CrossRef]
- Sui, T.; Fan, L.; Wen, Z.; Wang, J. Properties of Belite-Rich Portland Cement and Concrete in China. JCEA 2015, 9, 384–392. [Google Scholar] [CrossRef]
- Brand, A.S.; Gorham, J.M.; Bullard, J.W.; Juilland, P.; Gallucci, E. Dissolution rate spectra of β-dicalcium silicate in water of varying activity. Cem. Concr. Res. 2019, 118, 69–83. [Google Scholar] [CrossRef]
- Juilland, P.; Gallucci, E. Morpho-topological investigation of the mechanisms and kinetic regimes of alite dissolution. Cem. Concr. Res. 2015, 76, 180–191. [Google Scholar] [CrossRef]
- Naber, C.; Bellmann, F.; Sowoidnich, T.; Goetz-Neunhoeffer, F.; Neubauer, J. Alite dissolution and C-S-H precipitation rates during hydration. Cem. Concr. Res. 2019, 115, 283–293. [Google Scholar] [CrossRef]
- Chi, L.; Zhang, A.; Qiu, Z.; Zhang, L.; Wang, Z.; Lu, S.; Zhao, D. Hydration activity, crystal structural, and electronic properties studies of Ba-doped dicalcium silicate. Nanotechnol. Rev. 2020, 9, 1027–1033. [Google Scholar] [CrossRef]
- Salah Uddin, K.M.; Middendorf, B. Reactivity of Different Crystalline Surfaces of C3S During Early Hydration by the Atomistic Approach. Materials 2019, 12, 1514. [Google Scholar] [CrossRef]
- Izadifar, M.; Abadi, R.; Jam, A.N.; Rabczuk, T. Investigation into the effect of doping of boron and nitrogen atoms in the mechanical properties of single-layer polycrystalline graphene. Comput. Mater. Sci. 2017, 138, 435–447. [Google Scholar] [CrossRef]
- Izadifar, M.; Natzeck, C.; Emmerich, K.; Weidler, P.G.; Gohari, S.; Burvill, C.; Thissen, P. Unexpected Chemical Activity of a Mineral Surface: The Role of Crystal Water in Tobermorite. J. Phys. Chem. C 2022. [Google Scholar] [CrossRef]
- Izadifar, M.; Dolado, J.S.; Thissen, P.; Ayuela, A. Interactions between Reduced Graphene Oxide with Monomers of (Calcium) Silicate Hydrates: A First-Principles Study. Nanomaterials 2021, 11, 2248. [Google Scholar] [CrossRef] [PubMed]
- Izadifar, M.; Königer, F.; Gerdes, A.; Wöll, C.; Thissen, P. Correlation between Composition and Mechanical Properties of Calcium Silicate Hydrates Identified by Infrared Spectroscopy and Density Functional Theory. J. Phys. Chem. C 2019, 123, 10868–10873. [Google Scholar] [CrossRef]
- Izadifar, M.; Thissen, P.; Steudel, A.; Kleeberg, R.; Kaufhold, S.; Kaltenbach, J.; Schuhmann, R.; Dehn, F.; Emmerich, K. Comprehensive examination of dehydroxylation of kaolinite, disordered kaolinite, and dickite: Experimental studies and density functional theory. Clays Clay Miner. 2020, 68, 319–333. [Google Scholar] [CrossRef]
- Chenoweth, K.; van Duin, A.C.T.; Goddard, W.A. ReaxFF reactive force field for molecular dynamics simulations of hydrocarbon oxidation. J. Phys. Chem. A 2008, 112, 1040–1053. [Google Scholar] [CrossRef]
- van Duin, A.C.T.; Strachan, A.; Stewman, S.; Zhang, Q.; Xu, X.; Goddard, W.A. ReaxFF SiO Reactive Force Field for Silicon and Silicon Oxide Systems. J. Phys. Chem. A 2003, 107, 3803–3811. [Google Scholar] [CrossRef]
- Manzano, H.; Pellenq, R.J.M.; Ulm, F.-J.; Buehler, M.J.; van Duin, A.C.T. Hydration of calcium oxide surface predicted by reactive force field molecular dynamics. Langmuir 2012, 28, 4187–4197. [Google Scholar] [CrossRef]
- Fogarty, J.C.; Aktulga, H.M.; Grama, A.Y.; van Duin, A.C.T.; Pandit, S.A. A reactive molecular dynamics simulation of the silica-water interface. J. Chem. Phys. 2010, 132, 174704. [Google Scholar] [CrossRef]
- Senftle, T.P.; Hong, S.; Islam, M.M.; Kylasa, S.B.; Zheng, Y.; Shin, Y.K.; Junkermeier, C.; Engel-Herbert, R.; Janik, M.J.; Aktulga, H.M.; et al. The ReaxFF reactive force-field: Development, applications and future directions. Npj Comput Mater 2016, 2, 9396. [Google Scholar] [CrossRef]
- Salah Uddin, K.M.; Izadifar, M.; Ukrainczyk, N.; Koenders, E.; Middendorf, B. Dissolution of Portlandite in Pure Water: Part 1 Molecular Dynamics (MD) Approach. Materials 2022, 15, 1404. [Google Scholar] [CrossRef]
- Salah Uddin, K.M. Elucidation of Chemical Reaction Pathways in Cementitious Materials; Kassel University Press: Kassel, Germany, 2020. [Google Scholar] [CrossRef]
- Izadifar, M.; Ukrainczyk, N.; Salah Uddin, K.M.; Middendorf, B.; Koenders, E. Dissolution of β-C2S Cement Clinker: Part 2 Atomistic Kinetic Monte Carlo (KMC) Upscaling Approach. 2022, under revision.
- Izadifar, M.; Ukrainczyk, N.; Salah Uddin, K.M.; Middendorf, B.; Koenders, E. Dissolution of Portlandite in Pure Water: Part 2 Atomistic Kinetic Monte Carlo (KMC) Approach. Materials 2022, 15, 1442. [Google Scholar] [CrossRef] [PubMed]
- Aktulga, H.M.; Fogarty, J.C.; Pandit, S.A.; Grama, A.Y. Parallel reactive molecular dynamics: Numerical methods and algorithmic techniques. Parallel Comput. 2012, 38, 245–259. [Google Scholar] [CrossRef]
- Barducci, A.; Bonomi, M.; Parrinello, M. Metadynamics. WIREs Comput. Mol. Sci. 2011, 1, 826–843. [Google Scholar] [CrossRef]
- Laio, A.; Gervasio, F.L. Metadynamics: A method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. J. Phys. Chem. A 2008, 71, 126601. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar] [CrossRef]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef]
- Kylasa, S.B.; Aktulga, H.M.; Grama, A.Y. PuReMD-GPU: A reactive molecular dynamics simulation package for GPUs. J. Comput. Phys. 2014, 272, 343–359. [Google Scholar] [CrossRef]
- Midgley, C.M. The crystal structure of β dicalcium silicate. Acta Cryst. 1952, 5, 307–312. [Google Scholar] [CrossRef]
- Schneider, J.; Hamaekers, J.; Chill, S.T.; Smidstrup, S.; Bulin, J.; Thesen, R.; Blom, A.; Stokbro, K. ATK-ForceField: A new generation molecular dynamics software package. Model. Simul. Mater. Sci. Eng. 2017, 25, 85007. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Tuckerman, M.E.; Alejandre, J.; López-Rendón, R.; Jochim, A.L.; Martyna, G.J. A Liouville-operator derived measure-preserving integrator for molecular dynamics simulations in the isothermal–isobaric ensemble. J. Phys. A Math. Gen. 2006, 39, 5629–5651. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Laidler, K.J.; King, M.C. The development of transition-state theory. J. Phys. Chem. A 1983, 87, 2657–2664. [Google Scholar] [CrossRef]
- Martin, P.; Gaitero, J.J.; Dolado, J.S.; Manzano, H. New Kinetic Monte Carlo Model to Study the Dissolution of Quartz. ACS Earth Space Chem. 2021, 5, 516–524. [Google Scholar] [CrossRef]
- Manzano, H.; Durgun, E.; López-Arbeloa, I.; Grossman, J.C. Insight on Tricalcium Silicate Hydration and Dissolution Mechanism from Molecular Simulations. ACS Appl. Mater. Interfaces 2015, 7, 14726–14733. [Google Scholar] [CrossRef] [PubMed]
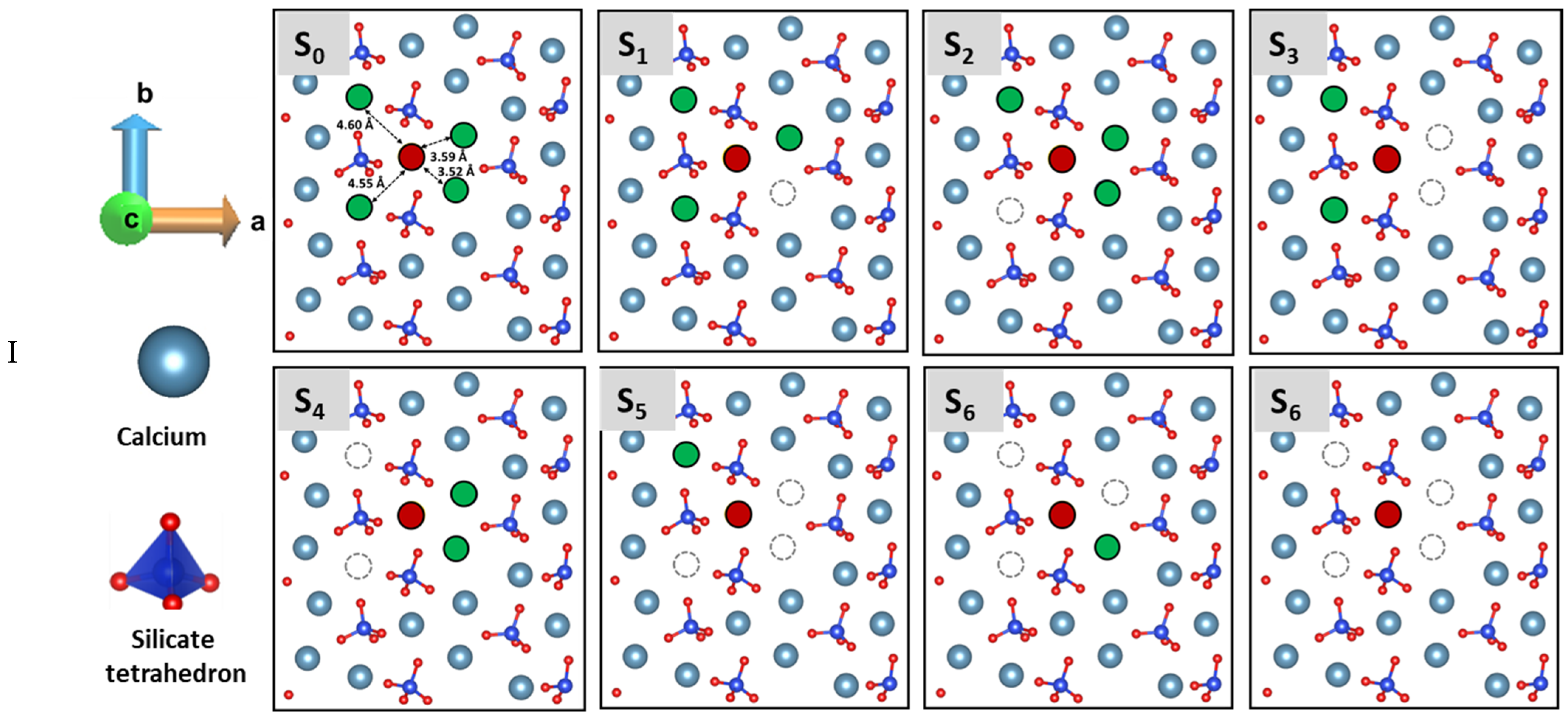

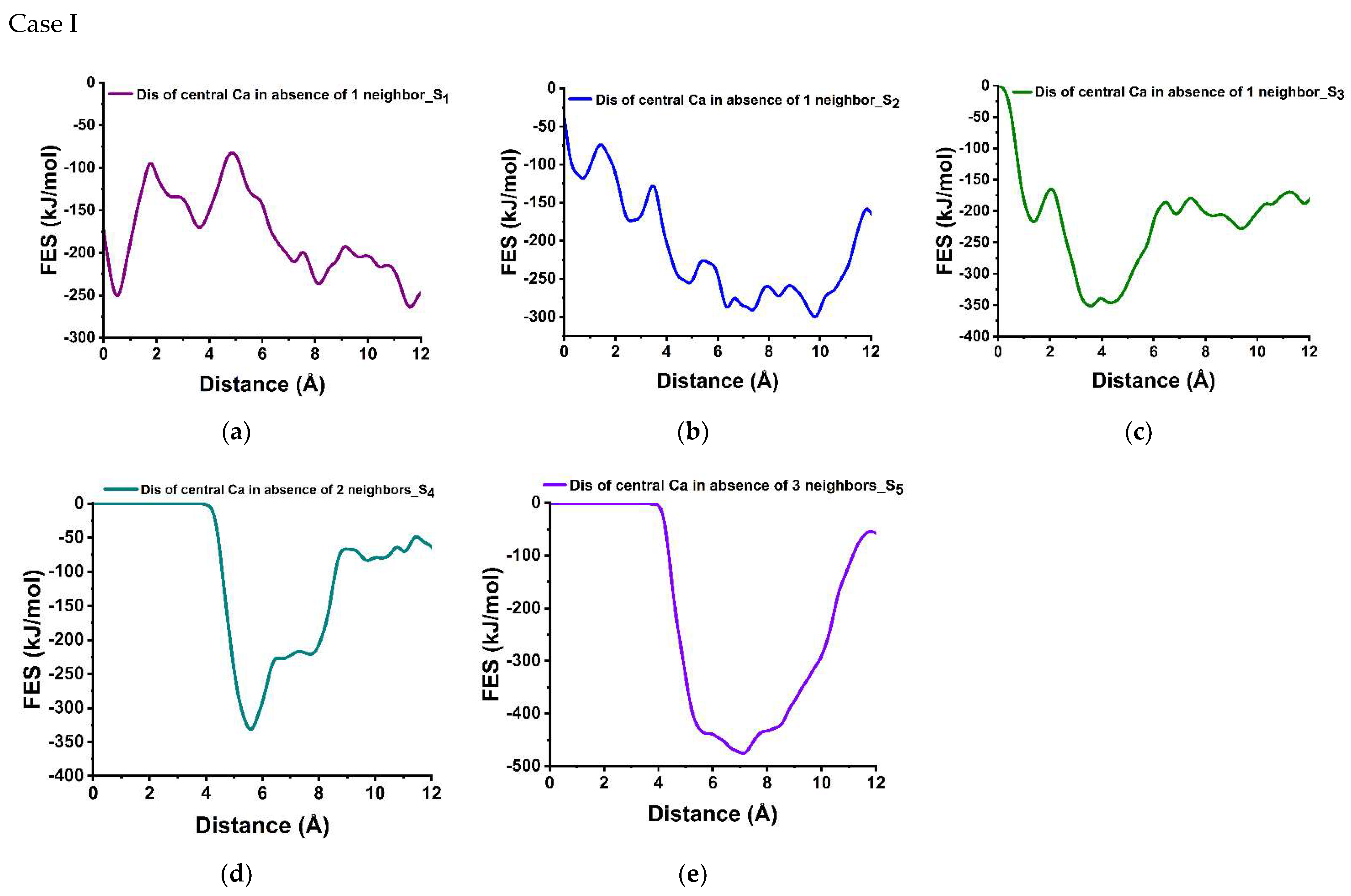
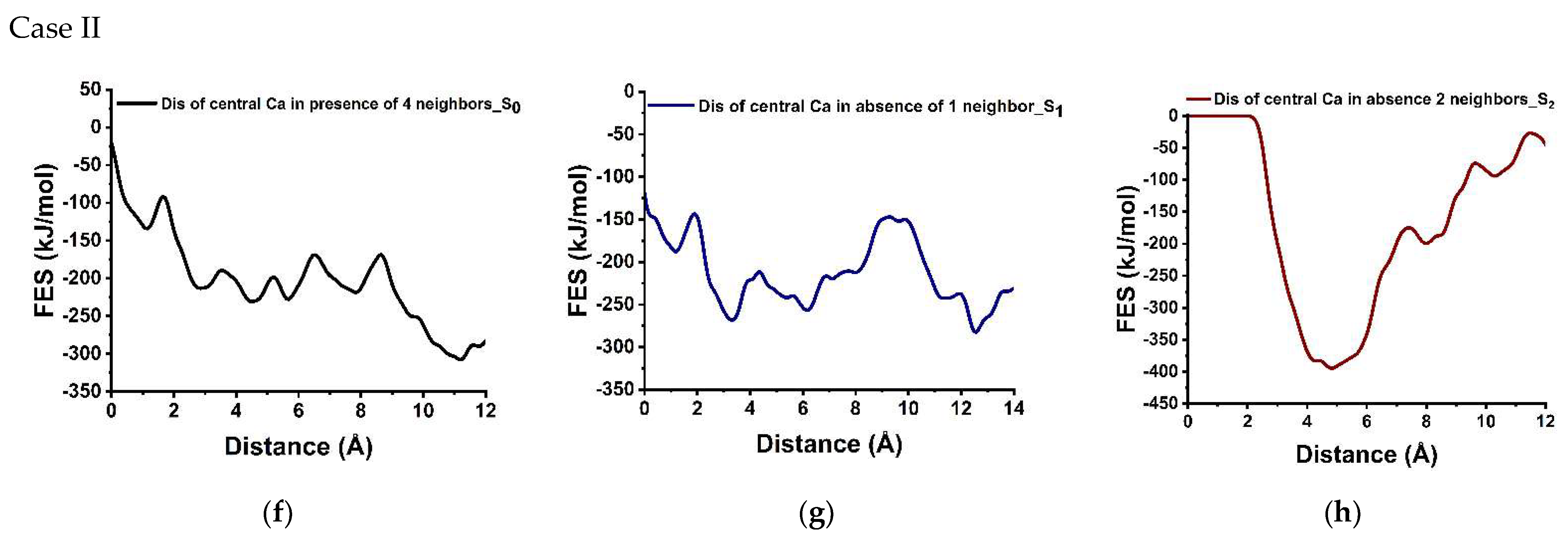

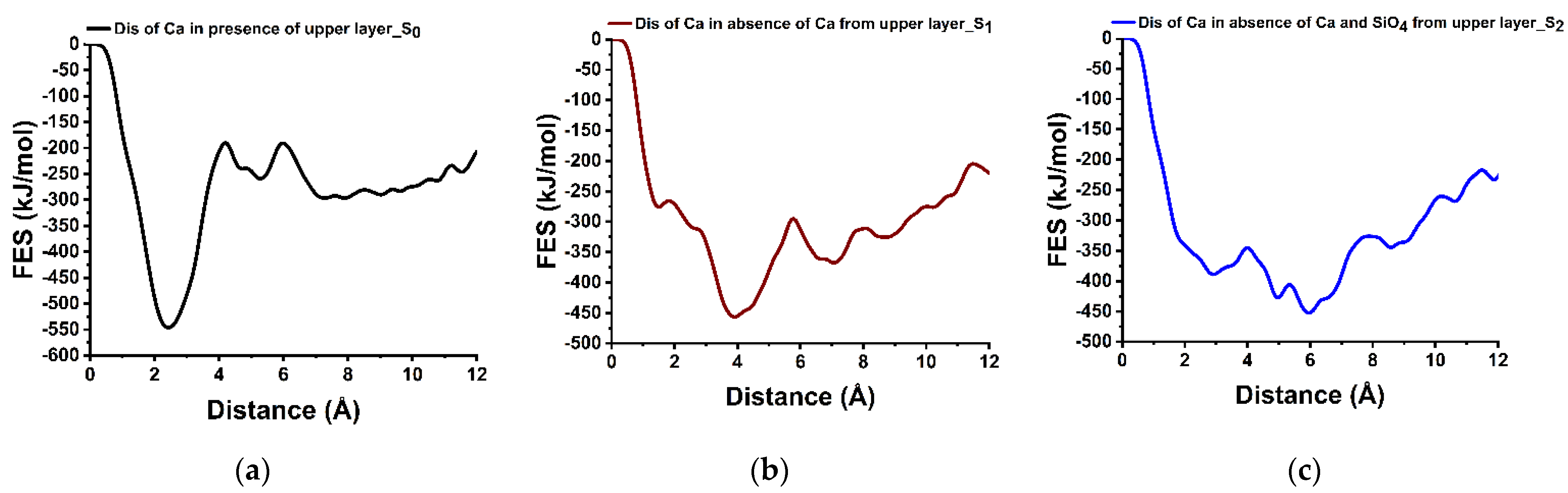
| Cases | Possible Scenarios of Dissolution of Central Ca or Silicate | Free Energy of Activation (ΔG*) kJ/mol | Free Energy Change (ΔG) kJ/mol | Thermodynamic Properties |
|---|---|---|---|---|
| I | S0: In presence of 4 neighbors | 164.30 | −78.00 | Exergonic |
| Ca dissolution | S1: In absence of 1 neighbor | 154.80 | −13.80 | Exergonic |
| S2: In absence of 1 neighbor | 44.00 | −182.00 | Exergonic | |
| S3: In absence of 2 neighbors | 52.00 | −130.16 | Exergonic | |
| S4 : In absence of 2 neighbors | 0.00 | * | ---- | |
| S5: In absence of 3 neighbors | 0.00 | * | ---- | |
| II | S0: In presence of 2 neighbors | 44.60 | −173.60 | Exergonic |
| Ca dissolution | S1: In absence of 1 neighbor | 42.40 | −94.60 | Exergonic |
| S2: In the absence of 2 neighbors | 0.00 | * | ---- | |
| III | S0: In presence of 2 neighbors | 89.00 | +31.00 | Endergonic |
| Silicate | S1: In absence of 1 neighbor | 67.70 | −144.60 | Exergonic |
| dissolution | S2: In the absence of 2 neighbors | 0.00 | * | ---- |
| IV | S0: In presence of upper layer Ca | 355.70 | +247.70 | Endergonic |
| Ca dissolution from 2nd layer | S1: In absence of upper layer Ca | 161.90 | +89.70 | Endergonic |
| S2: In absence of upper layer Ca and one silicate | 44.30 | −62.70 | Exergonic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salah Uddin, K.M.; Izadifar, M.; Ukrainczyk, N.; Koenders, E.; Middendorf, B. Dissolution of β-C2S Cement Clinker: Part 1 Molecular Dynamics (MD) Approach for Different Crystal Facets. Materials 2022, 15, 6388. https://doi.org/10.3390/ma15186388
Salah Uddin KM, Izadifar M, Ukrainczyk N, Koenders E, Middendorf B. Dissolution of β-C2S Cement Clinker: Part 1 Molecular Dynamics (MD) Approach for Different Crystal Facets. Materials. 2022; 15(18):6388. https://doi.org/10.3390/ma15186388
Chicago/Turabian StyleSalah Uddin, Khondakar Mohammad, Mohammadreza Izadifar, Neven Ukrainczyk, Eduardus Koenders, and Bernhard Middendorf. 2022. "Dissolution of β-C2S Cement Clinker: Part 1 Molecular Dynamics (MD) Approach for Different Crystal Facets" Materials 15, no. 18: 6388. https://doi.org/10.3390/ma15186388
APA StyleSalah Uddin, K. M., Izadifar, M., Ukrainczyk, N., Koenders, E., & Middendorf, B. (2022). Dissolution of β-C2S Cement Clinker: Part 1 Molecular Dynamics (MD) Approach for Different Crystal Facets. Materials, 15(18), 6388. https://doi.org/10.3390/ma15186388