
Dissolution of β-C2S Cement Clinker: Part 2 Atomistic Kinetic Monte Carlo (KMC) Upscaling Approach

 ,
,  ,
,  , and
, and 
Abstract
1. Introduction
2. Methods and Computational Models
2.1. Atomistic Model Preparation for Computation of Reaction Rates

2.2. Atomistic Kinetic Monte Carlo Upscaling Approach: Implementation in MATLAB
3. Results and Discussion
3.1. Dissolution Mechanism for β-C2S Crystal by Applying Periodic Boundary Conditions (PBCs) along the Y and Z Axes
3.2. Dissolution Mechanism for β-C2S Crystal by Applying Periodic Boundary Conditions (PBCs) along the Z Axis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Monteiro, P.J.M.; Miller, S.A.; Horvath, A. Towards sustainable concrete. Nat. Mater. 2017, 16, 698–699. [Google Scholar] [CrossRef]
- Mehta, P.K.; Monteiro, P.J.M. Concrete: Microstructure, Properties, and Materials; McGraw-Hill Education: Berkshire, UK, 2014. [Google Scholar]
- Worrell, E.; Price, L.; Martin, N.; Hendriks, C.; Meida, L.O. Carbon dioxide emissions from the global cement industry. Annu. Rev. Energy Environ. 2001, 26, 303–329. [Google Scholar] [CrossRef]
- Barcelo, L.; Kline, J.; Walenta, G.; Gartner, E. Cement and carbon emissions. Mater. Struct. 2014, 47, 1055–1065. [Google Scholar] [CrossRef]
- Cuesta, A.; Ayuela, A.; Aranda, M.A.G. Belite cements and their activation. Cem. Concr. Res. 2021, 140, 106319. [Google Scholar] [CrossRef]
- Salah Uddin, K.M.; Middendorf, B. Reactivity of Different Crystalline Surfaces of C3S During Early Hydration by the Atomistic Approach. Materials 2019, 12, 1514. [Google Scholar] [CrossRef]
- Salah Uddin, K.M. Elucidation of Chemical Reaction Pathways in Cementitious Materials; Kassel University Press: Kassel, Germany, 2020. [Google Scholar] [CrossRef]
- Odler, I. Strength of cement. Mater. Struct. 1991, 24, 143–157. [Google Scholar] [CrossRef]
- Kurdowski, W.; Duszak, S.; Trybalska, B. Belite produced by means of low-temperature synthesis. Cem. Concr. Res. 1997, 27, 51–62. [Google Scholar] [CrossRef]
- Chi, L.; Zhang, A.; Qiu, Z.; Zhang, L.; Wang, Z.; Lu, S.; Zhao, D. Hydration activity, crystal structural, and electronic properties studies of Ba-doped dicalcium silicate. Nanotechnol. Rev. 2020, 9, 1027–1033. [Google Scholar] [CrossRef]
- Scrivener, K.L.; John, V.M.; Gartner, E.M. Eco-efficient cements: Potential economically viable solutions for a low-CO2 cement-based materials industry. Cem. Concr. Res. 2018, 114, 2–26. [Google Scholar] [CrossRef]
- Izadifar, M.; Abadi, R.; Jam, A.N.; Rabczuk, T. Investigation into the effect of doping of boron and nitrogen atoms in the mechanical properties of single-layer polycrystalline graphene. Comput. Mater. Sci. 2017, 138, 435–447. [Google Scholar] [CrossRef]
- Izadifar, M.; Thissen, P.; Abadi, R.; Jam, A.N.; Gohari, S.; Burvill, C.; Rabczuk, T. Fracture toughness of various percentage of doping of boron atoms on the mechanical properties of polycrystalline graphene: A molecular dynamics study. Phys. E Low-Dimens. Syst. Nanostructures 2019, 114, 113614. [Google Scholar] [CrossRef]
- Salah Uddin, K.M.; Izadifar, M.; Ukrainczyk, N.; Koenders, E.; Middendorf, B. Dissolution of β-C2S Cement Clinker: Part 1 Molecular Dynamics (MD) Approach for Different Crystal Facets. Materials 2022, 15, 6388. [Google Scholar] [CrossRef] [PubMed]
- Izadifar, M.; Ukrainczyk, N.; Salah Uddin, K.; Middendorf, B.; Koenders, E. Dissolution of Portlandite in Pure Water: Part 2 Atomistic Kinetic Monte Carlo (KMC) Approach. Materials 2022, 15, 1442. [Google Scholar] [CrossRef]
- Salah Uddin, K.M.; Izadifar, M.; Ukrainczyk, N.; Koenders, E.; Middendorf, B. Dissolution of Portlandite in Pure Water: Part 1 Molecular Dynamics (MD) Approach. Materials 2022, 15, 1404. [Google Scholar] [CrossRef] [PubMed]
- Izadifar, M.; Natzeck, C.; Emmerich, K.; Weidler, P.G.; Gohari, S.; Burvill, C.; Thissen, P. Unexpected Chemical Activity of a Mineral Surface: The Role of Crystal Water in Tobermorite. J. Phys. Chem. C 2022, 126, 12405–12412. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Sheppard, D.; Xiao, P.; Chemelewski, W.; Johnson, D.D.; Henkelman, G. A generalized solid-state nudged elastic band method. J. Chem. Phys. 2012, 136, 74103. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Izadifar, M.; Thissen, P.; Steudel, A.; Kleeberg, R.; Kaufhold, S.; Kaltenbach, J.; Schuhmann, R.; Dehn, F.; Emmerich, K. Comprehensive examination of dehydroxylation of kaolinite, disordered kaolinite, and dickite: Experimental studies and density functional theory. Clays Clay Miner. 2020, 68, 319–333. [Google Scholar] [CrossRef]
- Izadifar, M.; Dolado, J.S.; Thissen, P.; Ayuela, A. Interactions between Reduced Graphene Oxide with Monomers of (Calcium) Silicate Hydrates: A First-Principles Study. Nanomaterials 2021, 11, 2248. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Izadifar, M.; Königer, F.; Gerdes, A.; Wöll, C.; Thissen, P. Correlation between Composition and Mechanical Properties of Calcium Silicate Hydrates Identified by Infrared Spectroscopy and Density Functional Theory. J. Phys. Chem. C 2019, 123, 10868–10873. [Google Scholar] [CrossRef]
- Martin, P.; Manzano, H.; Dolado, J.S. Mechanisms and dynamics of mineral dissolution: A new kinetic Monte Carlo model. Adv. Theory Simul. 2019, 2, 1900114. [Google Scholar] [CrossRef]
- Martin, P.; Gaitero, J.J.; Dolado, J.S.; Manzano, H. New Kinetic Monte Carlo Model to Study the Dissolution of Quartz. ACS Earth Space Chem. 2021, 5, 516–524. [Google Scholar] [CrossRef]
- Lasaga, A.C. Kinetic Theory in the Earth Sciences; Princeton University Press: Princeton, NJ, USA, 2014. [Google Scholar]





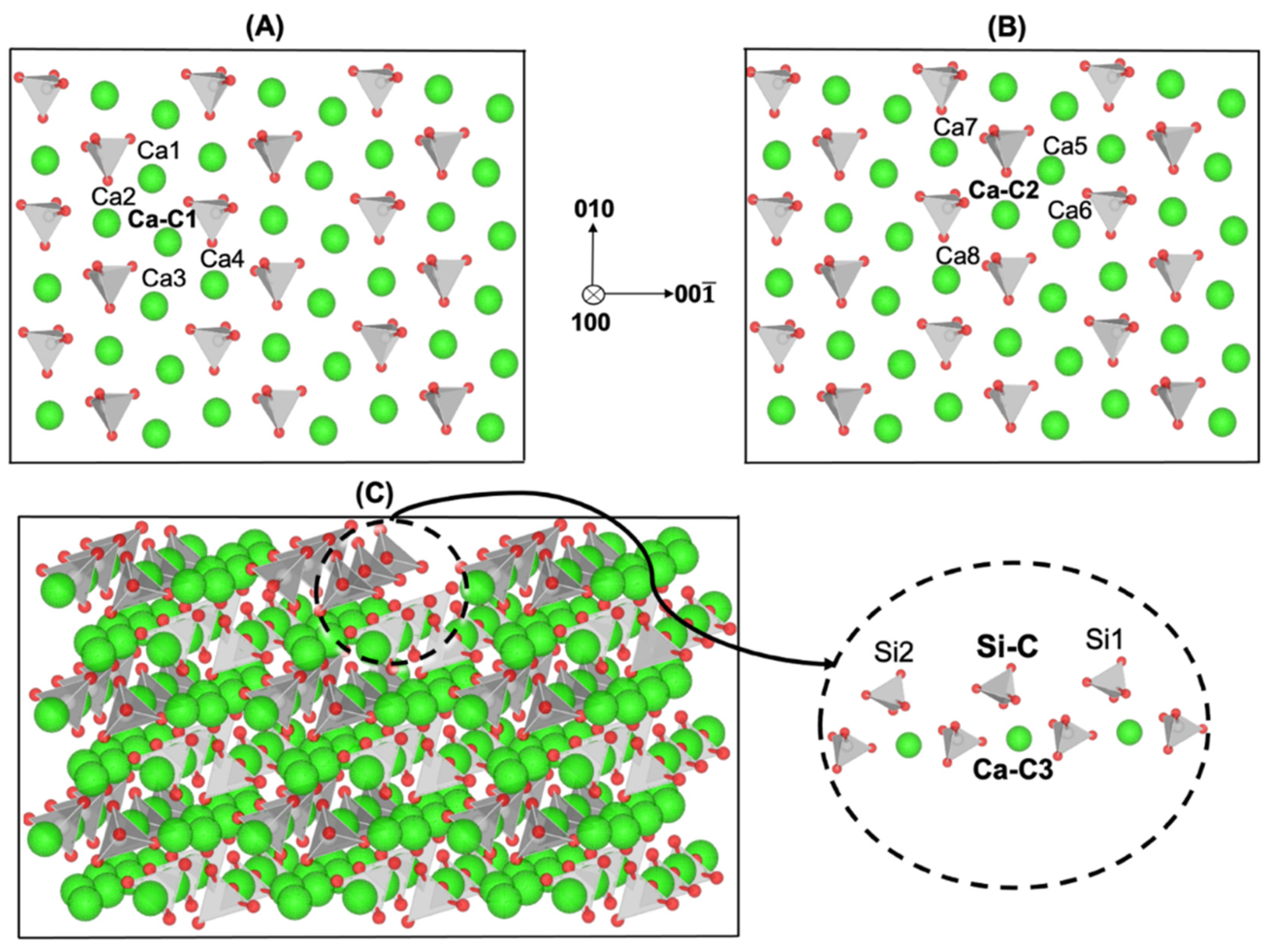{kind=link}
{kind=link}
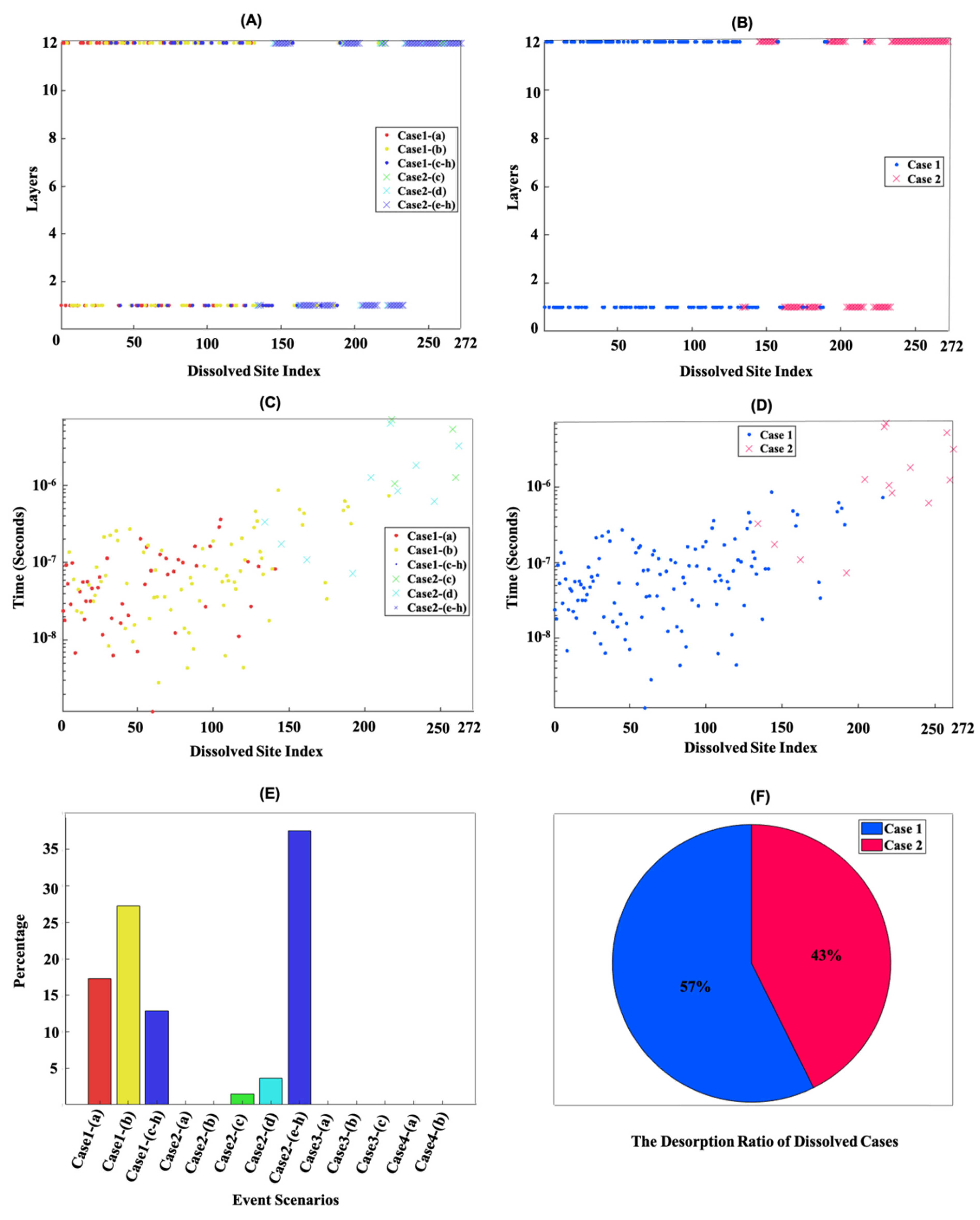{kind=link}
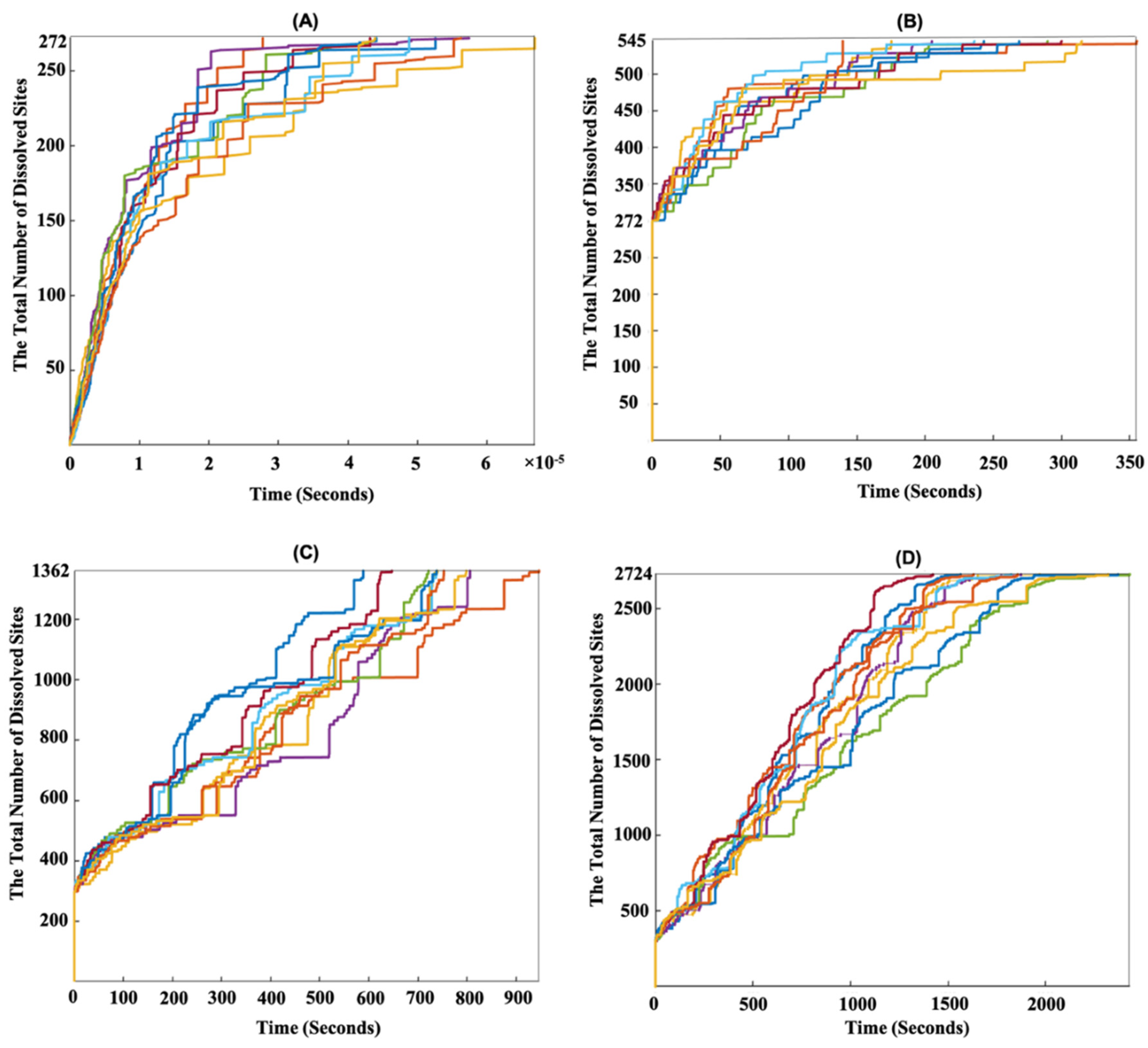{kind=link}
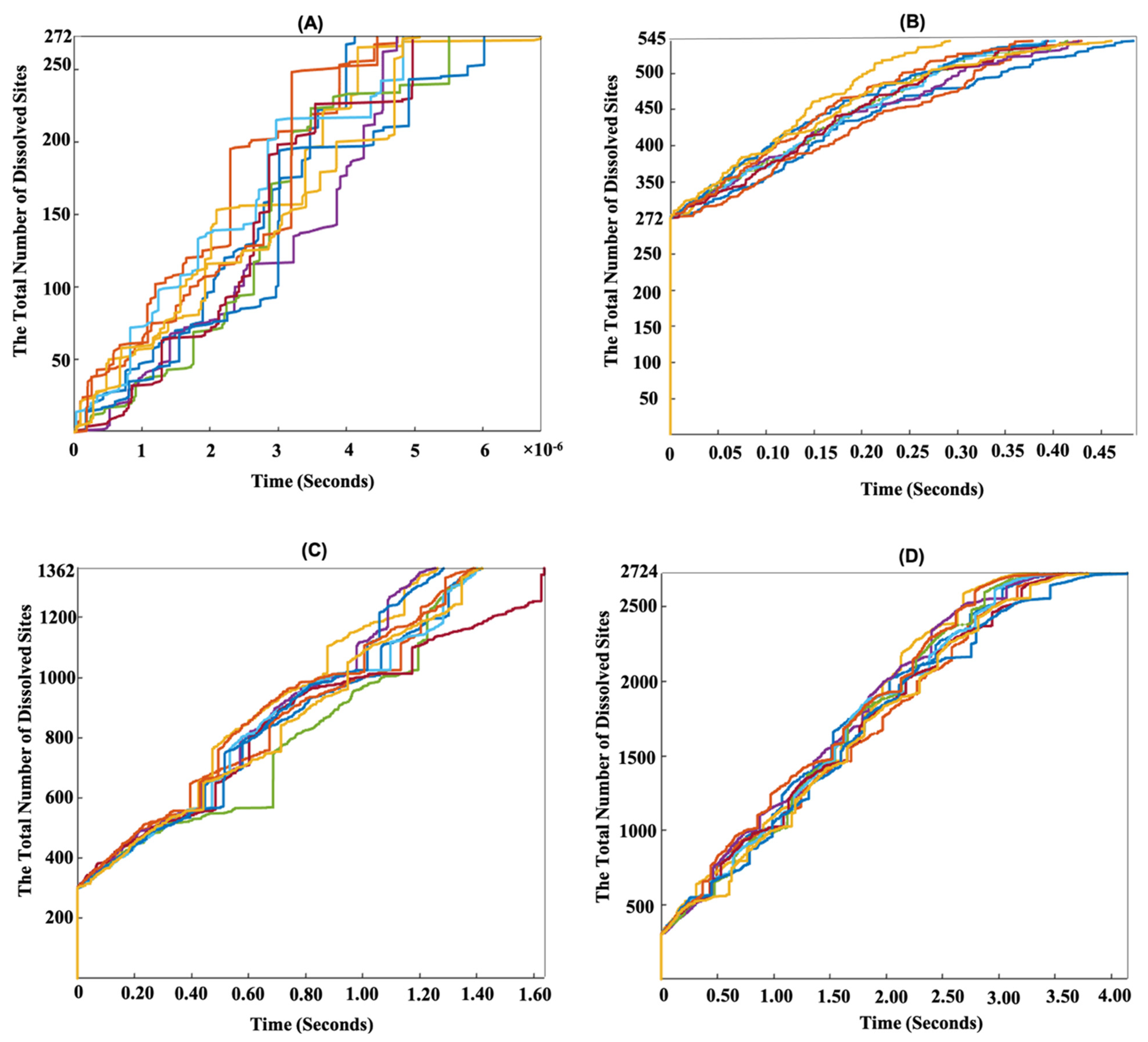{kind=link}
| Figure 1A | (a) | (b) | (c) | (d) | (e) | (f) | (g) | (h) |
|---|---|---|---|---|---|---|---|---|
| ΔG* (kJ/mol) [14] | 44.61 | 42.20 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| rD (s−1) | 1.0 × 105 | 2.5 × 105 |
| Figure 1B | (a) | (b) | (c) | (d) | (e) | (f) | (g) | (h) |
|---|---|---|---|---|---|---|---|---|
| ΔG* (kJ/mol) [14] | 164.30 | 154.80 | 44.06 | 52.06 | 0.00 | 0.00 | 0.00 | 0.00 |
| rD (s−1) | 9.97 × 10−17 | 4.61 × 10−15 |
| Figure 1C | (a) | (b) | (c) |
|---|---|---|---|
| ΔG* (kJ/mol) [14] | 89.00 | 67.70 | 0.00 |
| rD (s−1) | 1.50 × 10−3 | 8.47 |
| Figure 1C | (a) | (b) |
|---|---|---|
| ΔG* (kJ/mol) [14] | 161.90 | 44.30 |
| rD (s−1) | 2.62 × 10−16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izadifar, M.; Ukrainczyk, N.; Salah Uddin, K.M.; Middendorf, B.; Koenders, E. Dissolution of β-C2S Cement Clinker: Part 2 Atomistic Kinetic Monte Carlo (KMC) Upscaling Approach. Materials 2022, 15, 6716. https://doi.org/10.3390/ma15196716
Izadifar M, Ukrainczyk N, Salah Uddin KM, Middendorf B, Koenders E. Dissolution of β-C2S Cement Clinker: Part 2 Atomistic Kinetic Monte Carlo (KMC) Upscaling Approach. Materials. 2022; 15(19):6716. https://doi.org/10.3390/ma15196716
Chicago/Turabian StyleIzadifar, Mohammadreza, Neven Ukrainczyk, Khondakar Mohammad Salah Uddin, Bernhard Middendorf, and Eduardus Koenders. 2022. "Dissolution of β-C2S Cement Clinker: Part 2 Atomistic Kinetic Monte Carlo (KMC) Upscaling Approach" Materials 15, no. 19: 6716. https://doi.org/10.3390/ma15196716
APA StyleIzadifar, M., Ukrainczyk, N., Salah Uddin, K. M., Middendorf, B., & Koenders, E. (2022). Dissolution of β-C2S Cement Clinker: Part 2 Atomistic Kinetic Monte Carlo (KMC) Upscaling Approach. Materials, 15(19), 6716. https://doi.org/10.3390/ma15196716