
Structural Analysis of Lignin-Based Furan Resin


Abstract
:1. Introduction
2. Materials and Methods
2.1. General
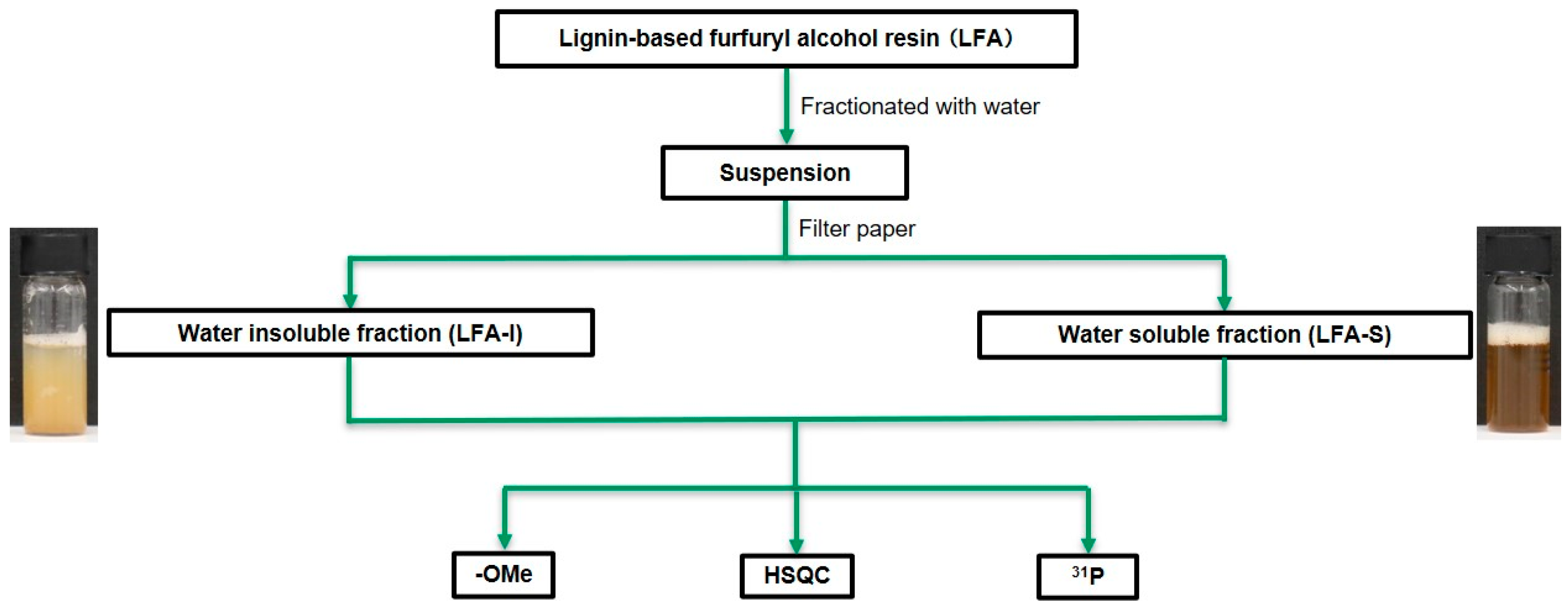
2.2. Fractionation of LFA Resins
2.3. Determination of Methoxyl Groups
2.4. NMR Analysis on Fractionated Bioresin
2.4.1. HSQC NMR
2.4.2. 31P NMR
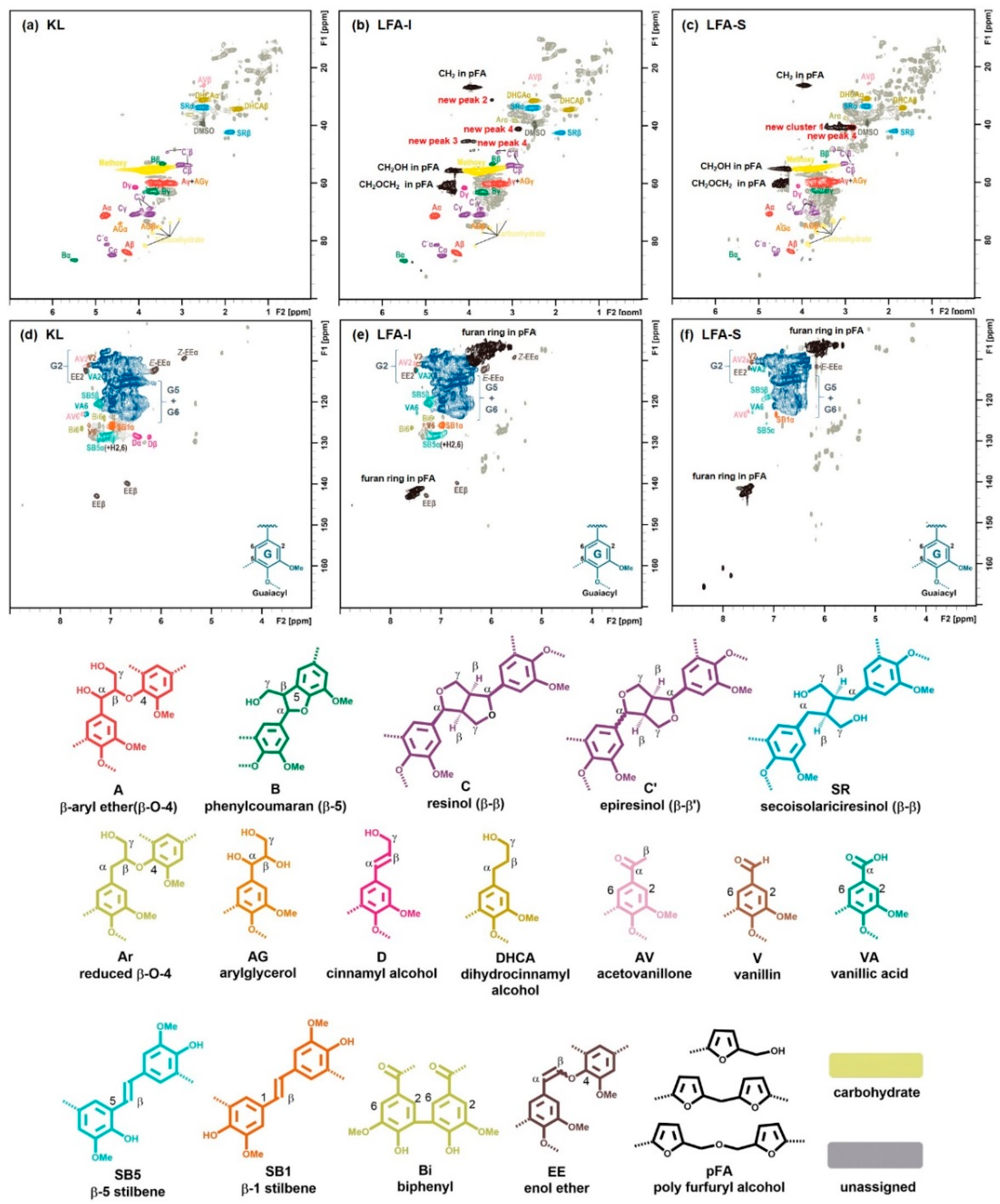
3. Results and Discussion
3.1. General
3.2. Material Balance
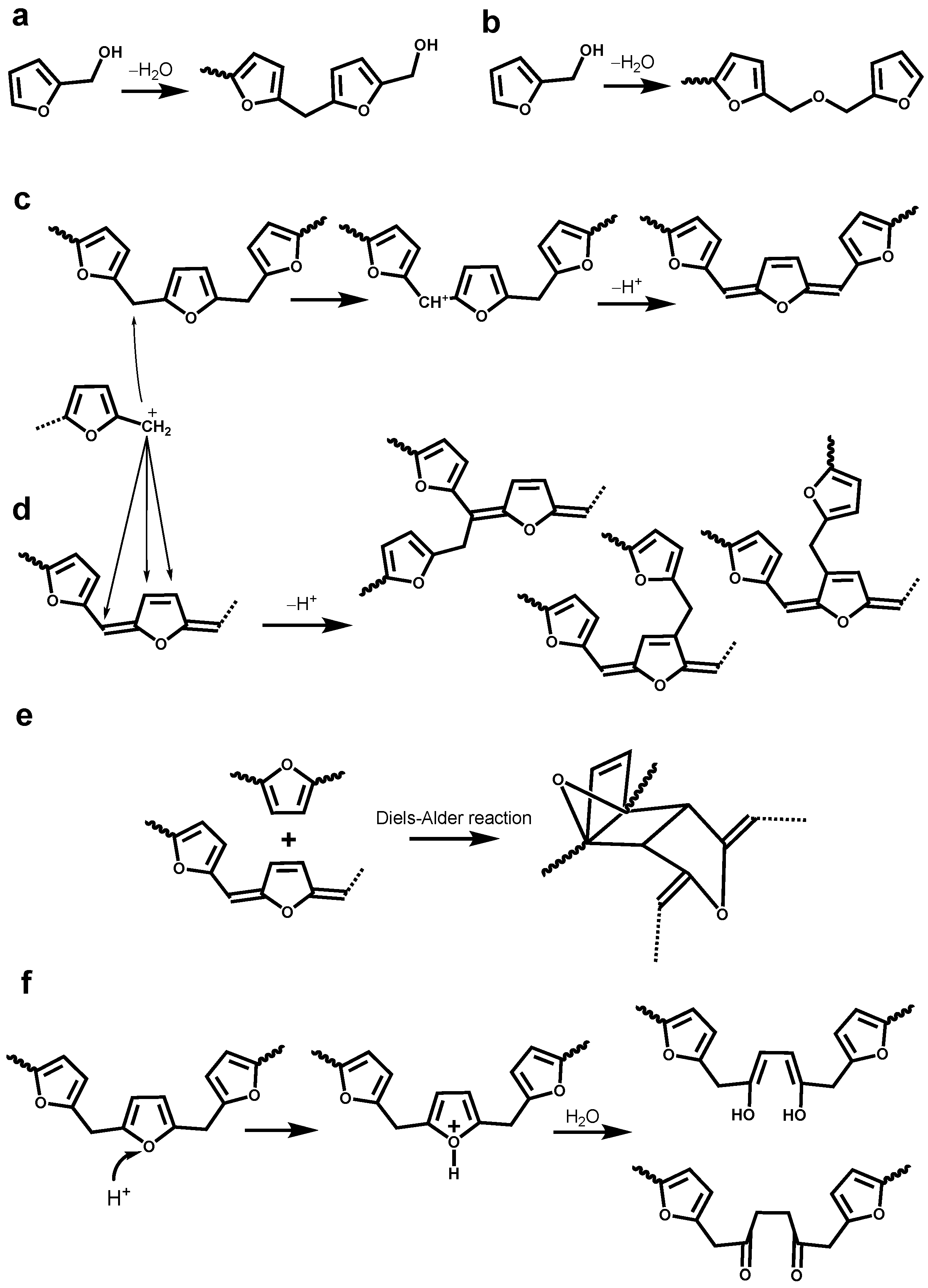
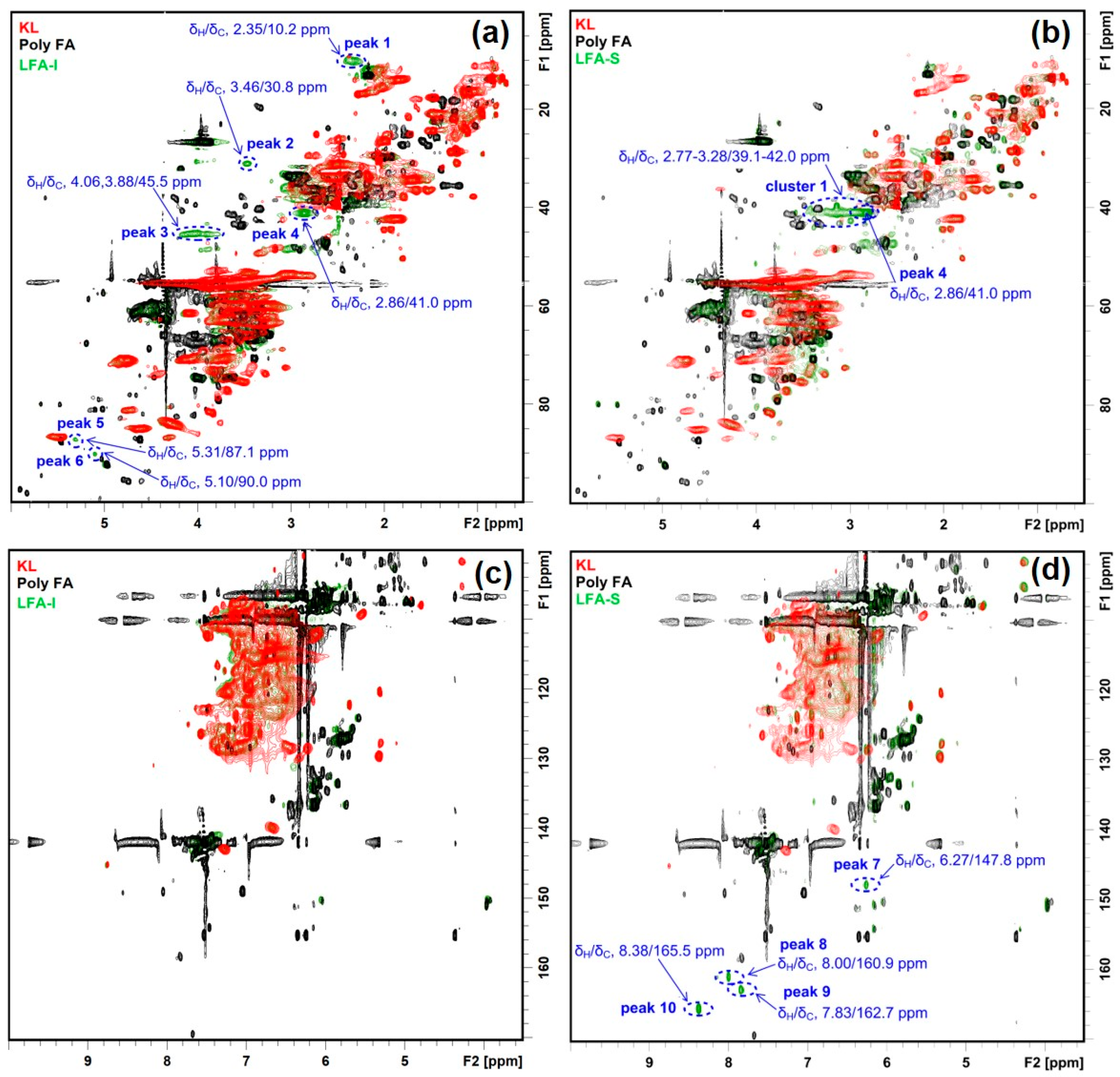
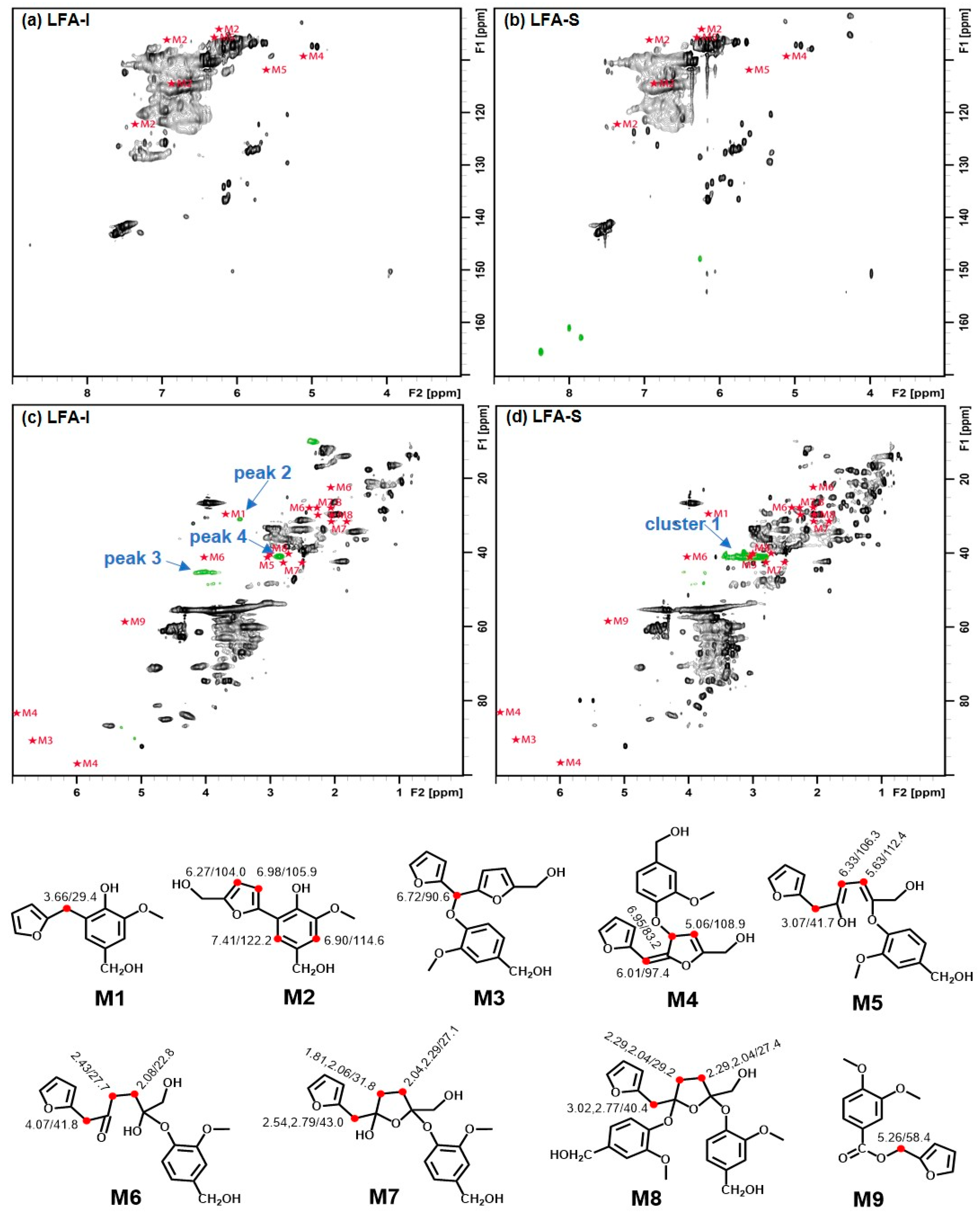
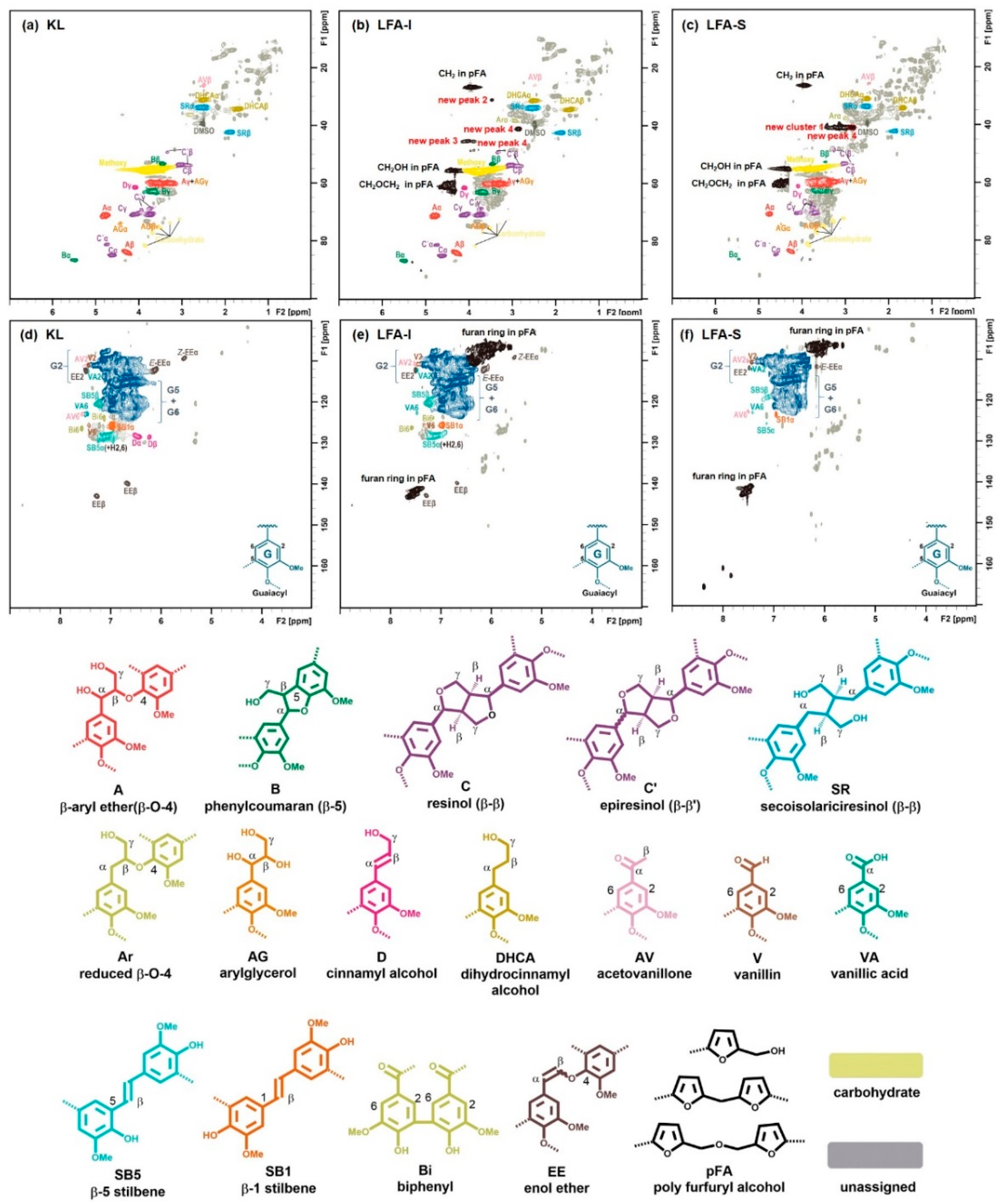
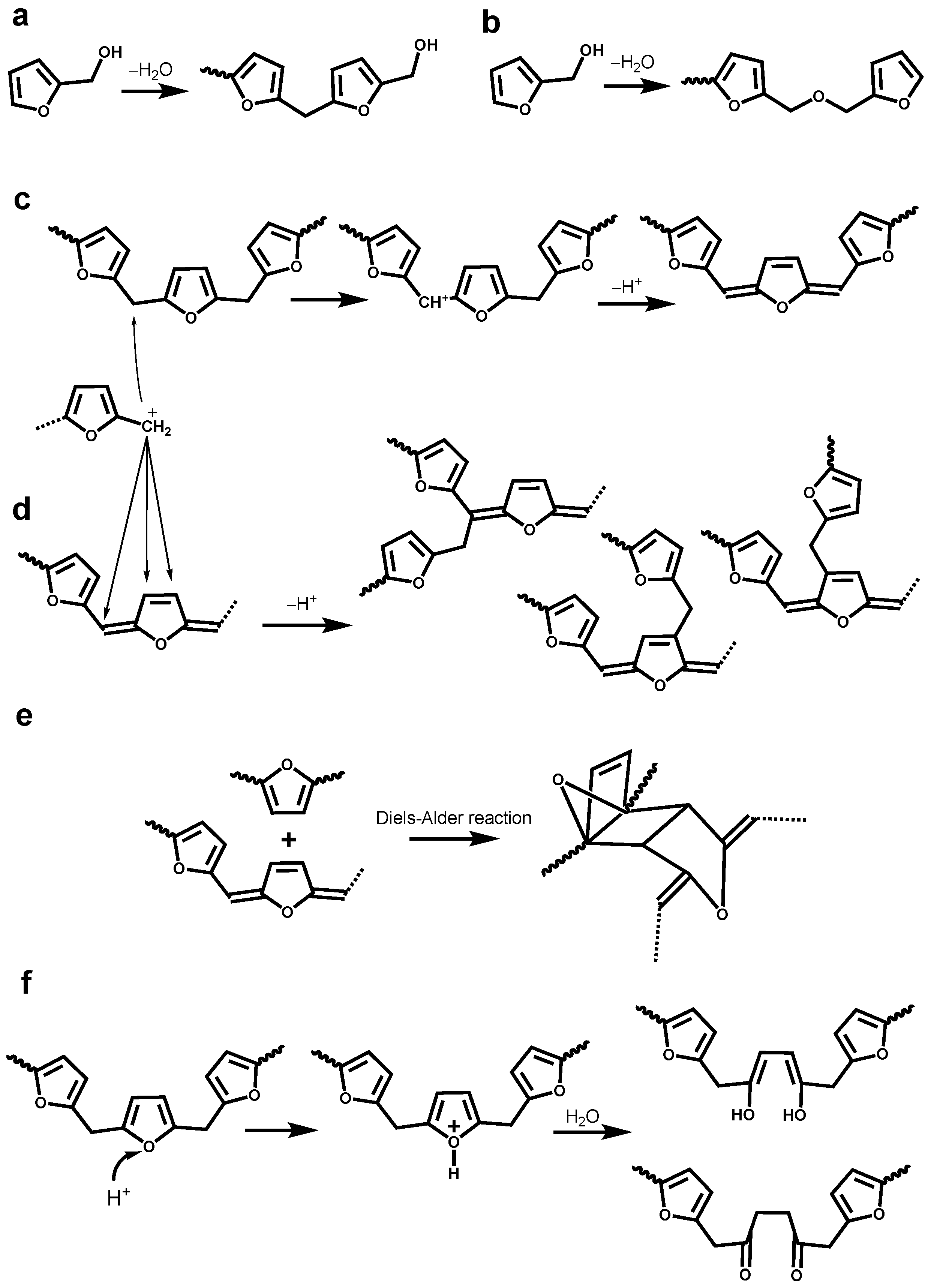
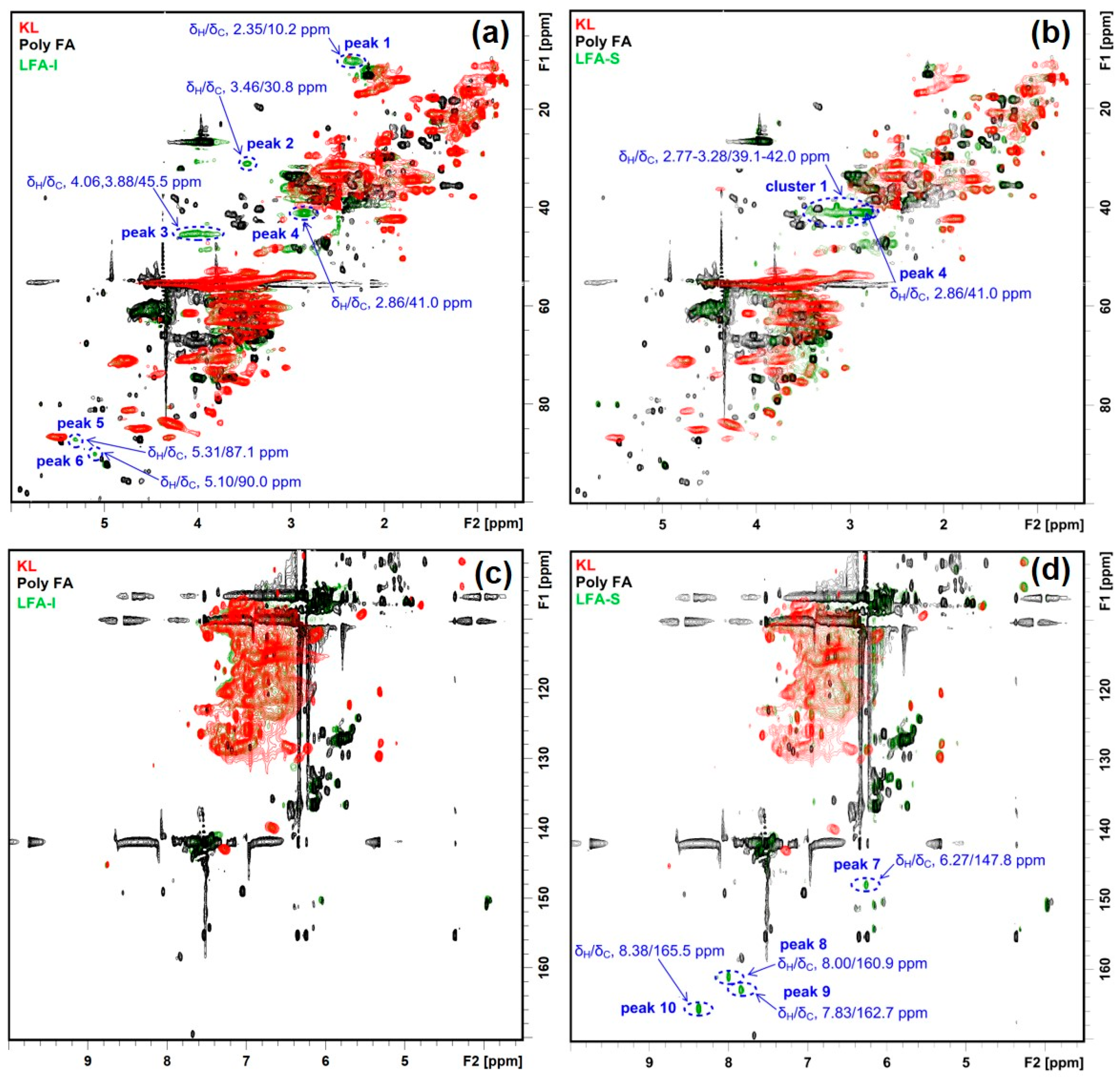
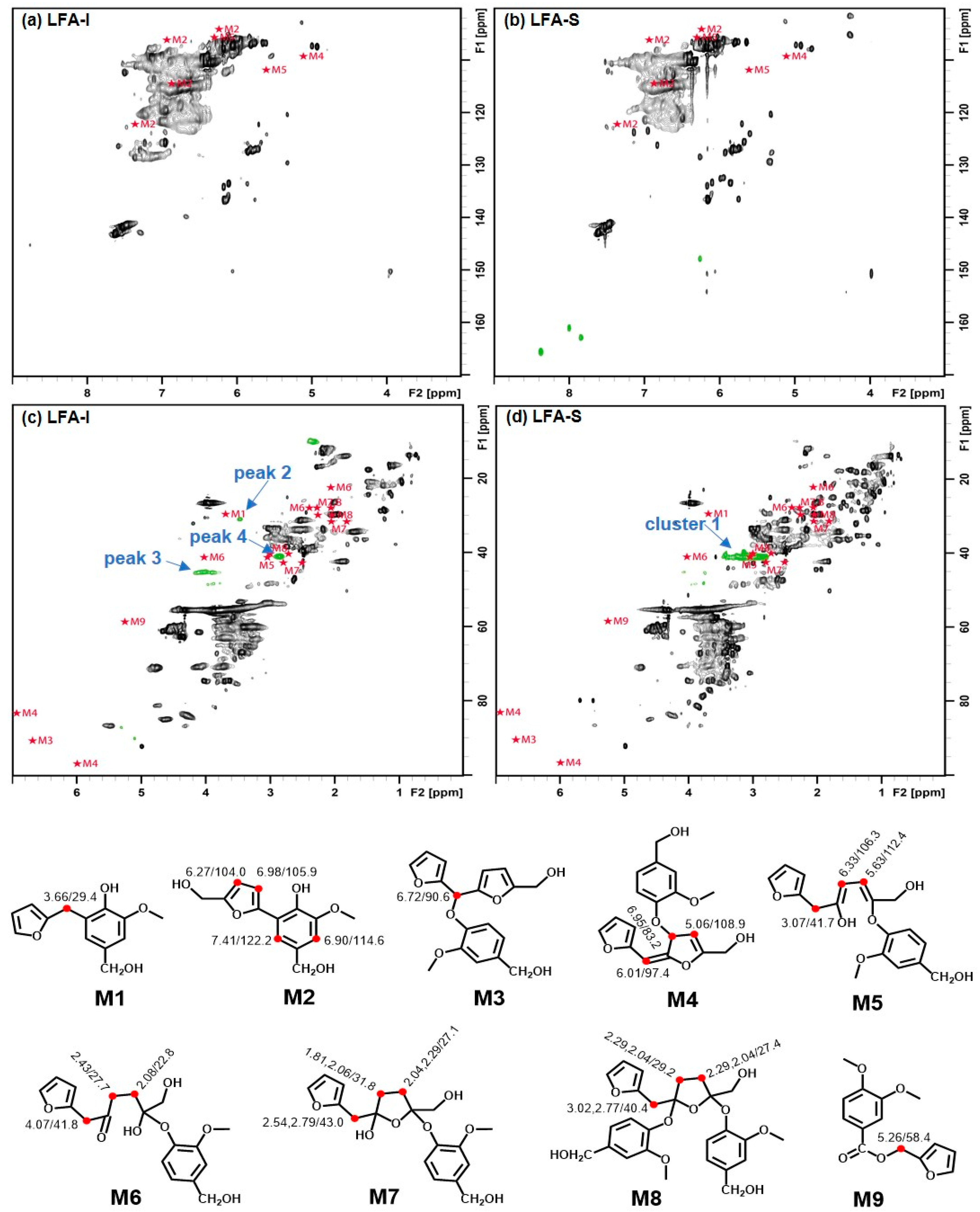
3.3. Identification of Newly Formed Structures in Bioresin
3.4. Tentative Assignment of New Signals
3.4.1. Aromatic Condensed Structures
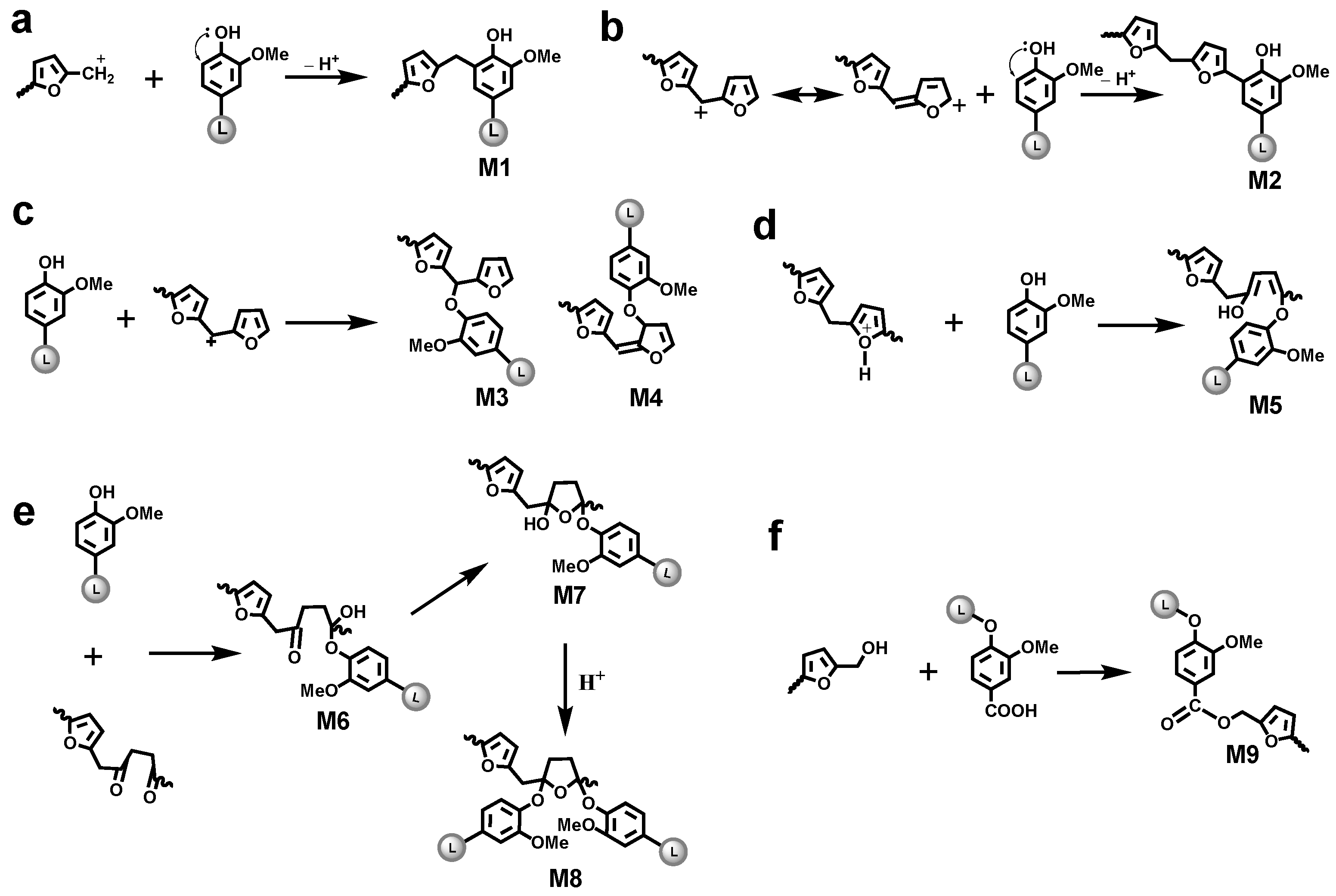
3.4.2. Aryl Ether Structures
3.4.3. Ester Structure
3.5. Tentative Structure of Bioresin
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ragauskas, A.J.; Beckham, G.T.; Biddy, M.J.; Chandra, R.; Chen, F.; Davis, M.F.; Davison, B.H.; Dixon, R.A.; Gilna, P.; Keller, M.; et al. Lignin Valorization: Improving Lignin Processing in the Biorefinery. Sciences 2014, 344, 1246843. [Google Scholar] [CrossRef]
- Dessbesell, L.; Paleologou, M.; Leitch, M.; Pulkki, R.; Xu, C. Global lignin supply overview and kraft lignin potential as an alternative for petroleum-based polymers. Renew. Sustain. Energy Rev. 2020, 123, 109768. [Google Scholar] [CrossRef]
- Gürbüz, E.I.; Gallo, J.M.R.; Alonso, D.M.; Wettstein, S.G.; Lim, W.Y.; Dumesic, J.A. Conversion of hemicellulose into furfural using solid acid catalysts in γ-valerolactone. Angew. Chem. Int. Ed. 2013, 52, 1270–1274. [Google Scholar] [CrossRef]
- Perez, R.F.; Fraga, M.A. Hemicellulose-derived chemicals: One-step production of furfuryl alcohol from xylose. Green Chem. 2014, 16, 3942–3950. [Google Scholar] [CrossRef]
- Gardziella, A. Furanharze. In Kunststoff-Handbuch, 2nd ed.; Woebcken, W., Ed.; Carl Hanser Verlag: München, Germany, 1988; Volume 10, pp. 70–84. ISBN 978-3-446-14418-7. [Google Scholar]
- Brydson, J.A. Furan Resins. In Plastics Materials, 7th ed.; Brydson, J.A., Ed.; Butterworth-Heinemann: Oxford, UK, 1999; pp. 810–813. ISBN 978-0-750-64132-6. [Google Scholar]
- Ibeh, C.C. Amino and Furan Resins. In Handbook of Thermoset Plastics, 2nd ed.; Goodman, S.H., Ed.; William Andrew Publishing: Norwich, NY, USA, 1998; pp. 72–96. ISBN 978-0-815-51421-3. [Google Scholar]
- Gardziella, A.; Hansen, A.; Kwasniok, A. Novel Low-Emission, Cold-Curing Binding Agents. U.S. Patent 5,288,774, 22 February 1994. [Google Scholar]
- Liang, J.; Zhang, J.; Du, G.; Feng, S.; Xi, X.; Lei, H. Lignin-based grinding wheels with aluminum oxide: Synthesis and characterization. BioResources 2018, 13, 1388–1400. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, W.; Zhou, X.; Liang, J.; Du, G.; Wu, Z. Lignin-based adhesive crosslinked by furfuryl alcohol–glyoxal and epoxy resins. Nord. Pulp Pap. Res. J. 2019, 34, 228–238. [Google Scholar] [CrossRef]
- Chung, H.; Washburn, N.R. Chemistry of lignin-based materials. Green Mater. 2012, 1, 137–160. [Google Scholar] [CrossRef]
- Balakshin, M.Y.; Capanema, E.A.; Zhu, X.; Sulaeva, I.; Potthast, A.; Rosenau, T.; Rojas, O.J. Spruce milled wood lignin: Linear, branched or cross-linked? Green Chem. 2020, 22, 3985–4001. [Google Scholar] [CrossRef]
- Balakshin, M.Y.; Capanema, E.A.; Chen, C.L.; Gracz, H.S. Elucidation of the structures of residual and dissolved pine kraft lignins using an HMQC NMR technique. J. Agric. Food Chem. 2003, 51, 6116–6127. [Google Scholar] [CrossRef]
- Constant, S.; Wienk, H.L.; Frissen, A.E.; de Peinder, P.; Boelens, R.; Van Es, D.S.; Grisel, R.J.; Weckhuysen, B.M.; Huijgen, W.J.; Gosselink, R.J.; et al. New insights into the structure and composition of technical lignins: A comparative characterization study. Green Chem. 2016, 18, 2651–2665. [Google Scholar] [CrossRef] [Green Version]
- Lancefield, C.S.; Wienk, H.L.; Boelens, R.; Weckhuysen, B.M.; Bruijnincx, P.C. Identification of a diagnostic structural motif reveals a new reaction intermediate and condensation pathway in kraft lignin formation. Chem. Sci. 2018, 9, 6348–6360. [Google Scholar] [CrossRef] [Green Version]
- Balakshin, M.Y.; Capanema, E.A. Comprehensive structural analysis of biorefinery lignins with a quantitative 13C NMR approach. RSC Adv. 2015, 5, 87187–87199. [Google Scholar] [CrossRef]
- Balakshin, M.Y.; Capanema, E.A.; Sulaeva, I.; Schlee, P.; Huang, Z.; Feng, M.; Borghei, M.; Rojas, O.J.; Potthast, A.; Rosenau, T. New opportunities in the valorization of technical lignins. ChemSusChem 2021, 14, 1016–1036. [Google Scholar] [CrossRef] [PubMed]
- Gandini, A.; Belgacem, M.N. Furans in polymer chemistry. Prog. Polym. Sci. 1997, 22, 1203–1379. [Google Scholar] [CrossRef]
- Szczurek, A.; Fierro, V.; Thébault, M.; Pizzi, A.; Celzard, A. Structure and properties of poly (furfuryl alcohol)-tannin polyHIPEs. Eur. Polym. J. 2016, 78, 195–212. [Google Scholar] [CrossRef]
- Choura, M.; Belgacem, N.M.; Gandini, A. Acid-catalyzed polycondensation of furfuryl alcohol: Mechanisms of chromophore formation and cross-linking. Macromolecules 1996, 29, 3839–3850. [Google Scholar] [CrossRef]
- Mansfield, S.D.; Kim, H.; Lu, F.; Ralph, J. Whole plant cell wall characterization using solution-state 2D NMR. Nat. Protoc. 2012, 7, 1579–1589. [Google Scholar] [CrossRef] [PubMed]
- Karhunen, P.; Rummakko, P.; Sipilä, J.; Brunow, G.; Kilpeläinen, I. Dibenzodioxocins; a novel type of linkage in softwood lignins. Tetrahedron Lett. 1995, 36, 169–170. [Google Scholar] [CrossRef]
- Zhang, L.; Gellerstedt, G. NMR observation of a new lignin structure, a spiro-dienone. Chem. Commun. 2001, 24, 2744–2745. [Google Scholar] [CrossRef]
- Zhang, L.; Gellerstedt, G. Quantitative 2D HSQC NMR determination of polymer structures by selecting suitable internal standard references. Magn. Reson. Chem. 2007, 45, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Capanema, E.A.; Balakshin, M.Y.; Chang, H.M.; Jameel, H. Quantitative analysis of technical lignins by a combination of 1H-13C HMQC and 13C NMR methods. In Proceedings of the International Conference on Pulping, Papermaking and Biotechnology 2008, Nanjing, China, 4–6 November 2008; pp. 647–651. [Google Scholar]
- Sette, M.; Wechselberger, R.; Crestini, C. Elucidation of lignin structure by quantitative 2D NMR. Chem. Eur. J. 2011, 17, 9529–9535. [Google Scholar] [CrossRef] [PubMed]
- Chen, C. Methods in Lignin Chemistry; Lin, S.Y., Dence, C.W., Eds.; Springer: Berlin/Heidelberg, Germany, 1992; pp. 465–472. ISBN 978-3-642-74067-1. [Google Scholar]
- Goto, H.; Koda, K.; Tong, G.; Matsumoto, Y.; Meshitsuka, G. Interference of carbohydrates in the determination of the methoxyl content of lignin in woody samples. J. Wood Chem. Technol. 2006, 26, 81–93. [Google Scholar] [CrossRef]
- Jin, Z.; Akiyama, T.; Chung, B.Y.; Matsumoto, Y.; Iiyama, K.; Watanabe, S. Changes in lignin content of leaf litters during mulching. Phytochemistry 2003, 64, 1023–1031. [Google Scholar] [CrossRef]
- Sarkanen, K.V.; Ludwig, C.H. Lignins: Occurrence, Formation, Structure, and Reactions; Wiley–Interscience: New York, NY, USA, 1971; pp. 43–94. ISBN 0-471-75422-6. [Google Scholar]
- Ralph, S.A.; Ralph, J.; Landucci, L.L. NMR Database of Lignin and Cell Wall Model Compounds. 2009. Available online: https://www.glbrc.org/databases_and_software/nmrdatabase/ (accessed on 28 October 2021).
- Chuang, I.S.; Maciel, G.E.; Myers, G.E. Carbon-13 NMR study of curing in furfuryl alcohol resins. Macromolecules 1984, 17, 1087–1090. [Google Scholar] [CrossRef]
- Maciel, G.E.; Chuang, I.S.; Myers, G.E. 13C NMR study of cured furfuryl alcohol resins using cross polarization and magic-angle spinning. Macromolecules 1982, 15, 1218–1220. [Google Scholar] [CrossRef]
- Nordstierna, L.; Lande, S.; Westin, M.; Karlsson, O.; Furó, I. Towards novel wood-based materials: Chemical bonds between lignin-like model molecules and poly (furfuryl alcohol) studied by NMR. Holzforschung 2008, 62, 709–713. [Google Scholar] [CrossRef]
- Principe, M.; Ortiz, P.; Martínez, R. An NMR study of poly (furfuryl alcohol) prepared with p-toluenesulphonic acid. Polym. Int. 2015, 48, 637–641. [Google Scholar] [CrossRef]
- Balakshin, M.Y.; Capanema, E. On the quantification of lignin hydroxyl groups with 31P and 13C NMR spectroscopy. J. Wood Chem. Technol. 2015, 35, 220–237. [Google Scholar] [CrossRef]
- Lourençon, T.V.; Alakurtti, S.; Virtanen, T.; Jääskeläinen, A.S.; Liitiä, T.; Hughes, M.; Magalhães, W.L.E.; Muniz, G.I.B.; Tamminen, T. Phenol-formaldehyde resins with suitable bonding strength synthesized from “less-reactive” hardwood lignin fractions. Holzforschung 2020, 74, 175–183. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | OMe Content (mmol/g Sample) | Lignin Content (Weight %) |
|---|---|---|
| KL | 4.12 | 100 |
| LFA-I | 3.52 | 85 |
| LFA-S | 1.84 | 44 |
| No. | Code | KL | LFA-I | LFA-S | Assignment |
|---|---|---|---|---|---|
| 1 | Methoxyl | 100.0 | 100.0 | 100.0 | —OCH3 |
| 2 | G2 | 93.0 | 91.3 | 91.9 | CH-2 in Guaiacyl unit |
| 3 | Aα | 6.9 | 6.4 | 6.0 | β-O-4 |
| 4 | Bα | 2.1 | 2.4 | 2.0 | β-5 |
| 5 | Cα | 1.8 | 1.9 | 1.8 | β-β |
| 6 | C’α | 1.3 | 1.4 | 1.3 | epiresinol and other benzyl-O-Alk ethers |
| 7 | SRβ | 2.6 | 2.5 | 3.2 | secoisolariciresinol |
| 8 | Arα | 0.8 | 1.0 | 1.1 | reduced β-O-4 |
| 9 | AGα | 1.2 | 1.1 | 1.0 | arylglycerol |
| 10 | Dγ | 1.5 | 1.2 | 1.2 | cinnamyl alcohol |
| 11 | DHCAβ | 4.1 | 4.3 | 4.6 | dihydrocinnamyl |
| 12 | AVβ | 0.3 | 0.2 | 0.2 | acetovanillone |
| 13 | V6 | 0.9 | 0.9 | 0.9 | vanillin |
| 14 | VA6 | 0.7 | 0.5 | 0.7 | vanillic acid |
| 15 | Bi6 | 0.7 | 1.0 | 0.8 | biphenyl |
| 16 | E-EEα | 2.4 | 3.1 | 3.3 | E-enol ether |
| 17 | Z-EEα | 1.0 | 0.8 | nd a | Z-enol ether |
| 18 | SB1α | 3.1 | 2.8 | 2.8 | β-1 stilbene |
| 19 | SB5β | 5.3 | 6.1 | 5.8 | β-5 stilbene |
| 20 | CH2 in pFA | nd a | 37.2 | 75.4 | CH2 linkage in pFA |
| 21 | CH2OH in pFA | nd a | 20.8 | 92.3 | terminal CH2OH in pFA |
| 22 | CH2OCH2 in pFA | nd a | 5.3 | 12.8 | CH2OCH2 linkage in pFA |
| 23 | peak 2 | nd a | 0.6 | nd a | condensed structure |
| 24 | peak 3 | nd a | 2.9 | nd a | aryl ether structure |
| 25 | peak 4 | nd a | 2.4 | - b | aryl ether structure |
| 26 | cluster 1 | nd a | nd a | 100.1 | aryl ether structure |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, X.; Bruijnaers, B.; Lourençon, T.V.; Balakshin, M. Structural Analysis of Lignin-Based Furan Resin. Materials 2022, 15, 350. https://doi.org/10.3390/ma15010350
Zhu X, Bruijnaers B, Lourençon TV, Balakshin M. Structural Analysis of Lignin-Based Furan Resin. Materials. 2022; 15(1):350. https://doi.org/10.3390/ma15010350
Chicago/Turabian StyleZhu, Xuhai, Bardo Bruijnaers, Tainise V. Lourençon, and Mikhail Balakshin. 2022. "Structural Analysis of Lignin-Based Furan Resin" Materials 15, no. 1: 350. https://doi.org/10.3390/ma15010350
APA StyleZhu, X., Bruijnaers, B., Lourençon, T. V., & Balakshin, M. (2022). Structural Analysis of Lignin-Based Furan Resin. Materials, 15(1), 350. https://doi.org/10.3390/ma15010350