
Diflunisal Targeted Delivery Systems: A Review



Abstract
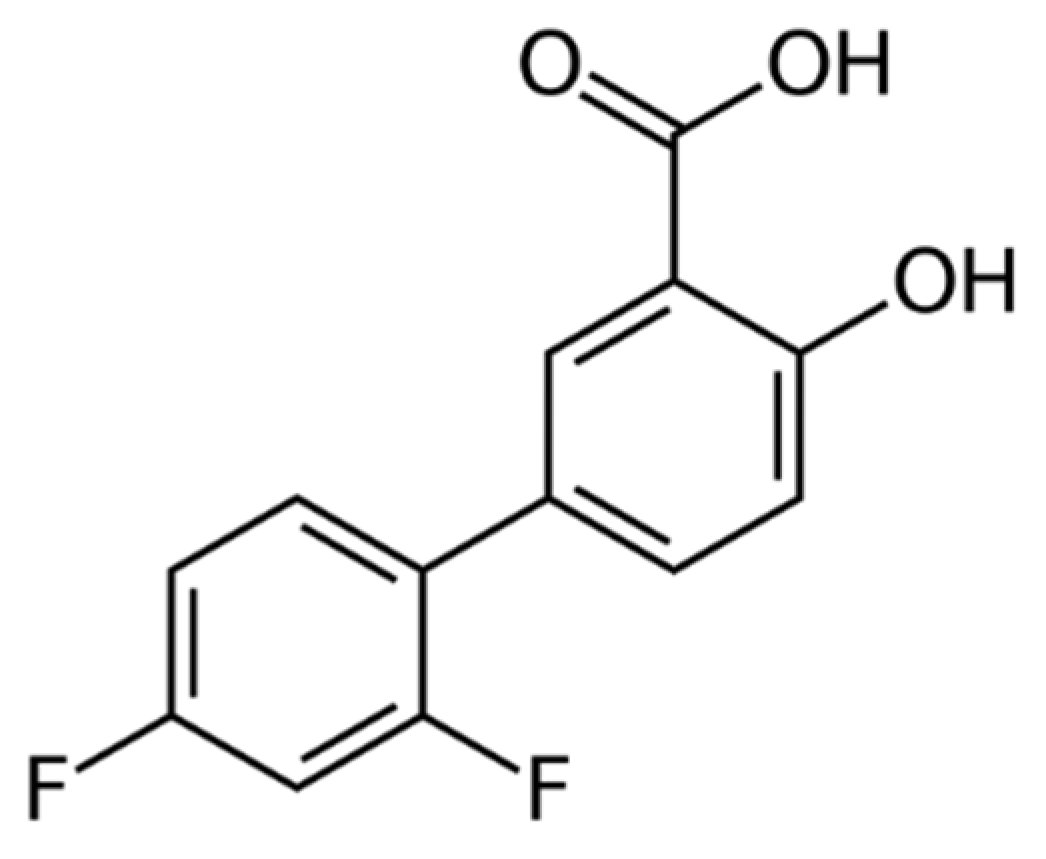
:1. Introduction
2. Drug Delivery Systems
2.1. Nanoparticles
2.2. Hydrogels and Oleogels
2.3. Complexes
2.4. Co-Crystals
2.5. Solid Dispersions
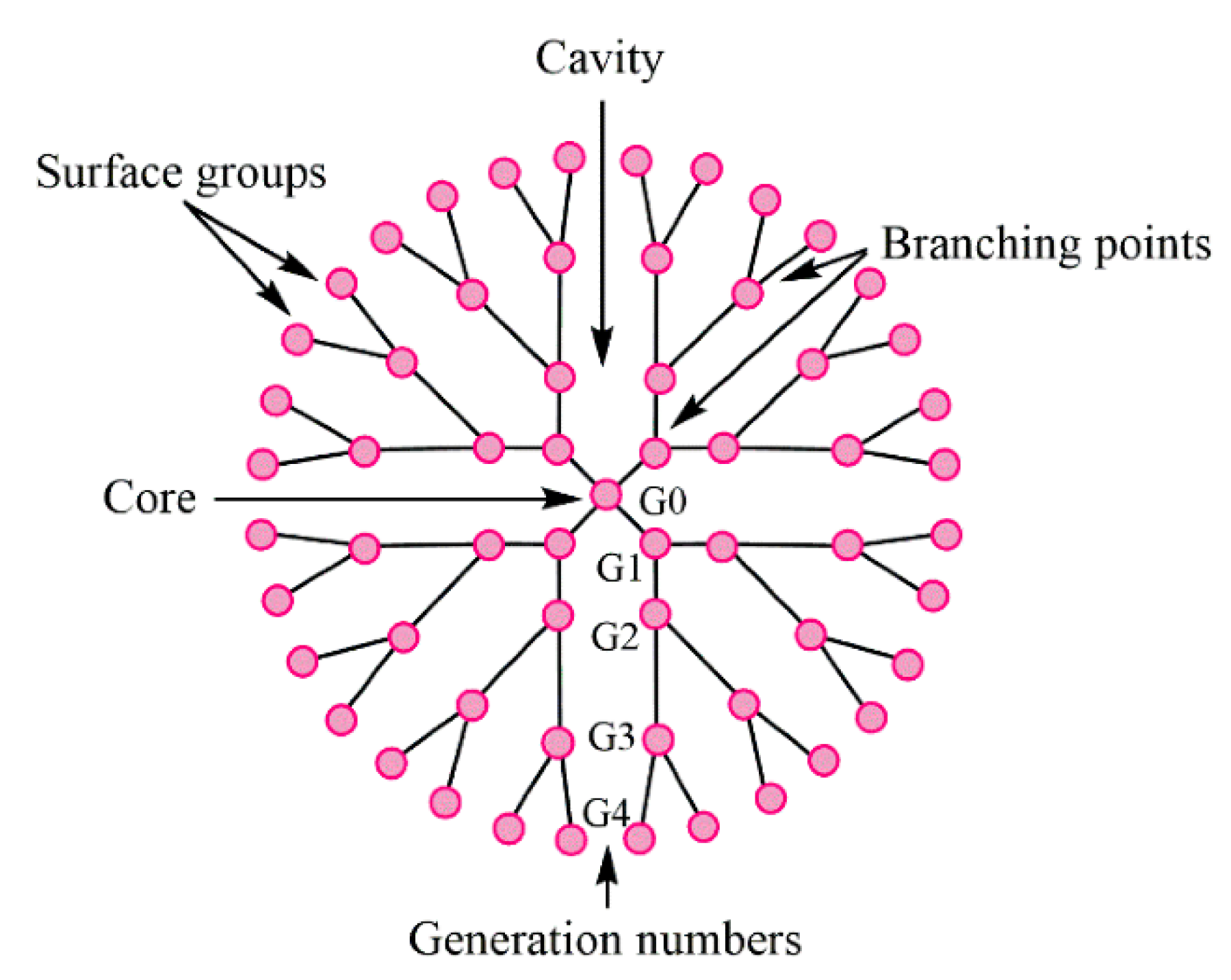
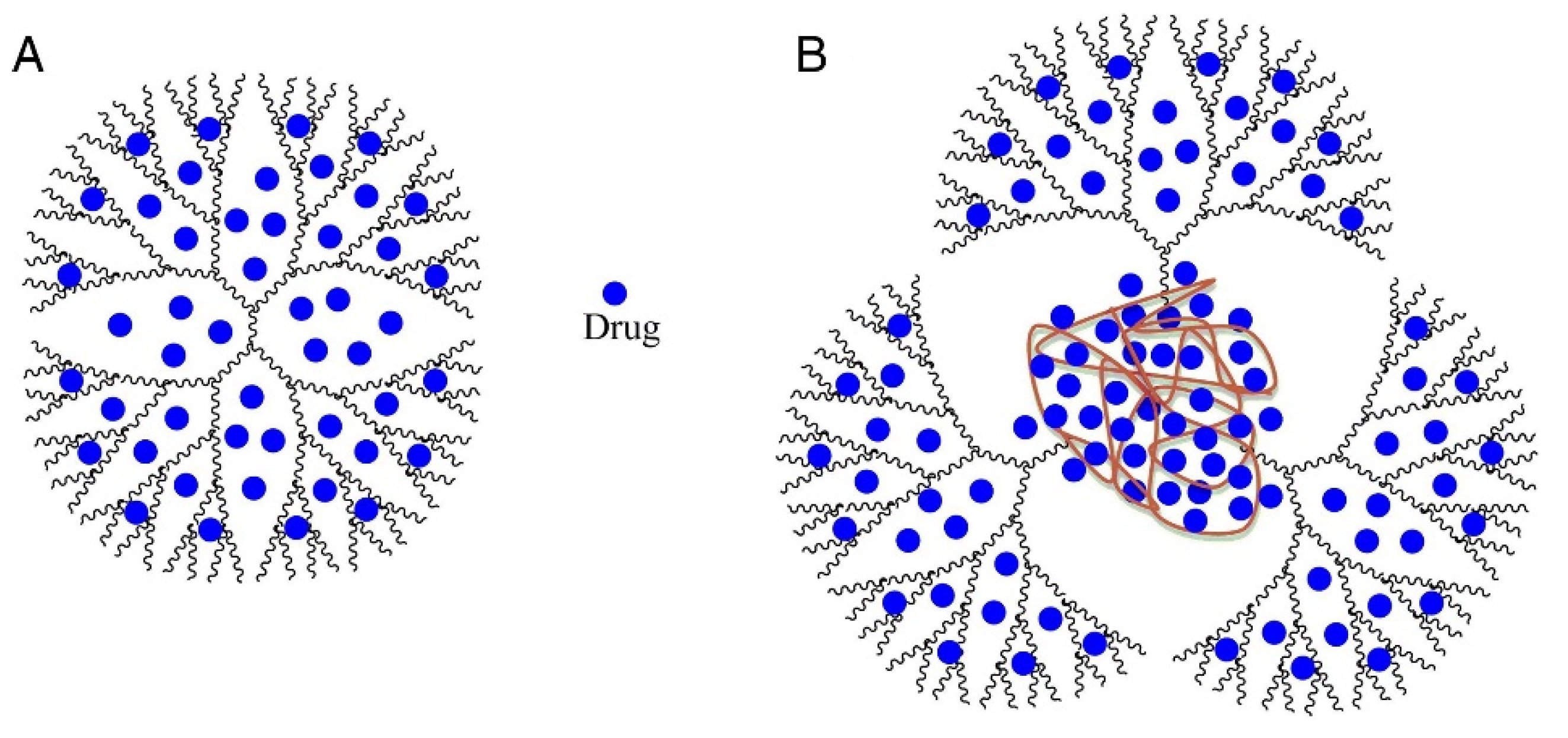
2.6. Dendrimers
3. Conclusions and Future Perspectives
- (1)
- No burst release effect was detected in all developed systems, which underlines the importance of drug delivery systems for diflunisal for prolonged use;
- (2)
- Most systems demonstrated a selective mode of action both in in vitro and in vivo studies;
- (3)
- Cyclodextrin and hydroxypropyl-β-cyclodextrin complexes as well as dendrimers and nanoparticles are the most effective drug delivery systems for diflunisal;
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hannah, J.; Ruyle, W.V.; Jones, H.; Matzuk, A.R.; Kelly, K.W.; Witzel, B.E.; Holtz, W.J.; Houser, R.W.; Shen, T.Y.; Sarett, L.H. Discovery of diflunisal. Br. J. Clin. Pharmacol. 1977, 4, 7S–13S. [Google Scholar] [CrossRef]
- Kreutz, W. Diflunisal for the Treatment of Cancer. U.S. Patent 20060293390, 28 December 2006. [Google Scholar]
- Entellas, C.M.; Insa, B.R.; Reig, B.N.; Gavalda, B.N. Therapy for Transthyretin-Associated Amyloidosis. U.S. Patent 2020390733A1, 17 December 2020. [Google Scholar]
- Tojo, K.; Sekijima, Y.; Kelly, J.W.; Ikeda, S. Diflunisal stabilizes familial amyloid polyneuropathy-associated transthyretin variant tetramers in serum against dissociation required for amyloidogenesis. Neurosci. Res. 2006, 56, 441–449. [Google Scholar] [CrossRef]
- Berk, J.L.; Suhr, O.B.; Sekijima, Y.; Yamashita, T.; Heneghan, M.; Zeldenrust, S.R.; Ando, Y.; Ikeda, S.; Gorevic, P.; Merlini, G.; et al. The Diflunisal Trial: Study accrual and drug tolerance. Amyloid 2012, 19 (Suppl. 1), 37–38. [Google Scholar] [CrossRef]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Diflunisal Trial, Consortium. Repurposing diflunisal for familial amyloid polyneuropathy: A randomized clinical trial. JAMA 2013, 310, 2658–2667. [Google Scholar] [CrossRef] [Green Version]
- Sekijima, Y.; Tojo, K.; Morita, H.; Koyama, J.; Ikeda, S. Safety and efficacy of long-term diflunisal administration in hereditary transthyretin (ATTR) amyloidosis. Amyloid 2015, 22, 79–83. [Google Scholar] [CrossRef]
- Ikram, A.; Donnelly, J.P.; Sperry, B.W.; Samaras, C.; Valent, J.; Hanna, M. Diflunisal tolerability in transthyretin cardiac amyloidosis: A single center’s experience. Amyloid 2018, 25, 197–202. [Google Scholar] [CrossRef]
- Castaño, A.; Helmke, S.; Alvarez, J.; Delisle, S.; Mathew, S.; Maurer, M.S. Diflunisal for ATTR Cardiac Amyloidosis. Congest. Heart Fail. 2012, 18, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, H.; Castano, A.; Alvarez, J.; Goldsmith, J.; Helmke, S.; Maurer, M.S. TTR (transthyretin) stabilizers are associated with improved survival in patients with TTR cardiac amyloidosis. Circ. Heart Fail. 2018, 11, e004769. [Google Scholar] [CrossRef] [PubMed]
- Yadav, J.D.; Othee, H.; Chan, K.A.; Man, D.C.; Belliveau, P.P.; Towle, J. Transthyretin Amyloid Cardiomyopathy-Current and Future Therapies. Ann. Pharmacother. 2021. [Google Scholar] [CrossRef] [PubMed]
- Benbrahim, M.; Norman, K.; Sanchorawala, V.; Siddiqi, O.K.; Hughes, D. A Review of Novel Agents and Clinical Considerations in Patients with ATTR Cardiac Amyloidosis. J. Cardiovasc. Pharmacol. 2021, 77, 544–548. [Google Scholar] [CrossRef]
- Kearney, P.M.; Baigent, C.; Godwin, J.; Halls, H.; Emberson, J.R.; Patrono, C. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ 2006, 332, 1302–1308. [Google Scholar] [CrossRef] [Green Version]
- Wallace, J.L. Pathogenesis of NSAID-induced gastroduodenal mucosal injury. Best Pract. Res. Clin. Gastroenterol. 2001, 15, 691–703. [Google Scholar] [CrossRef]
- Mukherjee, D.; Nissen, S.E.; Topol, E.J. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA 2001, 286, 954–959. [Google Scholar] [CrossRef]
- Shen, T.Y. Chemical and pharmacological properties of diflunisal. Pharmacotherapy 1983, 2, 3S–8S. [Google Scholar] [CrossRef]
- Greish, K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. Methods Mol. Biol. 2010, 624, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Nandwana, V.; Singh, A.; You, M.M.; Zhang, G.; Higham, J.; Zheng, T.S.; Li, Y.; Prasad, P.V.; Dravid, V.P. Magnetic lipid nanocapsules (MLNCs): Self-assembled lipid-based nanoconstruct for non-invasive theranostic applications. J. Mater. Chem. B 2018, 6, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, K.J.; Deshmane, S.V.; Biyani, K.R. Polymers in pharmaceutical drug delivery system: A review. Int. J. Pharm. Sci. Rev. Res. 2012, 14, 57–66. [Google Scholar]
- Deng, S.; Gigliobianco, M.R.; Censi, R.; Di Martino, P. Polymeric Nanocapsules as Nanotechnological Alternative for Drug Delivery System: Current Status, Challenges and Opportunities. Nanomaterials 2020, 10, 847. [Google Scholar] [CrossRef] [PubMed]
- Crecente-Campo, J.; Alonso, M.J. Engineering, on-demand manufacturing, and scaling-up of polymeric nanocapsules. Bioeng. Transl. Med. 2018, 4, 38–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Koker, S.; Hoogenboom, R.; De Geest, B.G. Polymeric multilayer capsules for drug delivery. Chem. Soc. Rev. 2012, 41, 2867–2884. [Google Scholar] [CrossRef]
- Ford, C.A.; Spoonmore, T.J.; Gupta, M.K.; Duvall, C.L.; Guelcher, S.A.; Cassat, J.E. Diflunisal-loaded poly(propylene sulfide)nanoparticles decrease S. aureus-mediated bone destruction during osteomyelitis. J. Orthop. Res. 2021, 39, 426–437. [Google Scholar] [CrossRef]
- Vanderburgh, J.P.; Kwakwa, K.A.; Werfel, T.A.; Merkel, A.R.; Gupta, M.K.; Johnson, R.W.; Guelcher, S.A.; Duvall, C.L.; Rhoades, J.A. Systemic delivery of a Gli inhibitor via polymeric nanocarriers inhibits tumor-induced bone disease. J. Control. Release 2019, 311, 257–272. [Google Scholar] [CrossRef]
- Amanpreet, K.; Shishu, G.; Katare, O.P. Formulation, characterisation and in vivo evaluation of lipid-based nanocarrier for topical delivery of diflunisal. J. Microencapsul. 2016, 33, 475–486. [Google Scholar] [CrossRef]
- Rochín-Wong, S.; Rosas-Durazo, A.; Zavala-Rivera, P.; Maldonado, A.; Martínez-Barbosa, M.E.; Vélaz, I.; Tánori, J. Drug Release Properties of Diflunisal from Layer-By-Layer Self-Assembled κ-Carrageenan/Chitosan Nanocapsules: Effect of Deposited Layers. Polymers 2018, 10, 760. [Google Scholar] [CrossRef] [Green Version]
- Mansour, H.M.; Sohn, M.; Al-Ghananeem, A.; DeLuca, P.P. Materials for Pharmaceutical Dosage Forms: Molecular Pharmaceutics and Controlled Release Drug Delivery Aspects. Int. J. Mol. Sci. 2010, 11, 3298–3322. [Google Scholar] [CrossRef] [Green Version]
- Naz, S.; Shamoon, M.; Wang, R.; Zhang, L.; Zhou, J.; Chen, J. Advances in Therapeutic Implications of Inorganic Drug Delivery Nano-Platforms for Cancer. Int. J. Mol. Sci. 2019, 20, 965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallaro, G.; Pierro, P.; Palumbo, F.S.; Testa, F.; Luigi Pasqua, L.; Aiello, R. Drug Delivery Devices Based on Mesoporous Silicate. Drug Deliv. 2004, 11, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Bindu, S.M.; Vadithya, A.; Chatterjee, A. As a Review on Hydrogels as Drug Delivery in the Pharmaceutical Field. Int. J. Pharm. Chem. Sci. 2012, 1, 642–661. [Google Scholar]
- Amin, S.; Rajabnezhad, S.; Kohli, K. Hydrogels as potential drug delivery systems. Sci. Res. Essays 2009, 3, 1175–1183. [Google Scholar]
- Larrañeta, E.; Stewart, S.; Ervine, M.; Al-Kasasbeh, R.; Donnelly, R.F. Hydrogels for Hydrophobic Drug Delivery. Classification, Synthesis and Applications. J. Funct. Biomater. 2018, 9, 13. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Mooney, D. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef] [PubMed]
- Rehman, K.; Zulfakar, M.H. Recent advances in gel Technologies for topical and transdermal drug delivery. Drug development and industrial pharmacy. Drug Dev. Ind. Pharm. 2014, 40, 433–440. [Google Scholar] [CrossRef]
- Figueroa-Pizano, M.D.; Vélaz, I.; Martínez-Barbosa, M.E. A Freeze-Thawing Method to Prepare Chitosan-Poly(vinyl alcohol) Hydrogels Without Crosslinking Agents and Diflunisal Release Studies. J. Vis. Exp. 2020, 155, e59636. [Google Scholar] [CrossRef]
- Sallam, M.A.; Motawaa, A.M.; Mortada, S.M. An insight on human skin penetration of diflunisal: Lipogel versus hydrogel microemulsion. Drug Dev. Ind. Pharm. 2013, 41, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Sallam, M.A.; Motawaa, A.M.; Mortada, S.M. A modern approach for controlled transdermal delivery of diflunisal: Optimization and in vivo evaluation. Drug Dev. Ind. Pharm. 2013, 39, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Iemma, F.; Spizzirri, U.G.; Puoci, F.; Muzzalupo, R.; Trombino, S.; Cassano, R.; Picci, N. pH-Sensitive hydrogels based on bovine serum albumin for oral drug delivery. Int. J. Pharm. 2006, 312, 151–157. [Google Scholar] [CrossRef]
- Jacob, S.; Nair, A.B. Cyclodextrin complexes: Perspective from drug delivery and formulation. Drug Dev. Res. 2018, 79, 201–217. [Google Scholar] [CrossRef]
- Jambhekar, S.S.; Breen, P. Cyclodextrins in pharmaceutical formulations I: Structure and physicochemical properties, formation of complexes, and types of complex. Drug Discov. Today 2016, 21, 356–362. [Google Scholar] [CrossRef]
- Muankaew, C.; Loftsson, T. Cyclodextrin-Based Formulations: A Non-Invasive Platform for Targeted Drug Delivery. Basic Clin. Pharmacol. Toxicol. 2017, 122, 46–55. [Google Scholar] [CrossRef] [Green Version]
- Lincoln, S.F.; Coates, J.H.; Doddridge, B.G.; Hounslow, A.M. The inclusion of the drug diflunisal by alpha-and beta-cyclodextrins. A nuclear magnetic resonance and ultraviolet spectroscopic study. J. Incl. Phenom. 1987, 5, 49–53. [Google Scholar] [CrossRef]
- Lincoln, S.F.; Hounslow, A.M.; Coates, J.H.; Villani, R.P.; Schiller, R.L. The inclusion of diflunisal by γ-cyclodextrin and permethylated β-cyclodextrin. A UV-visible and 19F nuclear magnetic resonance spectroscopic study. J. Incl. Phenom. 1988, 6, 183–191. [Google Scholar] [CrossRef]
- Lincoln, S.F.; Hounslow, A.M.; Coates, J.H.; Doddridge, B.G. The inclusion of diflunisal by α- and β-cyclodextrins. A 19F Nuclear magnetic resonance and spectrophotometric study. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1987, 83, 2697–2703. [Google Scholar] [CrossRef]
- Sideris, E.E.; Valsami, G.N.; Koupparis, M.A.; Macheras, P.E. Studies on the interaction of diflunisal ion with cyclodextrins using ion-selective electrode potentiometry. Eur. J. Pharm. Sci. 1999, 7, 271–278. [Google Scholar] [CrossRef]
- Bashir, M.; Syed, H.K.; Asghar, S.; Irfan, M.; Almalki, W.H.; Menshawi, S.A.; Khan, I.U.; Shah, P.A.; Khalid, I.; Ahmad, J.; et al. Effect of Hydrophilic Polymers on Complexation Efficiency of Cyclodextrins in Enhancing Solubility and Release of Diflunisal. Polymers 2020, 12, 1564. [Google Scholar] [CrossRef]
- Zhong, Z.; Yang, X.; Fu, X.B.; Yao, Y.F.; Guo, B.H.; Huang, Y.; Xu, J. Crystalline inclusion complexes formed between the drug diflunisal and block copolymers. Chin. Chem. Lett. 2017, 28, 1268–1275. [Google Scholar] [CrossRef]
- Cai, M.; Chen, G.; Qin, L.; Qu, C.; Dong, X.; Ni, J.; Yin, X. Metal Organic Frameworks as Drug Targeting Delivery Vehicles in the Treatment of Cancer. Pharmaceutics 2020, 12, 232. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zheng, L.; Yang, Y.; Qian, X.; Fu, T.; Li, X.; Yang, Z.; Yan, H.; Cui, C.; Tan, W. Metal-Organic Framework Nanocarriers for Drug Delivery in Biomedical Applications. NanoMicro Lett. 2020, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Wang, T.; Yuan, S.; Wang, M.; Qi, W.; Su, R.; He, Z. A tumor-sensitive biological metal-organic complex for drug delivery and cancer therapy. J. Mater. Chem. B 2020, 8, 7189–7196. [Google Scholar] [CrossRef]
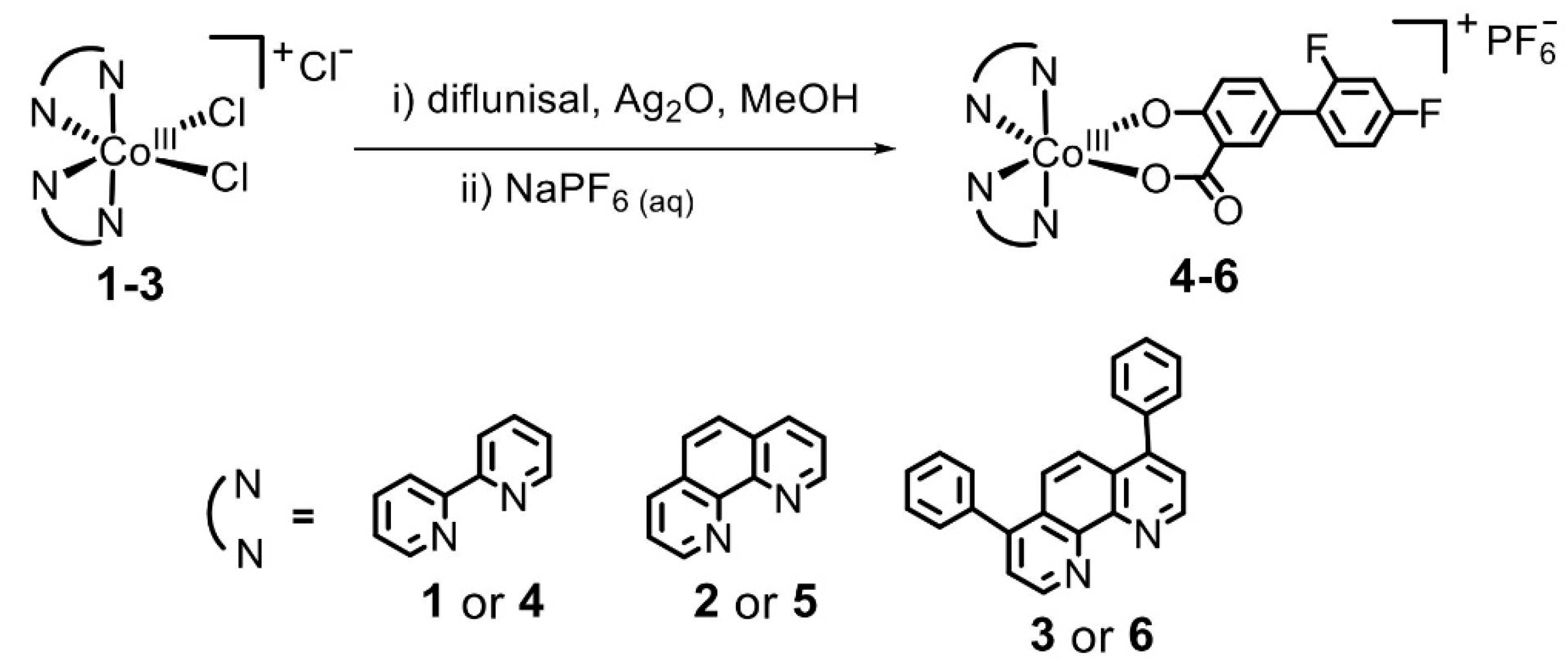
- Abe, D.; Eskandari, A.; Suntharalingam, K. Diflunisal-adjoined cobalt (III)-polypyridyl complexes as anti-cancer stem cell agents. Dalton Trans. 2018, 47, 13761–13765. [Google Scholar] [CrossRef]
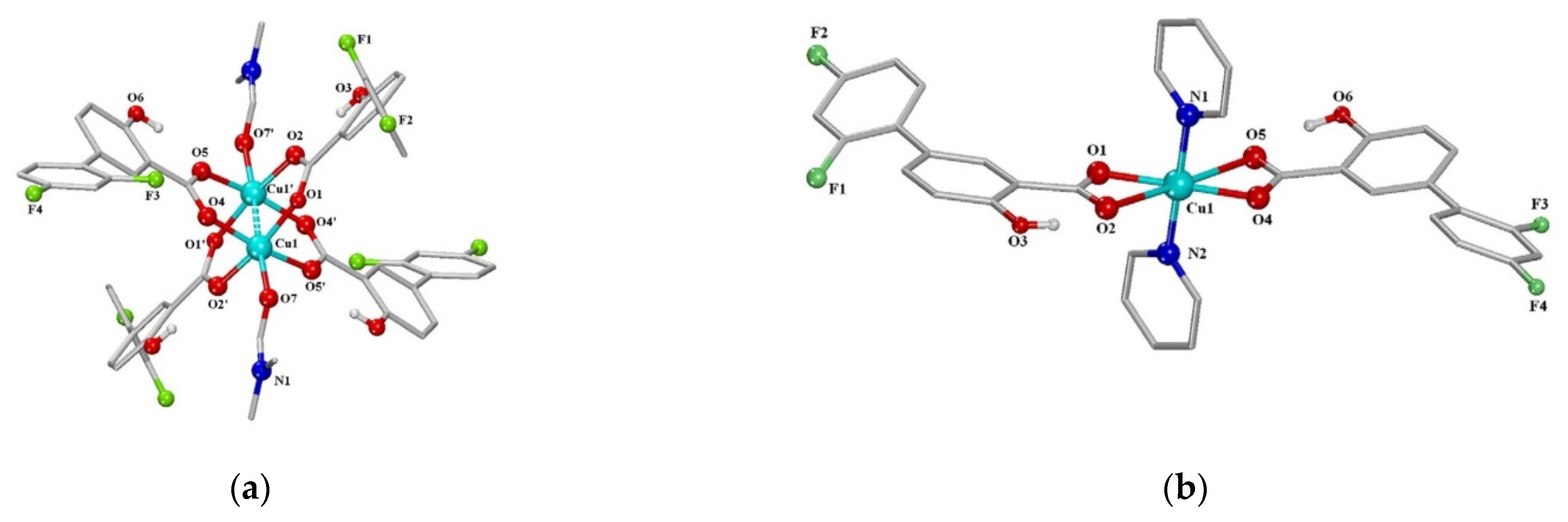
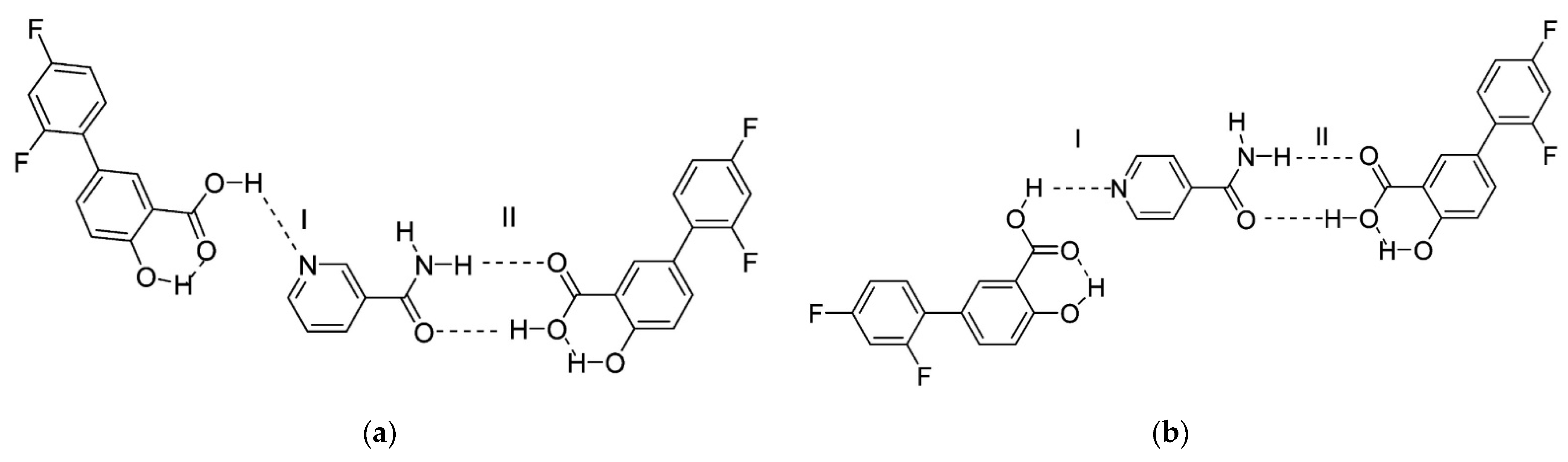
- Fountoulaki, S.; Perdih, F.; Turel, I.; Kessissoglou, D.P.; Psomas, G. Non-steroidal anti-inflammatory drug diflunisal interacting with Cu (II). Structure and biological features. J. Inorg. Biochem. 2011, 105, 1645–1655. [Google Scholar] [CrossRef]
- Bolla, G.; Nangia, A. Pharmaceutical cocrystals: Walking the talk. Chem. Commun. 2016, 52, 8342–8360. [Google Scholar] [CrossRef]
- Karagianni, A.; Malamatari, M.; Kachrimanis, K. Pharmaceutical Cocrystals: New Solid Phase Modification Approaches for the Formulation of APIs. Pharmaceutics 2018, 10, 18. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Sun, X.; Chen, J.; Cai, T. Pharmaceutical cocrystals: A review of preparations, physicochemical properties and applications. Acta Pharm. Sin. B 2021, 11, 2537–2564. [Google Scholar] [CrossRef]
- Tan, J.; Liu, J.; Ran, L. A Review of Pharmaceutical Nano-Cocrystals: A Novel Strategy to Improve the Chemical and Physical Properties for Poorly Soluble Drugs. Crystals 2021, 11, 463. [Google Scholar] [CrossRef]
- Évora, A.O.L.; Castro, R.A.E.; Maria, T.M.R.; Rosado, M.T.S.; Silva, M.R.; Beja, A.M.; Canotilho, J.; Eusébio, M.E.S. Pyrazinamide-Diflunisal: A New Dual-Drug Co-Crystal. Cryst. Growth Des. 2011, 11, 4780–4788. [Google Scholar] [CrossRef]
- Wang, L.; Tan, B.; Zhang, H.; Deng, Z. Pharmaceutical cocrystals of diflunisal with nicotinamide or isonicotinamide. Org. Process Res. Dev. 2013, 17, 1413–1418. [Google Scholar] [CrossRef]
- Wu, X.; Wang, Y.; Xue, J.; Liu, J.; Qin, J.; Hong, Z.; Du, Y. Solid phase drug-drug pharmaceutical co-crystal formed between pyrazinamide and diflunisal: Structural characterization based on terahertz/Raman spectroscopy combining with DFT calculation. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2020, 234, 118265. [Google Scholar] [CrossRef]
- Perlovich, G.L.; Manin, A.N.; Manin, N.G.; Surov, A.O.; Voronin, A.P. Co-Crystalline Form of Theophylline with Diflunisal or Diclofenac. RU2542100C1, 20 February 2015. [Google Scholar]
- Perlovich, G.L.; Manin, A.N.; Drozd, K.V. Diflunisal Co-Crystalline. RU2617849C1, 28 April 2017. [Google Scholar]
- Kim, K.; Lee, J.-Y.; Lee, M.; Song, C.K.; Choi, J.; Kim, D.-D. Solid Dispersions as a Drug Delivery System. J. Pharm. Investig. 2011, 41, 125–142. [Google Scholar] [CrossRef]
- Sharma, K.; Sahoo, J.; Agrawal, S.; Kumari, A. Solid dispersions: A technology for improving bioavailability. J. Anal. Pharm. Res. 2019, 8, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Najib, N.M.; Suleiman, M.S. Characterization of a diflunisal polyethylene glycol solid dispersion system. Int. J. Pharm. 1989, 51, 225–232. [Google Scholar] [CrossRef]
- Rodriguez-Espinosa, C.; Martinez-Oharriz, M.C.; Martin, C.; Goni, M.M.; Velaz, I.; Sanchez, M. Dissolution kinetics for coprecipitates of diflunisal with PVP K30. Eur. J. Drug Metab. Pharmacokinet. 1998, 23, 109–112. [Google Scholar] [CrossRef]
- Pignatello, R.; Ferro, M.; De Guidi, G.; Salemi, G.; Vandelli, M.A.; Guccione, S.; Geppi, M.; Forte, C.; Puglisi, G. Preparation, characterisation and photosensitivity studies of solid dispersions of diflunisal and Eudragit RS100® and RL100®. Int. J. Pharm. 2001, 218, 27–42. [Google Scholar] [CrossRef]
- Zein El Din, E.E.; El Maghraby, G.M.; Donia, A.A.; Mayah, S.I. Formulation and evaluation of a colon drug delivery system containing diflunisal. Bull. Pharm. Sci. Assiut 2014, 37, 33–49. [Google Scholar] [CrossRef] [Green Version]
- Chis, A.A.; Dobrea, C.; Morgovan, C.; Arseniu, A.M.; Rus, L.L.; Butuca, A.; Juncan, A.M.; Totan, M.; Vonica-Tincu, A.L.; Cormos, G.; et al. Applications and Limitations of Dendrimers in Biomedicine. Molecules 2020, 25, 3982. [Google Scholar] [CrossRef] [PubMed]
- Kalomiraki, M.; Thermos, K.; Chaniotakis, N. Dendrimers as tunable vectors of drug delivery systems and biomedical and ocular applications. Int. J. Nanomed. 2016, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Mittal, P.; Saharan, A.; Verma, R.; Altalbawy, F.M.A.; Alfaidi, M.A.; Batiha, G.E.; Akter, W.; Gautam, R.K.; Uddin, M.S.; Rahman, M.S. Dendrimers: A New Race of Pharmaceutical Nanocarriers. BioMed Res. Int. 2021, 2021, 8844030. [Google Scholar] [CrossRef]
- Cheng, Y.; Xu, T. Dendrimers as potential drug carriers. Part, I. Solubilization of non-steroidal anti-inflammatory drugs in the presence of polyamidoamine dendrimers. Eur. J. Med. Chem. 2005, 40, 1188–1192. [Google Scholar] [CrossRef]
- Cheng, Y.; Man, N.; Xu, T.; Fu, R.; Wang, X.; Wang, X.; Wen, L. Transdermal delivery of nonsteroidal anti-inflammatory drugs mediated by polyamidoamine (PAMAM) dendrimers. J. Pharm. Sci. 2007, 96, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Koc, F.E.; Senel, M. Solubility enhancement of Non-Steroidal AntiInflammatory Drugs (NSAIDS) using polypolypropylene oxide core PAMAM dendrimers. Int. J. Pharm. 2013, 451, 18–22. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DIF:CD (w/w) | Solubility (µg/mL) | |
|---|---|---|
| βCD | HPβCD | |
| 1:0 | 44.6 ± 0.02 | 44.6 ± 0.02 |
| 1:1 | 284.5 ± 0.5 | 765.5 ± 0.5 |
| 1:2 | 450.3 ± 0.6 | 907.5 ± 0.5 |
| 1:4 | 500.5 ± 0.5 | 940.4 ± 0.5 |
| 1:2 (2.5% PVA) | 789.6 ± 0.5 | 965.5 ± 0.5 |
| 1:2 (5.0% PVA) | 846.3 ± 0.6 | 1049.3 ± 0.6 |
| 1:2 (10.0% PVA) | 904.1 ± 0.5 | 1190.3 ± 0.6 |
| 1:2 (2.5% CMC-Na) | 692.1 ± 0.3 | 924.3 ± 0.6 |
| 1:2 (5.0% CMC-Na) | 723.6 ± 0.6 | 1000.5 ± 0.5 |
| 1:2 (10.0% CMC-Na) | 800.6 ± 0.5 | 1089.6 ± 0.6 |
| 1:2 (2.5% PXM-188) | 791.2 ± 0.7 | 998.5 ± 0.5 |
| 1:2 (5.0% PXM-188) | 894.4 ± 0.5 | 1181.6 ± 0.6 |
| 1:2 (10.0% PXM-188) | 930.0 ± 0.5 | 1259.5 ± 0.5 |
| № | System; Method; Size Obtained | Cell Line/In Vitro/In Vivo Models; Dose | Diflunisal Release, Biodistribution | Refs. |
|---|---|---|---|---|
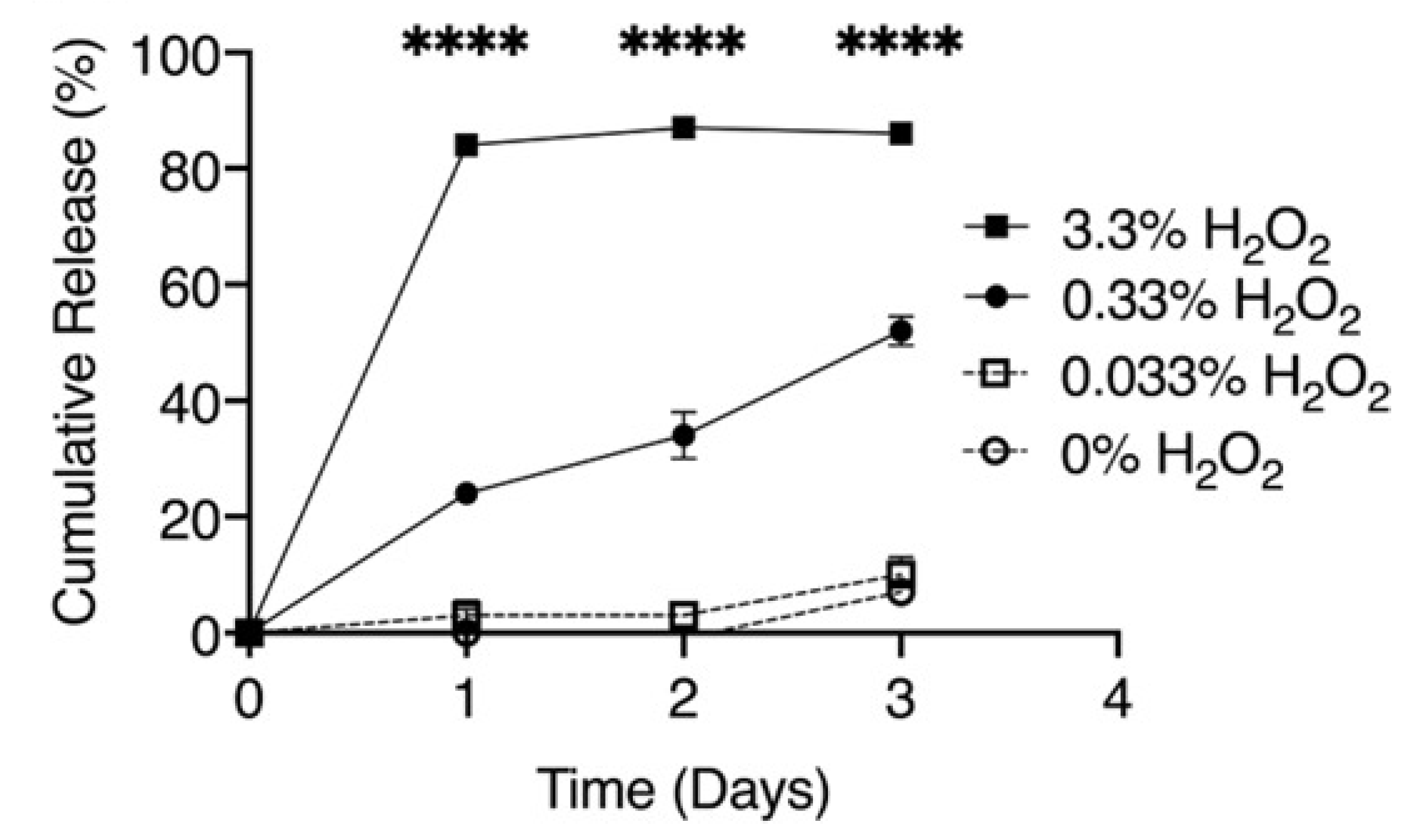
| 1 | Poly(propylene sulfide; oil-in-water emulsion method; 65.4 ± 0.4 nm | 10 μg/mL, parenterally; -murine preosteoblast MC3T3-E1 subclone 4 cell line; -colony of S. aureus from a tryptic soy agar; -inhibits the cytotoxicity of S. aureus supernatants; -decreases S. aureus-induced cortical bone loss during osteomyelitis (on Day 14); -had no effect on bacterial burdens. | -maximum release is reached at 33% of H2O2 at 24 h; -biodistribution (FVB/NJ mice with osteomyelitis of livers, kidneys, and spleens) up to 24 h post injection. | [23] |
| 2 | Carbopol 934, Glyceryl dibehenate (Compritol® ATO 888); microemulsification method; 124.0 ± 2.07 nm | -mice air pouch model; -in vivo pharmacodynamic studies; -better percentage suppression of oedema in mice ear oedema model (xylene induced) and rat hind paw oedema (carrageenan induced); -mean leukocyte count was reduced to 4500 ± 436 cells/mm3 in SLN gel from 173 800 ± 1950 cells/mm3 in positive control; -gastrointestinal and hepatic side effects were avoided; -anti-inflammatory efficacy of DIF SLN gel as compared with conventional cream; -did not cause any type of histopathology. | -permeation flux was maximum for solid lipid nanoparticles dispersion; -skin retention was maximum for solid lipid nanoparticles gel; -high-efficacy therapeutic effects were observed at a much less reduced dose as compared with conventional oral dose. | [25] |
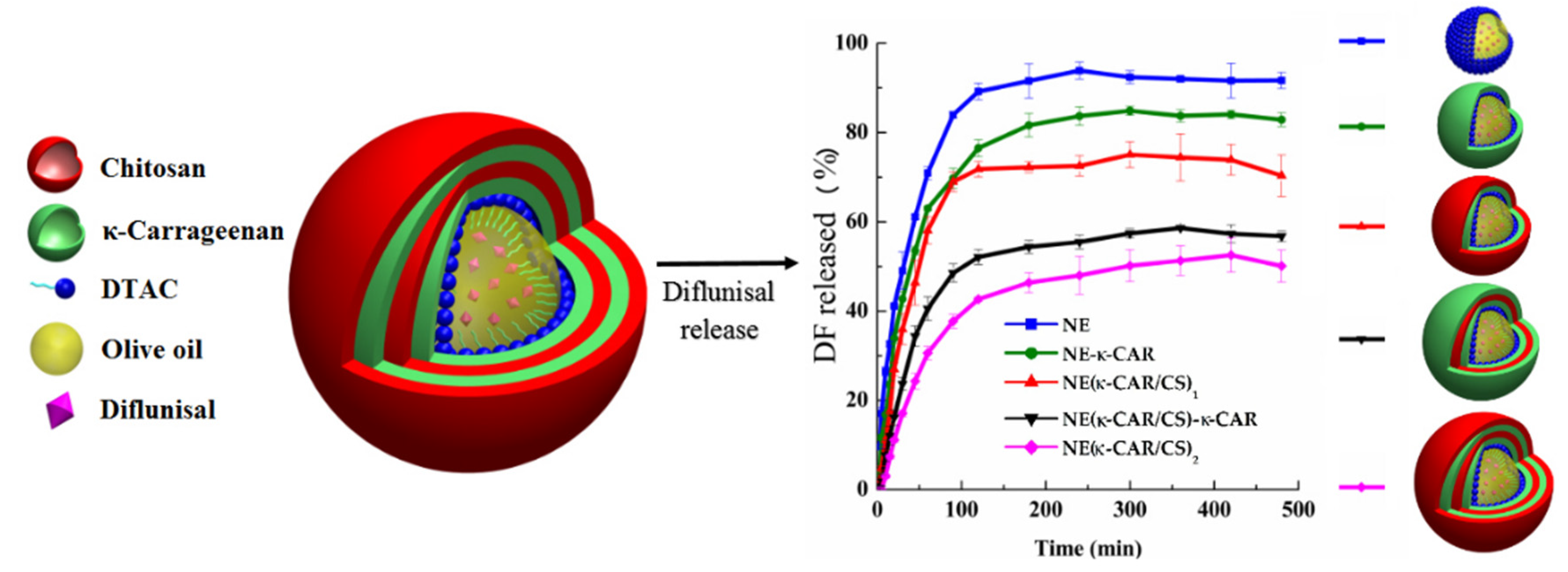
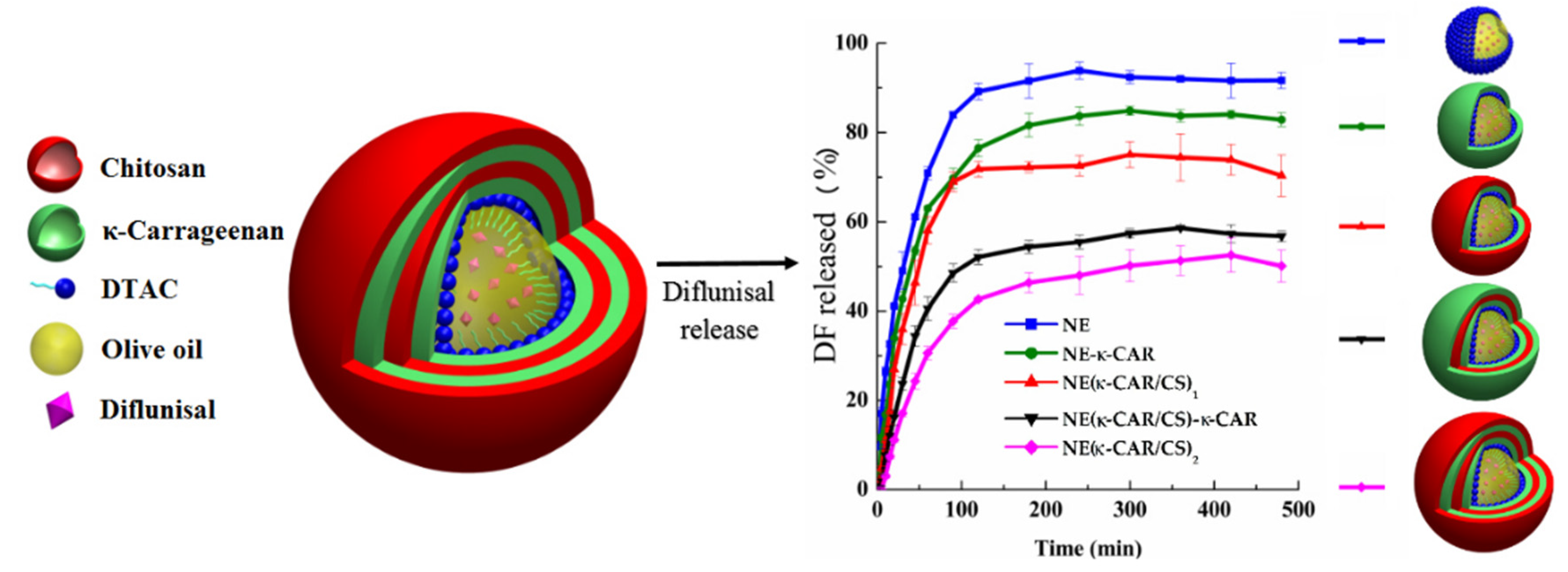
| 3 | k-Carrageenan and chitosan; layer-by-layer assembly technique; 300 nm | -nanocarriers with three and four coatings demonstrated Case II diflunisal transport mechanism and zero-order type of kinetics; | -the release profile is directly dependent on the number of layers; -maximum of cumulative release was reached at 100 min for all compositions with maximum as 95% for nanoemulsion and minimum as 45% for four (NE(k-CAR/CS)2) polyelectrolyte layers. | [26] |
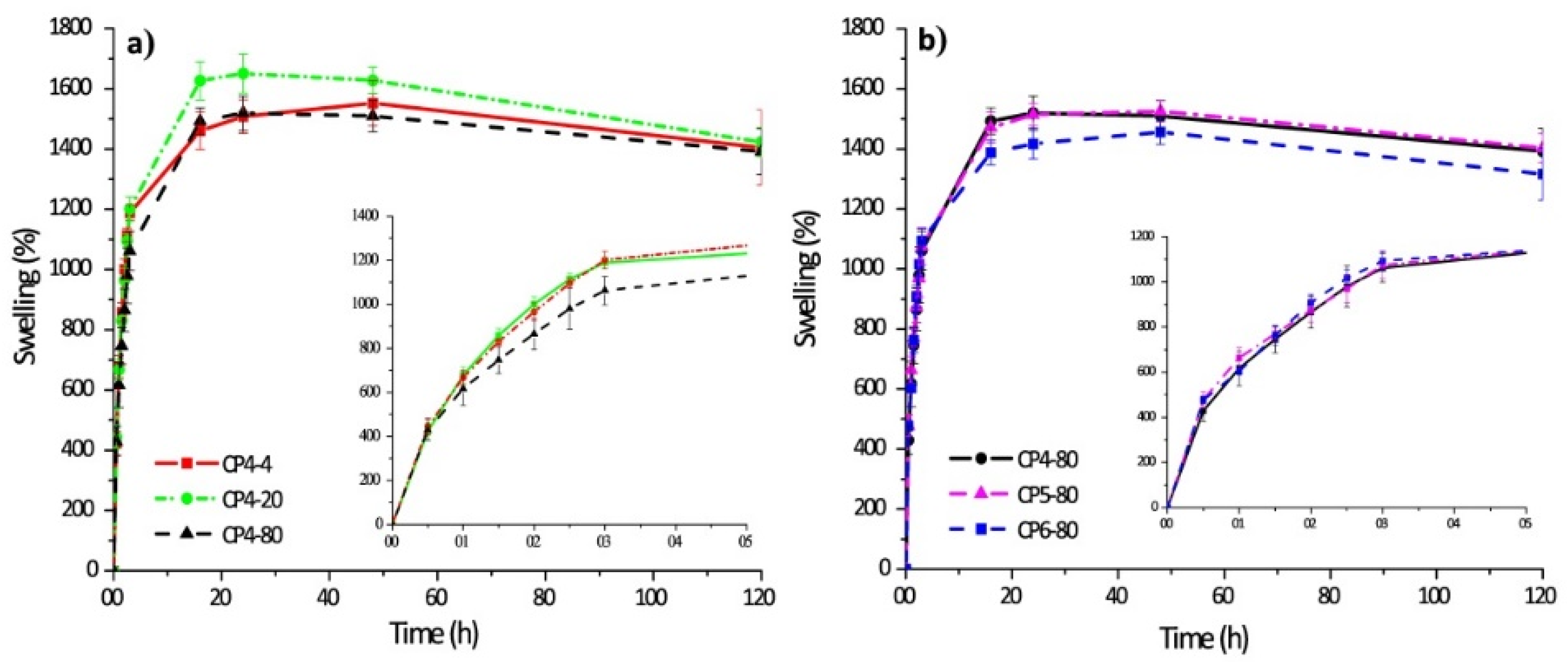
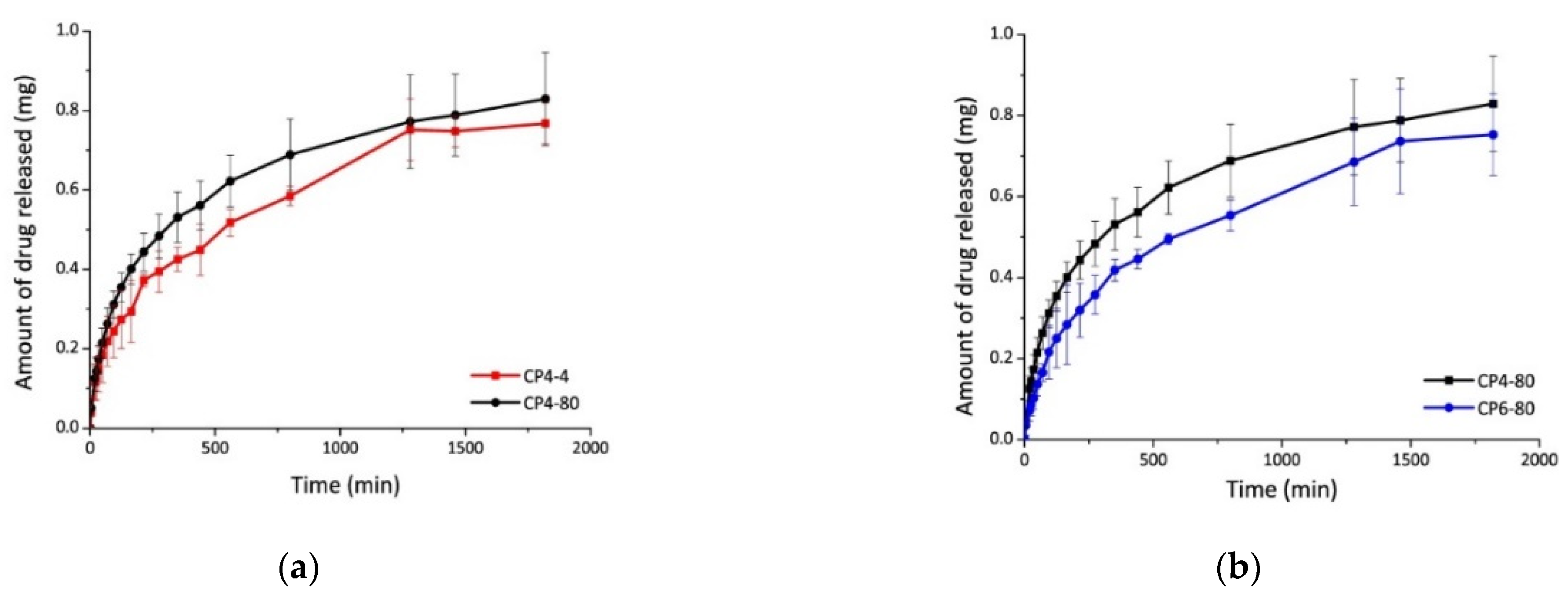
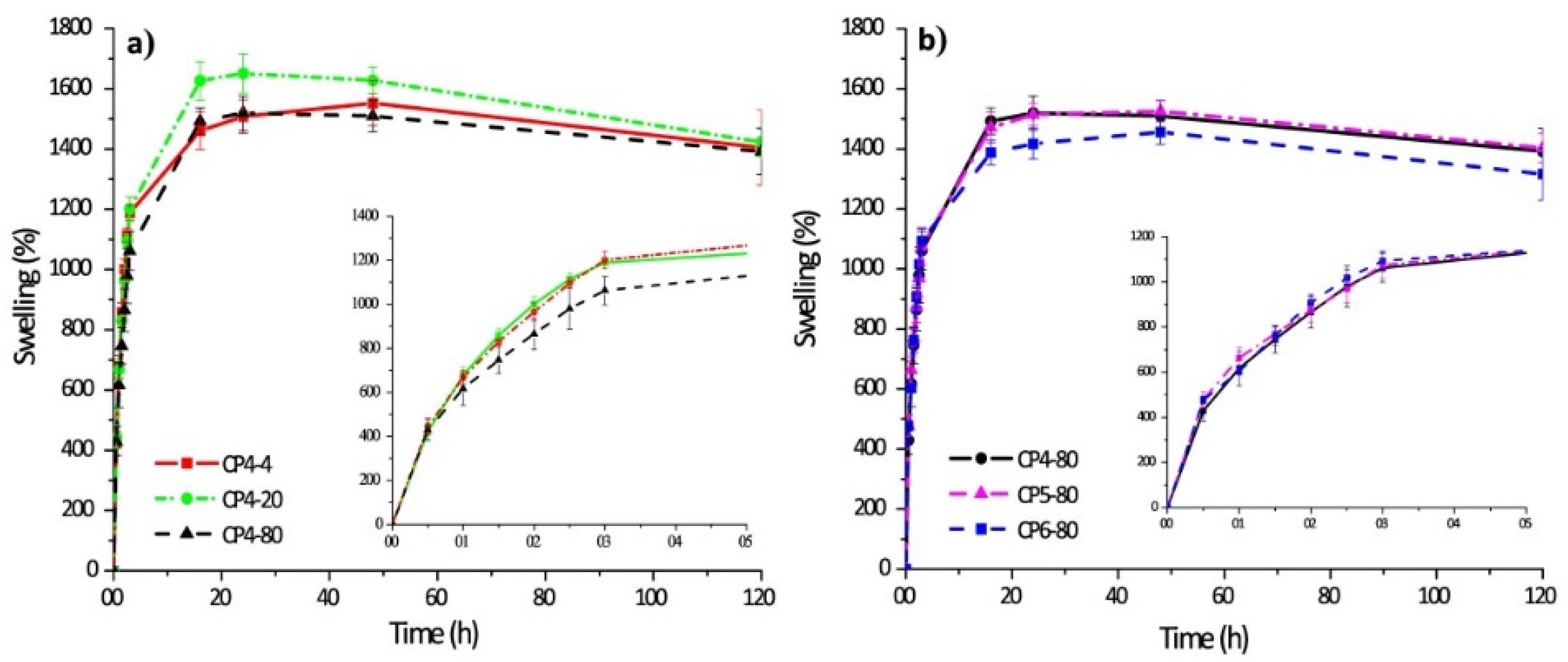
| 4 | Chitosan–poly(vinyl alcohol) hydrogels without crosslinking agents; the freeze–thawing method | -swelled 10-fold their initial weight, and after 20 h, hydrogel samples swelled up to 15-fold; -encapsulation efficiency was equal to 70% in all cases. | -the release of diflunisal from the hydrogels was for 30 h; -higher release profile is for sample CP4-80; -burst effect was not detected for any type of the hydrogels. | [35] |
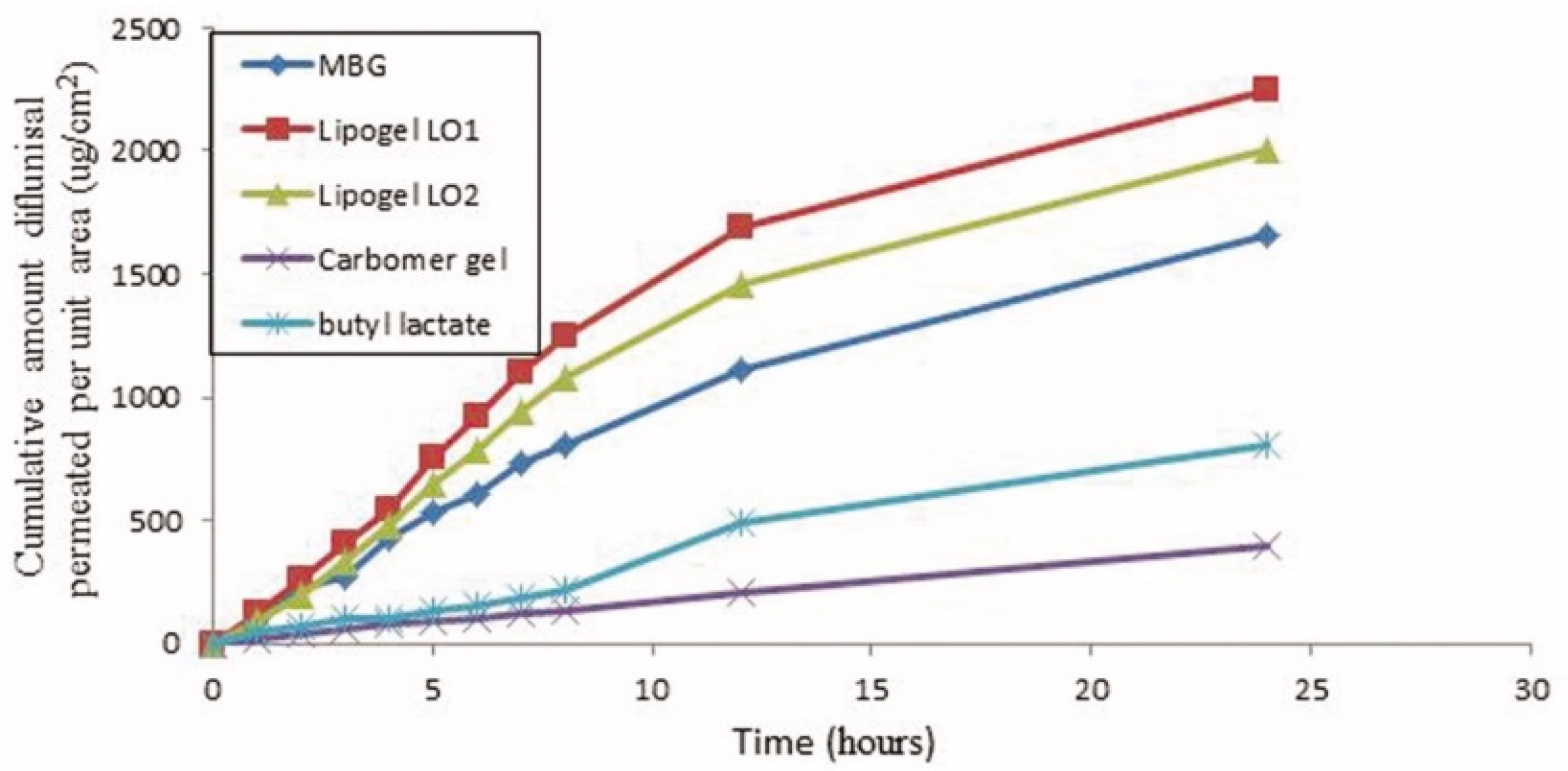
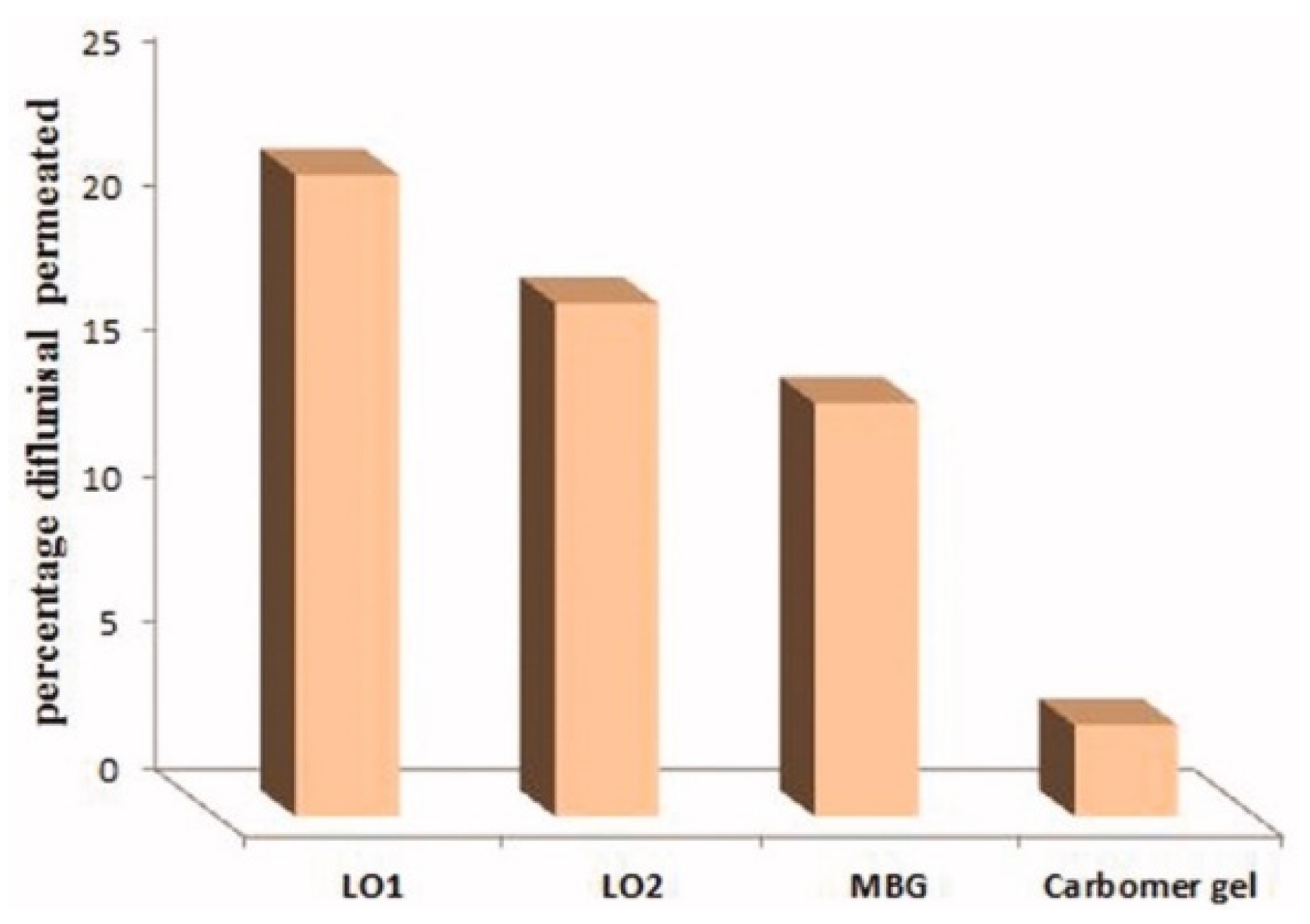
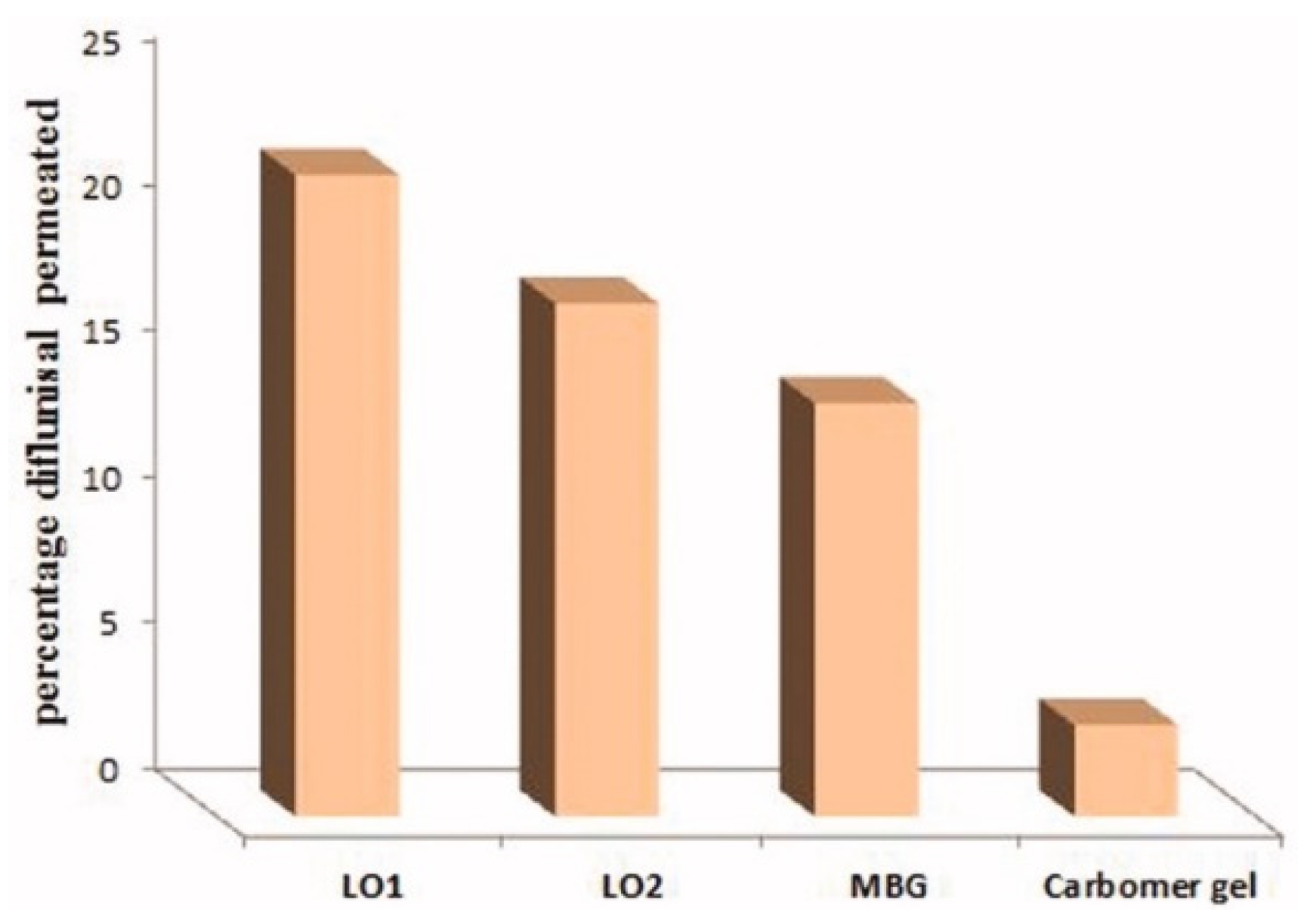
| 5 | 70% soya bean lecithin, 30% butyl lactate and 23% water; Lipoid S75 and Phospholipoin 85 G; lipogel form and hydrogel microemulsion; | -skin penetration; -gel has better spreadability and demonstrates a 5.07-fold increase in the transdermal flux as it was compared to Carbomer® 934 gel; -lipogel LO1 demonstrated the ultimate permeability level (210.8 µg cm−2 h−1) and advanced percentage diflunisal permeated; -the in vivo antihyperalgesia assay showed significant reduction of the licking time in the treated group compared to the control group. | − | [36,37] |
| 6 | pH-Sensitive hydrogels based on bovine serum albumin hydrophilic microspheres | − | -release profile depends on diflunisal–polymer matrix interacting and diffusional restriction related to degree of crosslinking in the microparticles; -at pH 6.8, the diflunisal released amount increased (w/w > 75% after 24 h). | [38] |
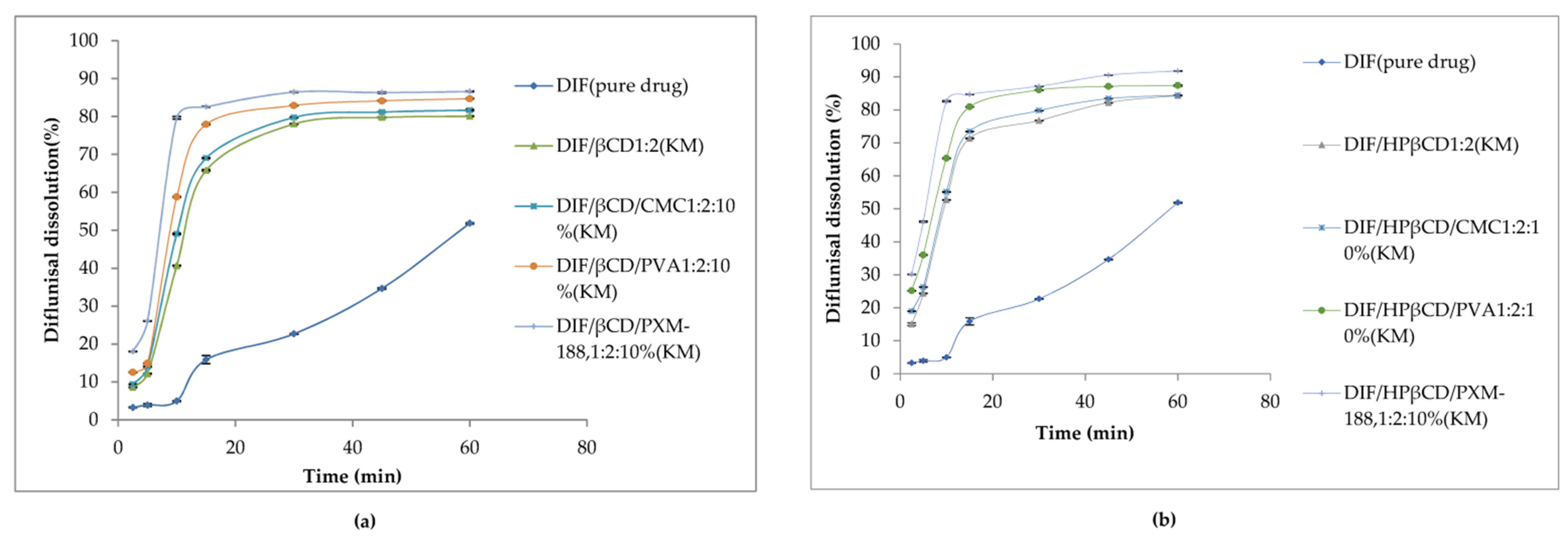
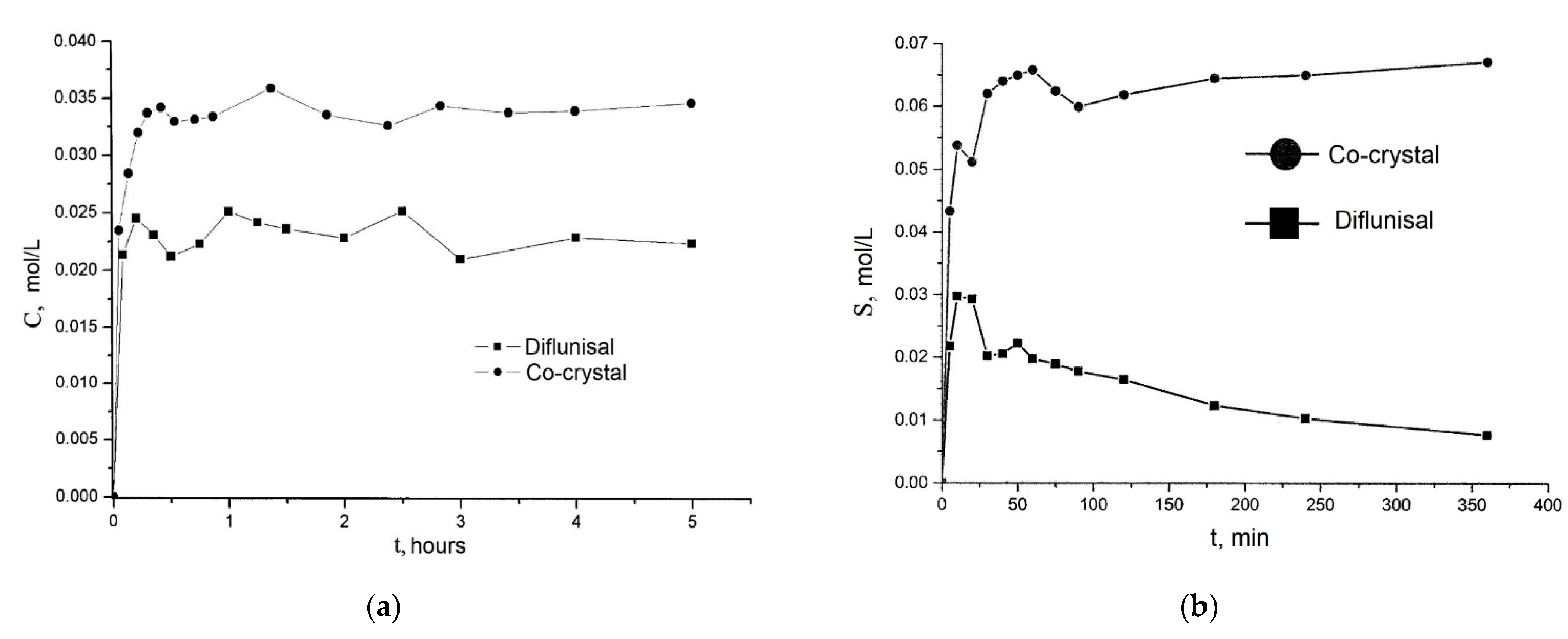
| 7 | β-Cyclodextrin (βCD), γ-cyclodextrin (γCD), and hydroxypropyl-β-cyclodextrin (HPβCD) | -hydrophilic polymers (carboxymethyl cellulose sodium, polyvinyl alcohol, and poloxamer-188 (PXM188)) were used, the effect of the polymer addition on the solubility and dissolution has been studied. -better solubility was for βCD and HPβCD inclusion complexes. -maximum of diflunisal solubility (1259.5 ± 0.5 µg/mL) was detected for the complex with hydroxypropyl β-cyclodextrin and Poloxamer-188. | -the diflunisal release from β-cyclodextrin and hydroxypropyl β-cyclodextrin complexes was higher than of pure diflunisal in 11–21 times; -hydrophilic polymers allow increasing the release rate of diflunisal in 15–28 times. | [45,46] |
| 8 | Cobalt(III)–polypyridyl complexes | -kill the cancer stem cells and the majority of bulk cancer cells even at low concentrations; -killing mechanism of cancer cells by composition number 5 includes the DNA damage and inhibition of COX-2; -against breast cancer cells HMLER and breast cancer stem cells-enriched HMLER-shEcad; -did not possess any toxicity toward normal skin fibroblast cells (line GM07575). | -differentially release the drug under acidic conditions; -complexes selectively release diflunisal/1,10-phenanthroline-bearing complex 5 displays selective potency toward hard-to-kill cancer stem cells (CSCs) (IC50 = 2.1 ± 0.1 μM) over bulk cancer (IC50 = 3.9 ± 0.2 μM) and normal cells (IC50 = 21.2 ± 1.3 μM). This complex induces CSC apoptosis by DNA damage and cyclooxygenase-2 inhibition. | [51] |
| 9 | Copper (II) complexes with O-donor ligand N,N-dimethylformamide or N-donor heterocyclic ligands (2,2′-bipyridine, 2,2′-bipyridylamine, 1,10-phenanthroline and pyridine) | -good binding ability to bovine and human serum as well as to calf–thymus DNA. | − | [52] |
| 10 | Poly(ethylene glycol) (PEG) | − | The better release was for the drug–polymer ratio of 1:7. | [64] |
| 11 | Eudragit RS100 and RL100 | − | Changes in the release profile, leading to slow and prolonged kinetic profile. | [66] |
| 12 | Eudragit RS100, S100, L100, and ethyl cellulose; pH-dependent, time dependent, and combined pH and time-dependent systems. | -the ratio 2:3:1 Eudragit S100/Eudragit RS100/diflunisal is the most successful; -colon-specific delivery for the treatment of a variety of diseases, such as nonspecific ulcerative colitis, cirrhosis disease, intestinal amoebiasis, colon tumor. | The release was 0.22 ± 0.03% of the drug included in it in the stomach pH and 26.29 ± 0.91% of the drug in the intestine pH and 77.59 ± 1.79% of the drug in the colon pH. | [67] |
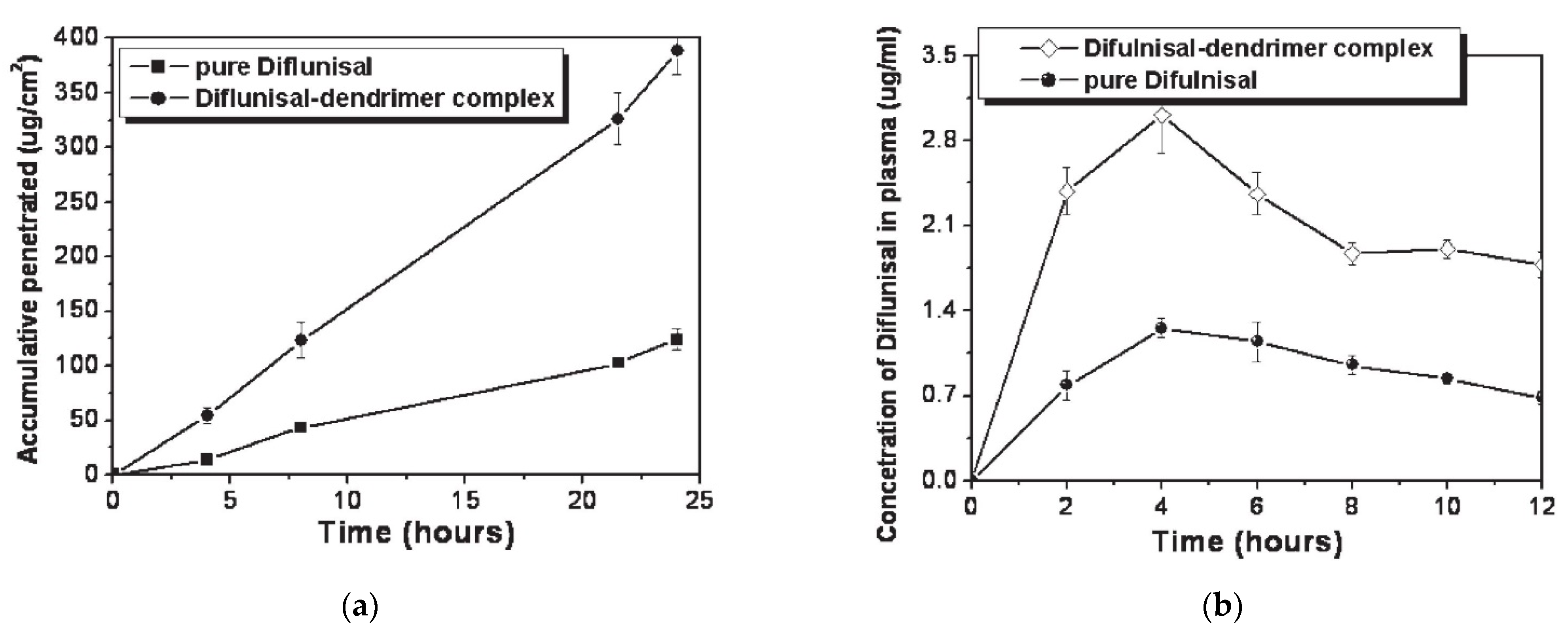
| 13 | Ethylenediamine (EDA) core polyamidoamine (PAMAM) dendrimers | -improve transdermal delivery of diflunisal and its pharmacokinetic and pharmacodynamics profiles; - enhance transdermal delivery of diflunisal; -diflunisal–PAMAM complexes lead to 2.48-fold increase in drug level. | − | [72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Snetkov, P.; Morozkina, S.; Olekhnovich, R.; Uspenskaya, M. Diflunisal Targeted Delivery Systems: A Review. Materials 2021, 14, 6687. https://doi.org/10.3390/ma14216687
Snetkov P, Morozkina S, Olekhnovich R, Uspenskaya M. Diflunisal Targeted Delivery Systems: A Review. Materials. 2021; 14(21):6687. https://doi.org/10.3390/ma14216687
Chicago/Turabian StyleSnetkov, Petr, Svetlana Morozkina, Roman Olekhnovich, and Mayya Uspenskaya. 2021. "Diflunisal Targeted Delivery Systems: A Review" Materials 14, no. 21: 6687. https://doi.org/10.3390/ma14216687
APA StyleSnetkov, P., Morozkina, S., Olekhnovich, R., & Uspenskaya, M. (2021). Diflunisal Targeted Delivery Systems: A Review. Materials, 14(21), 6687. https://doi.org/10.3390/ma14216687