
First Principles Calculations of Atomic and Electronic Structure of

 ,
, 

Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a0, b0, c0 (Å) | 5.179, 5.329, 7.370 | [50] |
| Y sites/cm3 | 1.97 × 1022 | |
| Bulk modulus, B (GPa) | 192 | [50] |
| Hardness (Knoop value) | 1310 | [13] |
| Melting point (°C) | 1875 | [13] |
| Thermal conductivity (W/cm-°C at 25°C) | 0.11 | [13] |
| Thermal expansion coefficient (10−6/°C, at 25°C) | 2.2 | [13] |
| Optical transmission (absorption coefficient < 1.0 cm−1) | 0.29–5.9 μ | [13] |
| Refractive index (6328 Å) (a = z, b = y, c = x) | na = 1.97 nb = 1.96 nc = 1.94 | [13] |
| Band gap energy, eV | 8.5–9.0 8.8 ± 0.2 | [8] [9] |
| Ti-ion related optical absorption bands (eV) | 7.08 (Ti3+-related) 5.76 (Ti4+-related) 5.27 (Ti4+-related) 5.39 (Ti3+ → Ti4+) 4.42 (Ti3+-related) 4.42 (2T2g → 4S, Ti3+) 4.20 (Ti4+ → Ti3+) 2.88 (2T2g → 2E, Ti3+) 2.87 (Ti3+) 2.81 (Ti3+) 2.53 (Ti3+) 2.52 (Ti3+) 2.50 (2T2g → 2E, Ti3+) | [9] [9] [9] [16] [9] [43] [9] [43] [16] [9] [9] [16] [43] |
| Ti-ion luminescence band | 3.02 (Ti4+) 2.10 (Ti3+, d-d transition) 2.06 (Ti3+, d-d transition) 1.99 eV (Ti3+, d-d transition) | [9] [43] [9] [16] |
| F center: optical absorption (eV) luminescence (eV) | 5.84 and 5.15 2.95 | [51] [51] |
| F+ center: optical absorption (eV)luminescence (eV) | 6.5, 5.63, and 4.3 3.49 | [51] [51] |
2. Computational Details
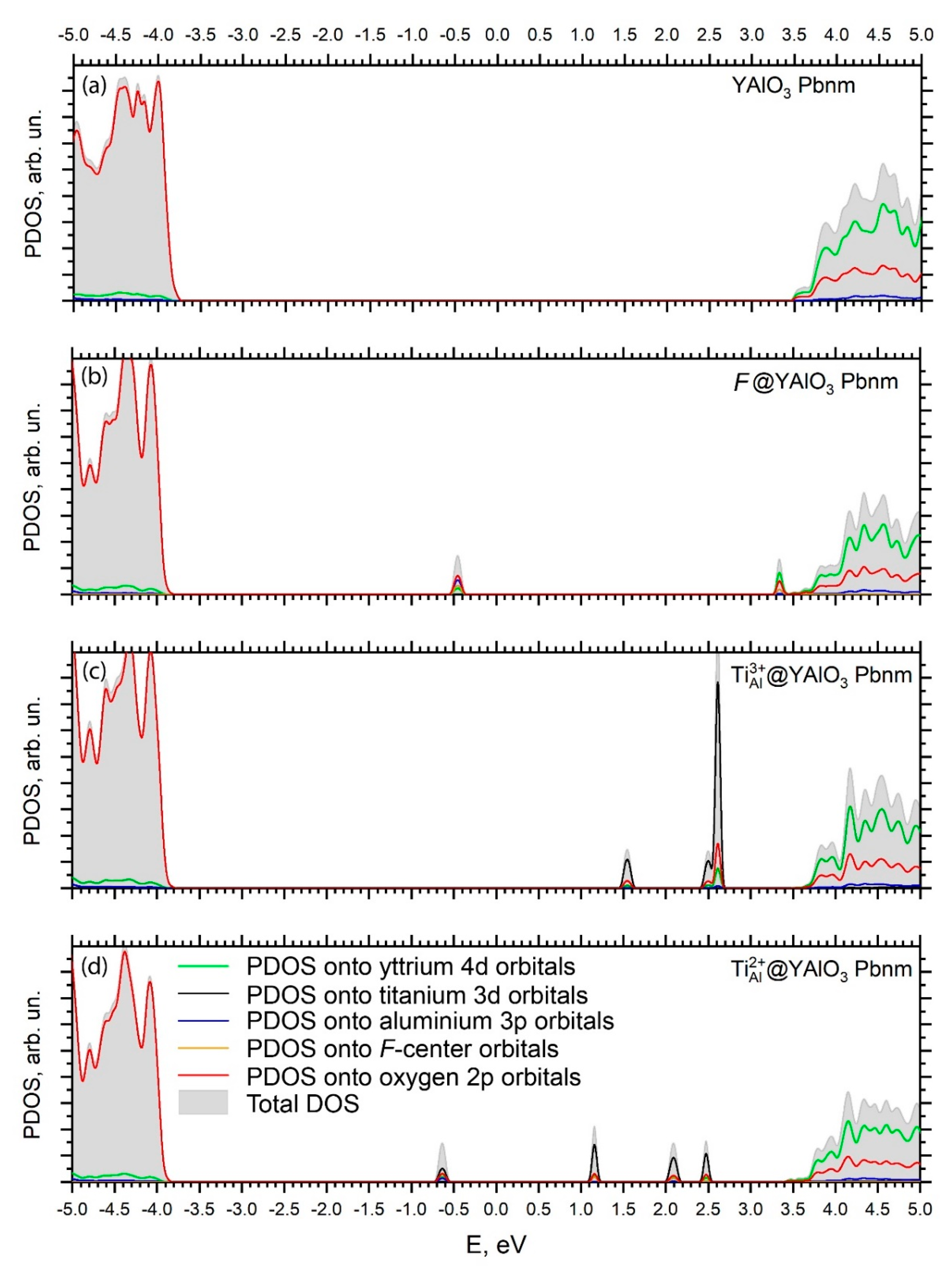
3. Results and Discussion
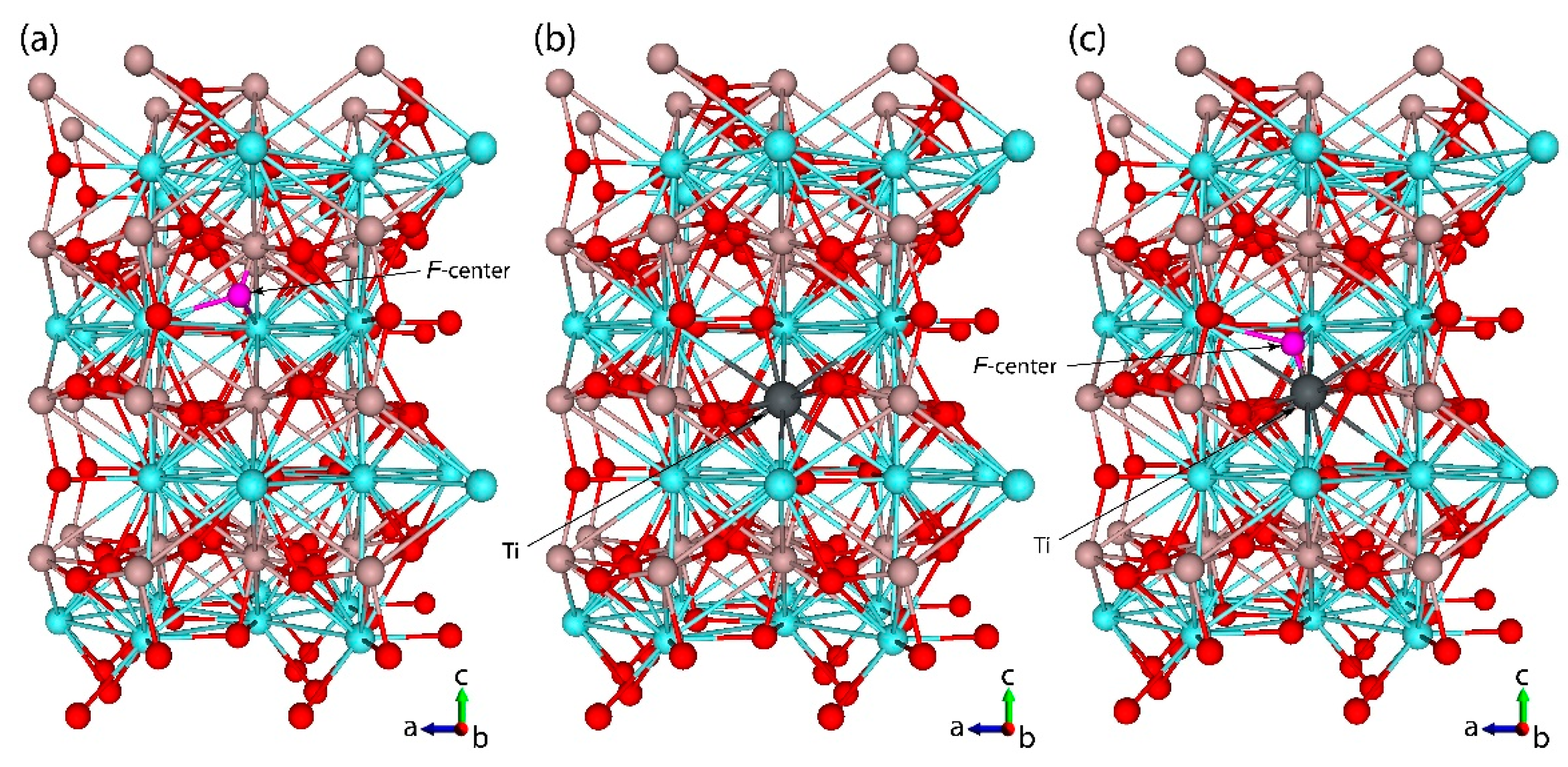
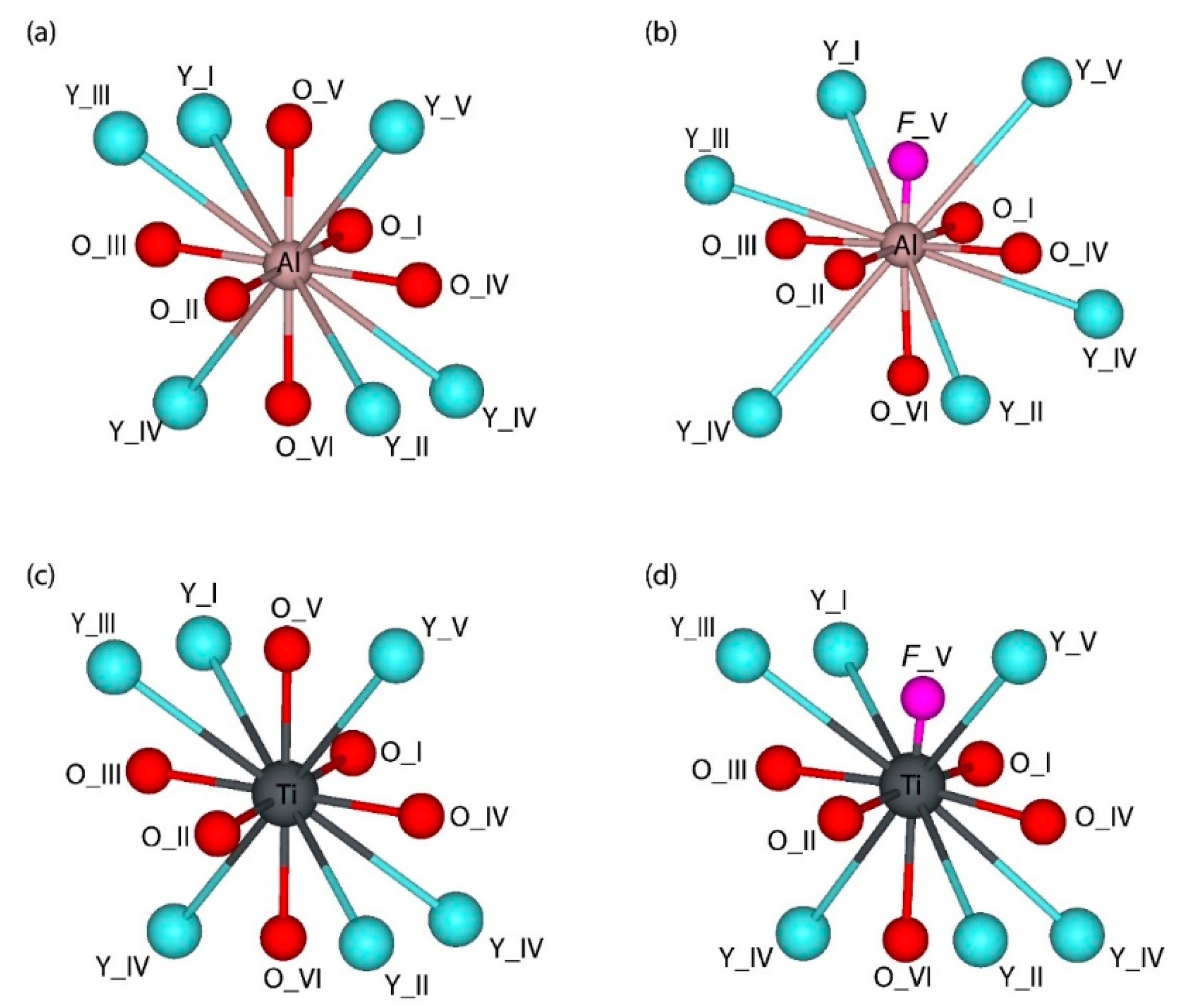
3.1. Structural Properties
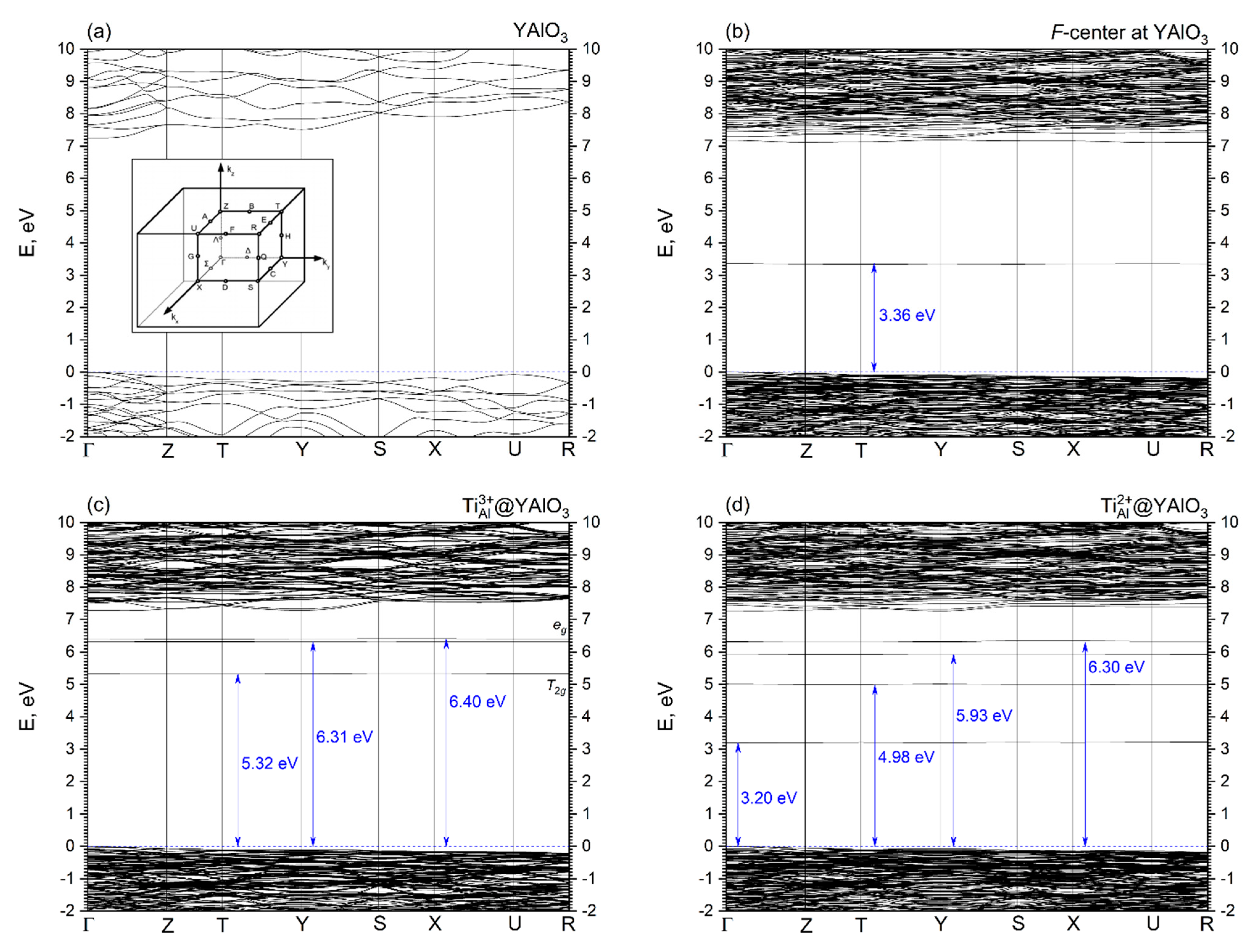
3.2. Electronic Properties
4. Conclusions
- (i)
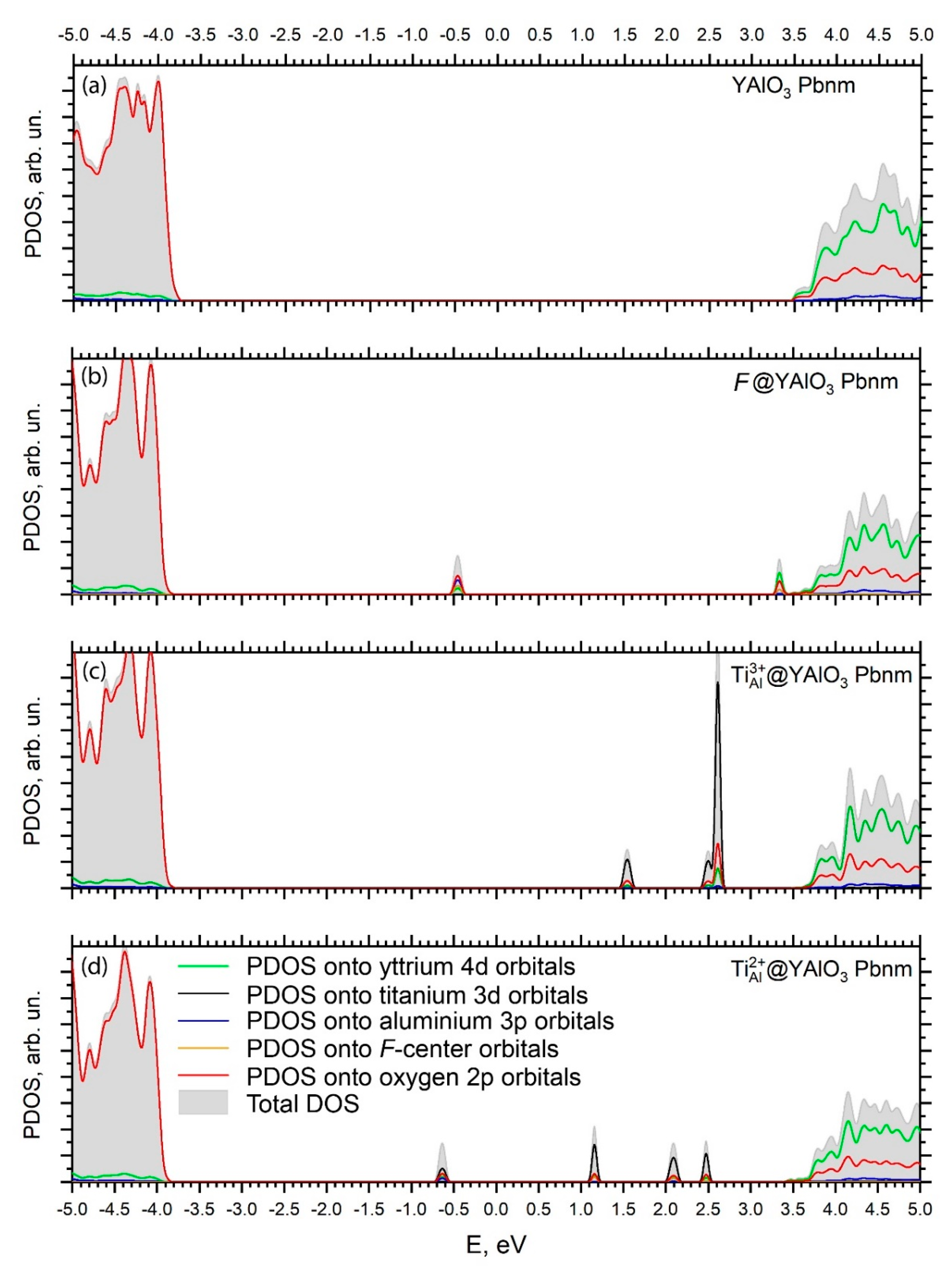
- Isovalent replacement of the Al3+ ion by the Ti3+ ion keeps the symmetry of the substitutional site; the only effect is a slight increase of the Ti–O distances as compared to the Al–O ones because of the difference in ionic radii of the Al3+ and Ti3+ ions. Our calculated position of the Ti3+ ground state in the YAP band gap (5.32 eV above the valence band top) agrees nicely with the value of about 5.21 eV derived by Rogers and Dorenbos in Ref. [77].
- (ii)
- Appearance of the oxygen vacancy or replacement of the Al3+ ion by the Ti2+ ion with simultaneous formation of the charge compensating defects lowers the symmetry of the substitutional site by modifying the angles between the chemical bonds in the AlO6 or TiO6 octahedra.
- (iii)
- Creation of the defects in the pristine YAlO3 structure leads to the formation of localized dispersionless defects’ energy levels in the host band gap. These levels are located in the central region of the band gap and closer to the CB bottom; they can significantly modify the host’s optical properties by causing additional defect-related absorption peaks in the optical spectra.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Przybylińska, H.; Ma, C.G.; Brik, M.G.; Kamińska, A.; Sybilski, P.; Wittlin, A.; Berkowski, M.; Zorenko, Y.; Gorbenko, V.; Wrzesinski, H.; et al. Electronic structure of Ce3+ multicenters in yttrium aluminum garnets. Appl. Phys. Lett. 2013, 102, 241112. [Google Scholar] [CrossRef] [Green Version]
- Polisadova, E.; Valiev, D.; Vaganov, V.; Oleshko, V.; Han, T.; Zhang, C.; Burachenko, A.; Popov, A.I. Time-resolved cathodoluminescence spectroscopy of YAG and YAG:Ce3+ phosphors. Opt. Mater. 2019, 96, 109289. [Google Scholar] [CrossRef]
- Karipbayev, Z.T.; Lisitsyn, V.M.; Mussakhanov, D.A.; Alpyssova, G.K.; Popov, A.I.; Polisadova, E.F.; Elsts, E.; Akilbekov, A.T.; Kukenova, A.B.; Kemere, M.; et al. Time-resolved luminescence of YAG:Ce and YAGG:Ce ceramics prepared by electron beam assisted synthesis. Nucl. Instrum. Methods B 2020, 479, 222–228. [Google Scholar] [CrossRef]
- Lisitsyn, V.M.; Tulegenova, A.T.; Lisitsyna, L.A.; Vaganov, V.A.; Soshchin, N.P.; Polisadova, E.F.; Abdullin, K.A.; Yangyang, J. Photo and cathodoluminescence of commercial YAG:Ce based phosphors in UV region. Nucl. Instrum. Methods B 2020, 478, 120–124. [Google Scholar] [CrossRef]
- Wang, Y.; Hrubiak, R.; Turczyński, S.; Pawlak, D.A.; Malinowski, M.; Włodarczyk, D.; Kosyl, K.M.; Paszkowicz, W.; Przyybylińska, H.; Wittlin, A.; et al. Spectroscopic properties and martensitic phase transition of Y4Al2O9: Ce single crystals under high pressure. Acta Mater. 2019, 165, 346–361. [Google Scholar] [CrossRef]
- Fetliński, B.; Turczyński, S.; Malinowski, M.; Szczepański, P. Down-Shifting in the YAM:Ce3++ Yb3+ System for Solar Cells. Materials 2021, 14, 2753. [Google Scholar] [CrossRef]
- Gomes, P.F.; Silva, T.C.; Maia, L.J.; Carvalho, J.F. Synthesis and visible down-and up-conversion emissions from Yb3+/Ho3+/Tm3+ Co-Doped Y4Al2O9 (YAM) nanocrystalline particles. J. Lumin. 2020, 227, 117554. [Google Scholar] [CrossRef]
- Lushchik, C.; Feldbach, E.; Frorip, A.; Kirm, M.; Lushchik, A.; Maaroos, A.; Martinson, I. Multiplication of electronic excitations in CaO and YAlO3 crystals with free and self-trapped excitons. J. Phys. Condens. Matter 1994, 6, 11177. [Google Scholar] [CrossRef]
- Basun, S.A.; Danger, T.; Kaplyanskii, A.A.; McClure, D.S.; Petermann, K.; Wong, W.C. Optical and photoelectrical studies of charge-transfer processes in YAlO3: Ti crystals. Phys. Rev. B 1996, 54, 6141. [Google Scholar] [CrossRef]
- Shimizu, Y.; Ueda, K.; Inaguma, Y. Photoluminescence excitation spectra of lanthanide doped YAlO3 in vacuum ultraviolet region. Opt. Mater. 2017, 66, 327–331. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Popov, A.I. Comparative Ab initio calculations for ABO3 perovskite (001), (011) and (111) as well as YAlO3 (001) surfaces and F centers. J. Nano-Electron. Phys. 2019, 11, 01001. [Google Scholar] [CrossRef] [Green Version]
- Zorenko, Y.V.; Voloshinovskiĭ, A.S.; Konstankevych, I.V. Luminescence of F+ and F centers in YAlO3. Opt. Spectrosc. 2004, 96, 532–537. [Google Scholar] [CrossRef]
- Weber, M.J.; Bass, M.; Andringa, K.; Monchamp, R.R.; Comperchio, E. Czochralski growth and properties of YAlO3 laser crystals. Appl. Phys. Lett. 1969, 15, 342–345. [Google Scholar] [CrossRef]
- Massey, G.A. Criterion for selection of cw laser host materials to increase available power in the fundamental mode. Appl. Phys. Lett. 1970, 17, 213–215. [Google Scholar] [CrossRef]
- Yamaga, M.; Henderson, B.; O’Donnell, K.P.; Rasheed, F.; Gao, Y.; Cockayne, B. Polarization of emission spectra from Ti3+-doped oxide crystals-II. Zero-phonon emission lines. Appl. Phys. B 1991, 52, 225–229. [Google Scholar] [CrossRef]
- Wegner, T.; Petermann, K. Excited state absorption of Ti3+: YAlO3⋆. Appl. Phys. B 1989, 49, 275–278. [Google Scholar] [CrossRef]
- Chosrovian, H.; Rentsch, S.; Stösser, R. ESR investigations and mechanisms of light-induced optical absorption in Ti3+: YAlO3 crystals. Phys. Status Solidi A 1992, 130, 463–473. [Google Scholar] [CrossRef]
- Moore, M.E.; Delzer, C.; Watts, J.; Musicó, B.L.; Xu, C.; Collette, R.M.; Rack, P.D.; Zhang, Y.; Melcher, C.L.; McConchie, S.; et al. Studying the effects of thermally diffusing Ce into the surface of YAlO3 for associated particle imaging. Nucl. Instrum. Methods B 2020, 473, 55–61. [Google Scholar] [CrossRef]
- Nakanishi, K.; Yamamoto, S.; Kamada, K.; Yoshikawa, A. Performance evaluation of YAlO3 scintillator plates with different Ce concentrations. Appl. Radiat. Isot. 2021, 168, 109483. [Google Scholar] [CrossRef] [PubMed]
- Dorenbos, P. Fundamental Limitations in the Performance of Ce3+–, Pr3+–, and Eu2+–Activated Scintillators. IEEE Trans. Nucl. Sci. 2010, 57, 1162–1167. [Google Scholar] [CrossRef]
- Baryshevsky, V.G.; Korzhik, M.V.; Moroz, V.I.; Pavlenko, V.B.; Fyodorov, A.A.; Smirnova, S.A.; Egorycheva, O.A.; Kachanov, V.A. YAlO3:Ce-fast-acting scintillators for detection of ionizing radiation. Nucl. Instrum. Methods B 1991, 58, 291–293. [Google Scholar] [CrossRef]
- Vedda, A.; Martini, M.; Meinardi, F.; Chval, J.; Dusek, M.; Mares, J.A.; Mihokova, E.; Nikl, M. Tunneling process in thermally stimulated luminescence of mixed LuxY1−xAlO3:Ce crystals. Phys. Rev. B 2000, 61, 8081. [Google Scholar] [CrossRef]
- Zorenko, Y.; Gorbenko, V.; Konstankevych, I.; Voznjak, T.; Savchyn, V.; Nikl, M.; Mares, J.A.; Nejezchleb, K.; Mikhailin, V.; Kolobanov, V.; et al. Peculiarities of luminescence and scintillation properties of YAP:Ce and LuAP:Ce single crystals and single crystalline films. Radiat. Meas. 2007, 42, 528–532. [Google Scholar] [CrossRef]
- De Queiroz, T.B.; Ferrari, C.R.; Ulbrich, D.; Doyle, R.; De Camargo, A.S.S. Luminescence characteristics of YAP:Ce scintillator powders and composites. Opt. Mater. 2010, 32, 1480–1484. [Google Scholar] [CrossRef]
- Zhuravleva, M.; Novoselov, A.; Yoshikawa, A.; Pejchal, J.; Nikl, M.; Fukuda, T. Crystal growth and scintillation properties of Pr-doped YAlO3. Opt. Mater. 2007, 30, 171–173. [Google Scholar] [CrossRef]
- Gieszczyk, W.; Mrozik, A.; Bilski, P.; Vistovskyy, V.; Voloshinovskii, A.; Paprocki, K.; Zorenko, T.; Zorenko, Y. Scintillation and Energy-Storage Properties of Micro-Pulling-Down Grown Crystals of Sc3+-and La3+-Doped YAlO3 Perovskite. Crystals 2020, 10, 385. [Google Scholar] [CrossRef]
- Rathaiah, M.; Kucera, M.; Pejchal, J.; Beitlerova, A.; Kucerkova, R.; Nikl, M. Epitaxial growth, photoluminescence and scintillation properties of Gd3+ co-doped YAlO3:Ce3+ films. Radiat. Meas. 2019, 121, 86–90. [Google Scholar] [CrossRef]
- Sreebunpeng, K.; Janthon, P.; Chewpraditkul, W.; Szczesniak, T.; Nikl, M.; Yoshikawa, A. Scintillation characteristics of YAlO3:Pr perovskite single crystals. Opt. Mater. 2020, 108, 110161. [Google Scholar] [CrossRef]
- Zhydachevskii, Y.; Durygin, A.; Suchocki, A.; Matkovskii, A.; Sugak, D.; Bilski, P.; Warchol, S. Mn-doped YAlO3 crystal: A new potential TLD phosphor. Nucl. Instrum. Methods B 2005, 227, 545–550. [Google Scholar] [CrossRef]
- Zhydachevskii, Y.; Durygin, A.; Suchocki, A.; Matkovskii, A.; Sugak, D.; Loutts, G.B.; Noginov, M.A. Radiation and thermally induced effects in YAlO3:Mn crystals. J. Lumin. 2004, 109, 39–49. [Google Scholar] [CrossRef]
- Marciniak, L.; Trejgis, K. Luminescence lifetime thermometry with Mn3+–Mn4+ co-doped nanocrystals. J. Mater. Chem. C 2018, 6, 7092–7100. [Google Scholar] [CrossRef]
- Zhydachevskyy, Y.; Martynyuk, N.; Popov, A.I.; Sugak, D.; Bilski, P.; Ubizskii, S.; Berkowski, M.; Suchocki, A. Thermally induced fading of Mn-doped YAP nanoceramics. J. Phys. Conf. Ser. 2018, 987, 012009. [Google Scholar] [CrossRef]
- Afanassyev, D.; Ubizskii, S.; Zhydachevskyy, Y.; Luchechko, A.; Popov, A.I.; Suchocki, A. Time-resolved pulsed OSL of ceramic YAP:Mn phosphors. Integr. Ferroelectr. 2019, 196, 24–31. [Google Scholar] [CrossRef]
- Piskunov, S.; Isakoviča, I.; Popov, A.I. Atomic structure of manganese-doped yttrium orthoaluminate. Nucl. Instrum. Methods B 2018, 434, 6–8. [Google Scholar] [CrossRef]
- Kück, S. Laser-related spectroscopy of ion-doped crystals for tunable solid-state lasers. Appl. Phys. B 2001, 72, 515–562. [Google Scholar] [CrossRef]
- Liu, B.; Wang, Q.; Xu, X.; Xu, J.; Zhu, F.; Zhou, H. Polarized spectral properties of Sm: YAlO3 single crystal for reddish-orange laser. Opt. Mater. 2020, 99, 109510. [Google Scholar] [CrossRef]
- Shimizu, Y.; Ueda, K. Ultraviolet emission from YAlO3:Gd3+ thin film electroluminescent devices fabricated on perovskite-type oxide substrates. Opt. Mater. 2019, 91, 371–375. [Google Scholar] [CrossRef]
- Brik, M.G.; Sildos, I.; Berkowski, M.; Suchocki, A. Spectroscopic and crystal field studies of YAlO3 single crystals doped with Mn ions. J. Phys. Condens. Matter 2008, 21, 025404. [Google Scholar] [CrossRef]
- Wang, Y.; Włodarczyk, D.; Li, L.; Wittlin, A.; Przybylińska, H.; Sybilski, P.; Zhydachevskii, Y.; Ma, C.-G.; Brik, M.G.; Malinowski, M.; et al. Electronic structure of Ce3+ in yttrium and lutetium orthoaluminate crystals and single crystal layers. J. Alloy. Compd. 2017, 723, 157–163. [Google Scholar] [CrossRef]
- Brik, M.G.; Sildos, I.; Kiisk, V. Calculations of physical properties of pure and doped crystals: Ab initio and semi-empirical methods in application to YAlO3:Ce3+ and TiO2. J. Lumin. 2011, 131, 396–403. [Google Scholar] [CrossRef]
- Brik, M.G.; Ma, C.G.; Piasecki, M.; Suchocki, A. Locating impurity and defect levels in the host band gap by first-principles calculations: Pure and Ce3+-doped YAlO3. Opt. Mater. 2021, 113, 110843. [Google Scholar] [CrossRef]
- Danger, T.; Petermann, K.; Schwentner, N.; Sliwinski, G.; Wong, W.C. UV-spectroscopy and band structure of Ti:YAlO3. J. Lumin. 1997, 72, 171–173. [Google Scholar] [CrossRef]
- Baryshnikov, V.I.; Kvapil, J.; Sarukura, N.; Segawa, Y. Especialities of Ti3+ excitation in YAlO3 crystals. J. Lumin. 1997, 72, 157–158. [Google Scholar] [CrossRef]
- Grunwaldt, J.D.; Gunβer, W.; Danger, T.; Petermann, K.; Reller, A. Determination of a new titanium site in Ti:YAlO3. J. Phys. Chem. Solids 1994, 55, 699–705. [Google Scholar] [CrossRef]
- Ando, M.; Sakaguchi, T.; Yamanaka, A.; Kawabe, Y.; Hanamura, E. Electroluminescence of Oxygen-Deficient YAlO3 Crystals with Dopants. Jpn. J. Appl. Phys. 2006, 45, 3659. [Google Scholar] [CrossRef]
- Lin, J.Z.; Wu, S.Y.; Fu, Q.; Zhang, H.M. Studies of the local structure and the g factors for the orthorhombic Ti3+ center in YAP. Mod. Phys. Lett. B 2007, 21, 737–743. [Google Scholar] [CrossRef]
- van Vuuren, A.J.; Saifulin, M.M.; Skuratov, V.A.; O’Connell, J.H.; Aralbayeva, G.; Dauletbekova, A.; Zdorovets, M. The influence of stopping power and temperature on latent track formation in YAP and YAG. Nucl. Instrum. Methods B 2019, 460, 67–73. [Google Scholar] [CrossRef]
- Piskunov, S.; Isakoviča, I.; Popov, A.I. Electronic structure of MnAl3+-and MnAl2+-doped YAlO3: Prediction from the first principles. Opt. Mater. 2018, 85, 162–166. [Google Scholar] [CrossRef]
- Piskunov, S.; Isakoviča, I.; Putnina, M.; Popov, A.I. Ab initio calculations of the electronic structure for Mn2+-doped YAlO3 crystals. Low Temp. Phys. 2020, 46, 1160–1164. [Google Scholar] [CrossRef]
- Ross, N.L.; Zhao, J.; Angel, R.J. High-pressure single-crystal X-ray diffraction study of YAlO3 perovskite. J. Solid State Chem. 2004, 177, 1276–1284. [Google Scholar] [CrossRef]
- Popov, A.I.; Kotomin, E.A.; Maier, J. Basic properties of the F-type centers in halides, oxides and perovskites. Nucl. Instrum. Methods B 2010, 268, 3084–3089. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef] [PubMed]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; et al. Crystal’17 User’s Manual. 2018. Available online: https://www.crystal.unito.it/manuals/crystal17.pdf (accessed on 24 August 2021).
- Peintinger, M.F.; Oliveira, D.V.; Bredow, T. Consistent Gaussian basis sets of triple-zeta valence with polarization quality for solid-state calculations. J. Comput. Chem. 2013, 34, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef] [Green Version]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. II. Overlap populations, bond orders, and covalent bond energies. J. Chem. Phys. 1955, 23, 1841–1846. [Google Scholar] [CrossRef]
- Ross, N.L. Distortion of GdFeO3-type perovskites with pressure: A study of YAlO3 to 5 GPa. Phase Transit. 1996, 58, 27–41. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Ning, L.; Cheng, W.; Zhou, C.; Duan, C.; Zhang, Y. Energetic, Optical, and Electronic Properties of Intrinsic Electron-Trapping Defects in YAlO3: A Hybrid DFT Study. J. Physiclal Chem. C 2014, 118, 19940–19947. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Nikiforov, S.V.; Kortov, V.S.; Petrov, M.O. Luminescent and dosimetric properties of ultrafine magnesium oxide ceramics after high dose irradiation. Radiat. Meas. 2016, 90, 252–256. [Google Scholar] [CrossRef]
- Kortov, V.S.; Milman, I.I.; Moiseykin, E.V.; Nikiforov, S.V.; Ovchinnikov, M.M. Deep-trap competition model for TL in α-Al2O3:C heating stage. Radiat. Prot. Dosim. 2006, 119, 41–44. [Google Scholar] [CrossRef]
- Gonzalez, R.; Monge, M.A.; Santiuste, J.E.M.; Pareja, R.; Chen, Y.; Kotomin, E.; Kukla, M.M.; Popov, A.I. Photoconversion of F-type centers in thermochemically reduced MgO single crystals. Phys. Rev. B 1999, 59, 4786–4790. [Google Scholar] [CrossRef] [Green Version]
- Kotomin, E.A.; Popov, A.I.; Stashans, A.A. Novel model for F+ to F photoconversion in corundum crystals. J. Phys. Condens. Matter 1994, 6, L569–L573. [Google Scholar] [CrossRef]
- Usseinov, A.B.; Zhukovskii, Y.F.; Kotomin, E.A.; Akilbekov, A.T.; Zdorovets, M.V.; Baubekova, G.M.; Karipbayev, Z.T. Transition levels of acceptor impurities in ZnO crystals by DFT-LCAO calculations. J. Phys. Conf. Ser. 2018, 1115, 042064. [Google Scholar] [CrossRef]
- Uklein, A.V.; Multian, V.V.; Kuz’micheva, G.M.; Linnik, R.P.; Lisnyak, V.V.; Popov, A.I.; Gayvoronsky, V.Y. Nonlinear optical response of bulk ZnO crystals with different content of intrinsic defects. Opt. Mater. 2018, 84, 738–747. [Google Scholar] [CrossRef]
- Osinkin, D.A.; Khodimchuk, A.V.; Porotnikova, N.M.; Bogdanovich, N.M.; Fetisov, A.V.; Ananyev, M.V. Rate-Determining Steps of Oxygen Surface Exchange Kinetics on Sr2Fe1.5Mo0.5O6−δ. Energies 2020, 13, 250. [Google Scholar] [CrossRef] [Green Version]
- Porotnikova, N.M.; Eremin, V.A.; Farlenkov, A.S.; Kurumchin, E.K.; Sherstobitova, E.A.; Kochubey, D.I.; Ananyev, M.V. Effect of AO Segregation on Catalytical Activity of La0.7A0.3MnO3±δ (A = Ca, Sr, Ba) Regarding Oxygen Reduction Reaction. Catal. Lett. 2018, 148, 2839–2847. [Google Scholar] [CrossRef]
- Kotomin, E.A.; Piskunov, S.; Zhukovskii, Y.F.; Eglitis, R.I.; Gopejenko, A.; Ellis, D.E. The electronic properties of an oxygen vacancy at ZrO2-terminated (001) surfaces of a cubic PbZrO3: Computer simulations from the first principles. Phys. Chem. Chem. Phys. 2008, 10, 4258–4263. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, J.; Illas, F.; Lopez, N.; Kotomin, E.A.; Zhukovskii, Y.F.; Piskunov, S.; Maier, J.; Hermansson, K. First principles simulations of F centers in cubic SrTiO3. Phys. Status Solidi C 2005, 2, 153–158. [Google Scholar] [CrossRef]
- Zhumatayeva, I.Z.; Kenzhina, I.E.; Kozlovskiy, A.L.; Zdorovets, M.V. The study of the prospects for the use of Li0. 15Sr0. 85TiO3 ceramics. J. Mater. Sci. Mater. Electron. 2020, 31, 6764–6772. [Google Scholar] [CrossRef]
- Kozlovskiy, A.L.; Kenzhina, I.E.; Zdorovets, M.V.; Saiymova, M.; Tishkevich, D.I.; Trukhanov, S.V.; Trukhanov, A.V. Synthesis, phase composition and structural and conductive properties of ferroelectric microparticles based on ATiOx (A = Ba, Ca, Sr). Ceram. Int. 2019, 45, 17236–17242. [Google Scholar] [CrossRef]
- Kato, S.; Nakajima, N.; Yasui, S.; Yasuhara, S.; Fu, D.; Adachi, J.; Nitani, H.; Takeichi, Y.; Anspoks, A. Dielectric response of BaTiO3 electronic states under AC fields via microsecond time-resolved X-ray absorption spectroscopy. Acta Materialia 2021, 207, 116681. [Google Scholar] [CrossRef]
- Alekseev, V.; Liber, V.; Starikov, A.; Anspoks, A.; Auzins, E.; Klotins, E.; Kotleris, J. High efficiency angular deflection of the laser beam: PLZT/intracavity array. Ferroelectrics 1992, 131, 301–306. [Google Scholar] [CrossRef]
- Anspoks, A.; Bocharov, D.; Purans, J.; Rocca, F.; Sarakovskis, A.; Trepakov, V.; Dejneka, A.; Itoh, M. Local structure studies of SrTi16O3 and SrTi18O3. Phys. Scr. 2014, 89, 044002. [Google Scholar] [CrossRef]
- Rogers, E.G.; Dorenbos, P. Vacuum energy referred Ti3+/4+ donor/acceptor states in insulating and semiconducting inorganic compounds. J. Lumin. 2014, 153, 40–45. [Google Scholar] [CrossRef]
| Lattice Constants and Volume | Experiment, Ref. [50] | Calc. (HSE06), Ref [60] | This Study | ||||||
|---|---|---|---|---|---|---|---|---|---|
| a0, Å | 5.179 | 5.179 | 5.189 | ||||||
| b0, Å | 5.329 | 5.342 | 5.317 | ||||||
| c0, Å | 7.370 | 7.367 | 7.388 | ||||||
| V, Å3 | 203.49 | 203.82 | 203.86 | ||||||
| Fractional Coordinates (In Units of the Lattice Constants) | |||||||||
| Atoms: | x | y | z | x | y | z | x | y | z |
| Y | −0.012 | 0.053 | 0.25 | −0.012 | 0.055 | 0.25 | −0.011 | 0.050 | 0.25 |
| Al | 0 | 0.5 | 0 | 0 | 0.5 | 0 | 0 | 0.5 | 0 |
| O1 | 0.084 | 0.477 | 0.25 | 0.084 | 0.478 | 0.25 | 0.082 | 0.479 | 0.25 |
| O2 | 0.705 | 0.295 | 0.044 | 0.706 | 0.294 | 0.042 | 0.708 | 0.292 | 0.044 |
| YAlO3 | YAlO3:F | YAlO3:Ti3+ | YAlO3:Ti2+ | |
|---|---|---|---|---|
| lAl,Ti-O_I | 1.92 | 1.92 | 2.01 | 1.94 |
| lAl,Ti-O_II | 1.92 | 1.92 | 2.01 | 1.96 |
| lAl,Ti-O_III | 1.90 | 1.90 | 1.99 | 2.00 |
| lAl,Ti-O_IV | 1.90 | 1.90 | 1.99 | 2.00 |
| lAl,Ti-O_V,F | 1.90 | 1.22 | 1.97 | 1.21 |
| lAl,Ti-O_VI | 1.90 | 1.92 | 1.97 | 2.08 |
| lAl,Ti-Y_I | 3.02 | 2.98 | 3.02 | 2.98 |
| lAl,Ti-Y_II | 3.02 | 3.04 | 3.02 | 3.10 |
| lAl,Ti-Y_III | 3.15 | 3.12 | 3.17 | 3.15 |
| lAl,Ti-Y_IV | 3.15 | 3.16 | 3.17 | 3.25 |
| lAl,Ti-Y_V | 3.24 | 3.17 | 3.30 | 3.14 |
| lAl,Ti-Y_VI | 3.24 | 3.26 | 3.30 | 3.33 |
| α0_I-Al,Ti-O_II | 180.0 | 178.6 | 180.0 | 175.4 |
| α0_III-Al,Ti-O_IV | 180.0 | 178.5 | 180.0 | 172.8 |
| α0_V,F-Al,Ti-O_VI | 180.0 | 174.4 | 180.0 | 174.7 |
| αY_I-Al,Ti-Y_II | 180.0 | 178.9 | 180.0 | 175.4 |
| αY_III-Al,Ti-Y_IV | 180.0 | 179.0 | 180.0 | 175.8 |
| αY_V-Al,Ti-Y_VI | 180.0 | 179.4 | 180.0 | 176.8 |
| YAlO3 | YAlO3:F | YAlO3:Ti3+ | YAlO3:Ti2+ | |
|---|---|---|---|---|
| QY_I | 2.46 | 2.42 | 2.45 | 2.40 |
| QY_II | 2.46 | 2.46 | 2.45 | 2.46 |
| QY_III | 2.46 | 2.45 | 2.45 | 2.45 |
| QY_IV | 2.46 | 2.46 | 2.45 | 2.46 |
| QY_V | 2.46 | 2.45 | 2.46 | 2.43 |
| QY_VI | 2.46 | 2.46 | 2.46 | 2.46 |
| QAl,Ti | 1.77 | 1.26 | 1.56 | 1.07 |
| QO_I | −1.42 | −1.44 | −1.39 | −1.35 |
| QO_II | −1.42 | −1.43 | −1.39 | −1.37 |
| QO_III | −1.40 | −1.43 | −1.37 | −1.38 |
| QO_IV | −1.40 | −1.44 | −1.37 | −1.39 |
| QO_V,F | −1.40 | −0.53 | −1.35 | −0.66 |
| QO_VI | −1.40 | −1.41 | −1.35 | −1.38 |
| PAl,Ti-O_I | 0.28 | 0.38 | 0.16 | 0.11 |
| PAl,Ti-O_II | 0.28 | 0.37 | 0.16 | 0.15 |
| PAl,Ti-O_III | 0.27 | 0.34 | 0.15 | 0.05 |
| PAl,Ti-O_IV | 0.27 | 0.35 | 0.15 | 0.09 |
| PAl,Ti-O_V,F | 0.27 | −0.62 | 0.16 | −1.44 |
| PAl,Ti-O_VI | 0.27 | 0.18 | 0.16 | −0.24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piskunov, S.; Gopejenko, A.; Pankratov, V.; Isakoviča, I.; Ma, C.-G.; Brik, M.G.; Piasecki, M.; Popov, A.I.
First Principles Calculations of Atomic and Electronic Structure of
Piskunov S, Gopejenko A, Pankratov V, Isakoviča I, Ma C-G, Brik MG, Piasecki M, Popov AI.
First Principles Calculations of Atomic and Electronic Structure of
Piskunov, Sergei, Aleksejs Gopejenko, Vladimir Pankratov, Inta Isakoviča, Chong-Geng Ma, Mikhail G. Brik, Michal Piasecki, and Anatoli I. Popov.
2021. "First Principles Calculations of Atomic and Electronic Structure of
Piskunov, S., Gopejenko, A., Pankratov, V., Isakoviča, I., Ma, C.-G., Brik, M. G., Piasecki, M., & Popov, A. I.
(2021). First Principles Calculations of Atomic and Electronic Structure of