
Acenaphthoquinoxaline Derivatives as Dental Photoinitiators of Acrylates Polymerization


Abstract
:
1. Introduction
- diffusion of the radical-ion pair.
- intramolecular proton transfer of the radical-ion pair that is retained in the solvent cage.
- monomers and polyfunctional monomers, e.g., trimethylolpropane triacrylate (TMPTA), pentaerythritol triacrylate (PETA), 1,6-hexanediol diacrylate (HDDA), bisphenol A diacrylate (bis DMA);
- a photoinitiator, e.g., camphorquinone;
- a co-initiator, usually aromatic amines, e.g., N,N-dimethyl-p-toluidine;
- reinforcing fillers, e.g., quartz, silicate glass, silicon nitrides, etc., coated with 3-(trimethoxysilanyl)propan-1-ol acrylate. This additive is used to improve the adhesion of the filler to the polymer matrix;
- inhibitors that prevent premature polymerization during storage;
- light stabilizers that prevent the filling from changing color;
- compounds that allow to match the color of the filling to the natural color of the patient’s teeth.
2. Materials and Methods
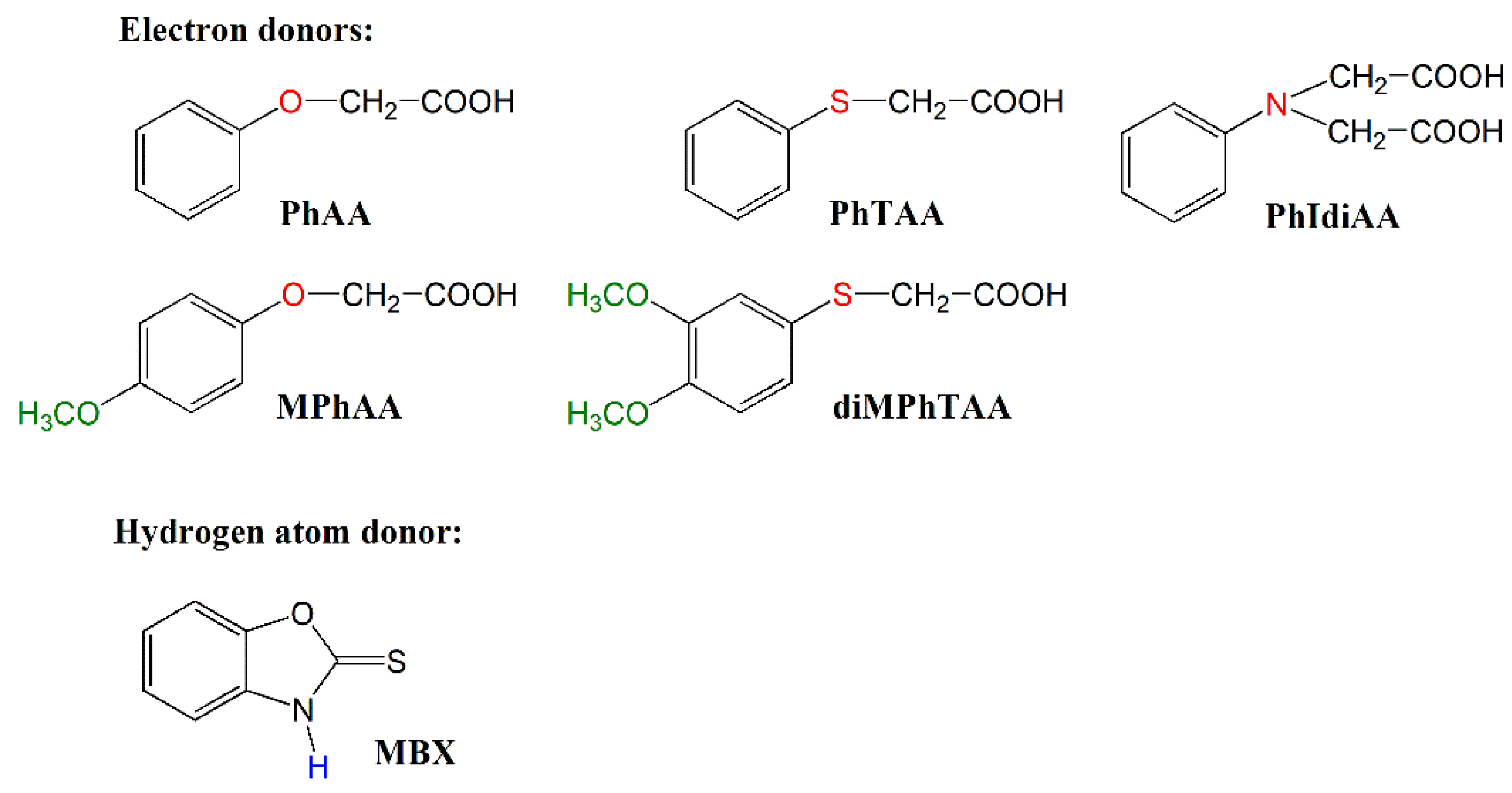
2.1. Reagents
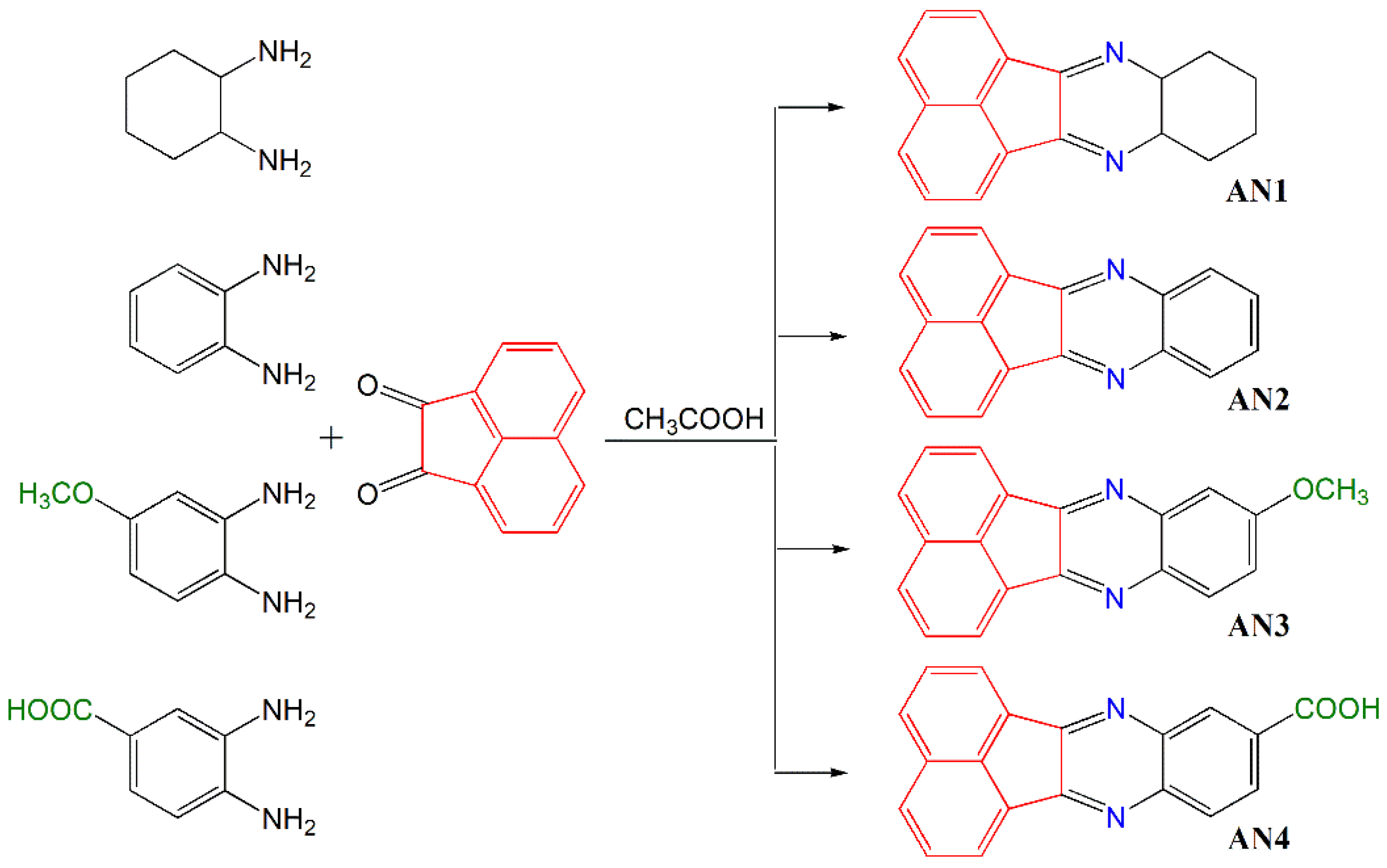
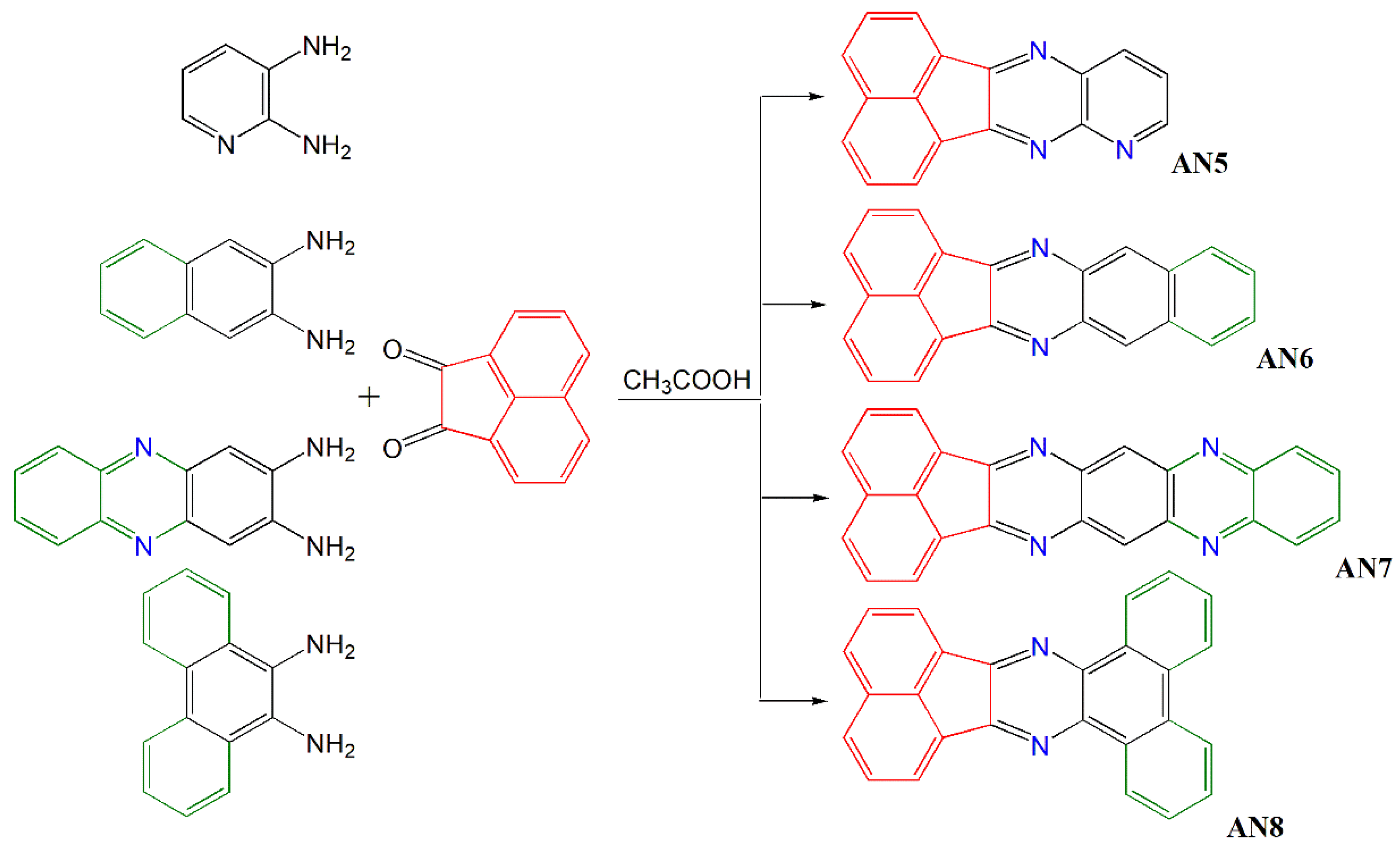
2.2. General Procedure for Synthesis of Acenaphthoquinoxaline Derivatives
- 8,9,10,11-Tetrahydroacenaphto[1,2-b]quinoxaline—AN1
- Acenaphto[1,2-b]quinoxaline—AN2
- 9-Methoxyacenaphto[1,2-b]quinoxaline—AN3
- Acenaphto[1,2-b]quinoxaline-9-carboxylic acid—AN4
- Acenaphto[1,2-b]pirydo[2,3-e]pyrazine—AN5
- Acenaphto[1,2-b]benzo[g]quinoxaline—AN6
- Acenaphto[1′,2′:5,6]pyrazine[2,3-b]phenazine—AN7
- Dibenzoacenaphto[1,2-b]quinoxaline—AN8
2.3. Methods
3. Results
3.1. Molecular Design and Synthetic Procedures
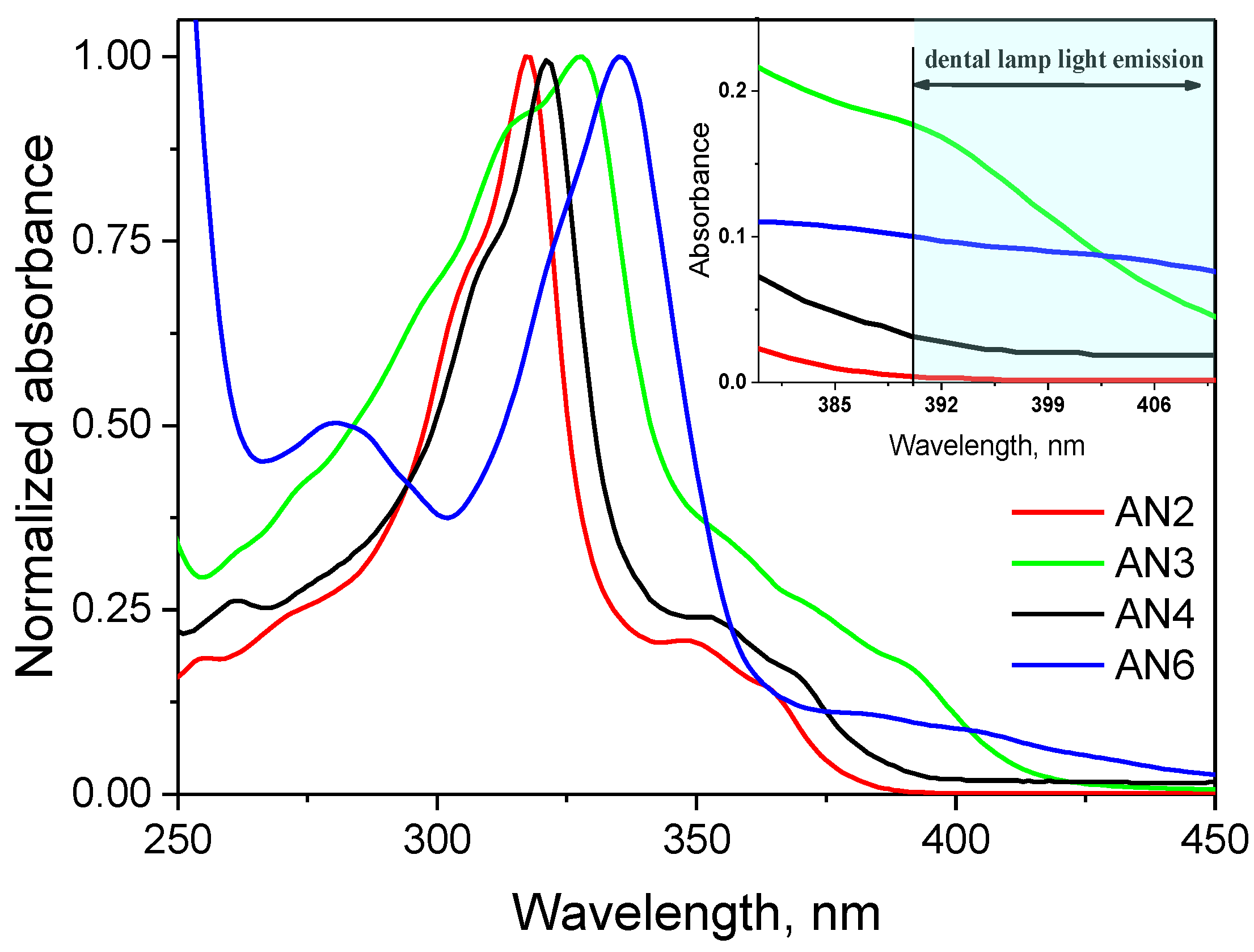
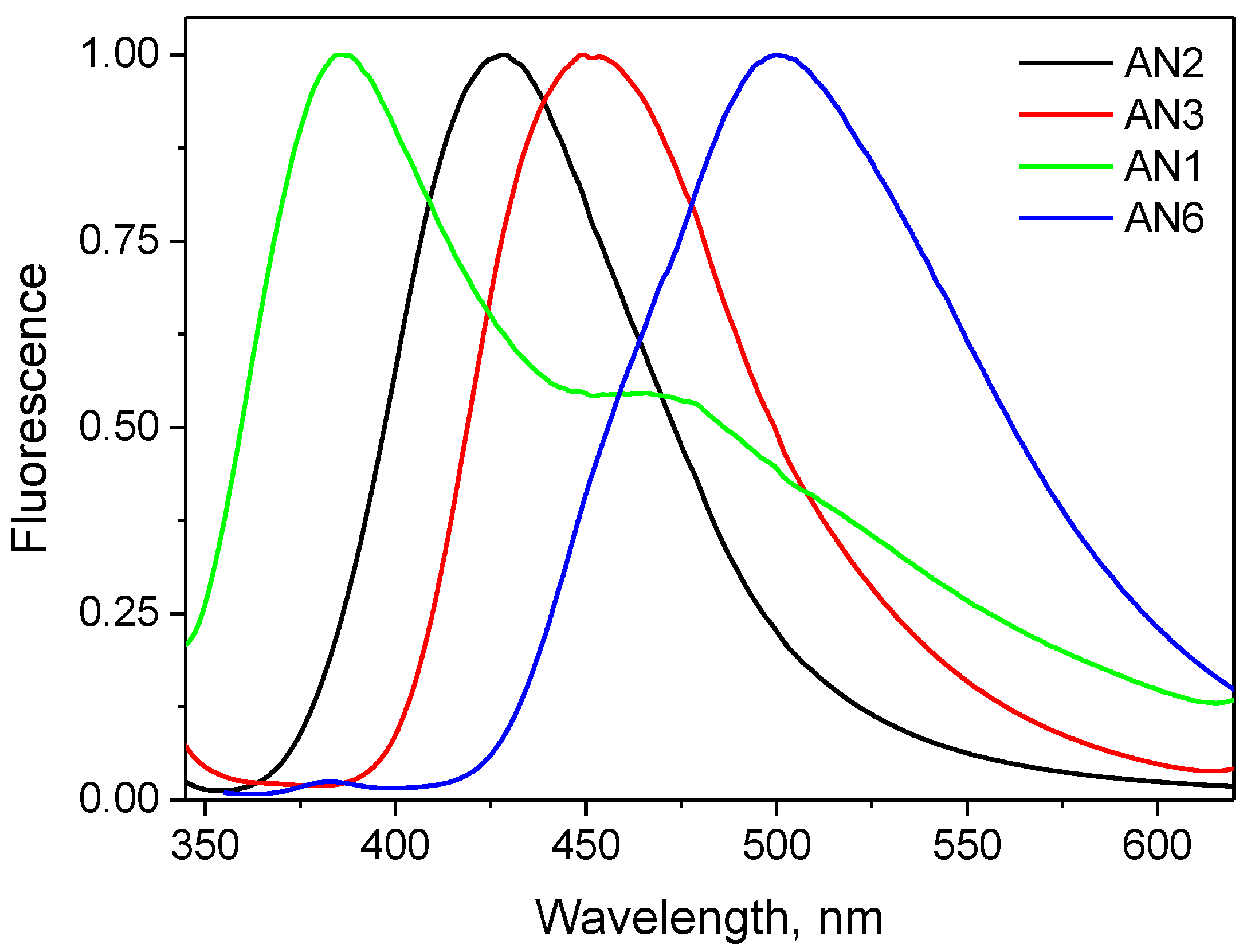
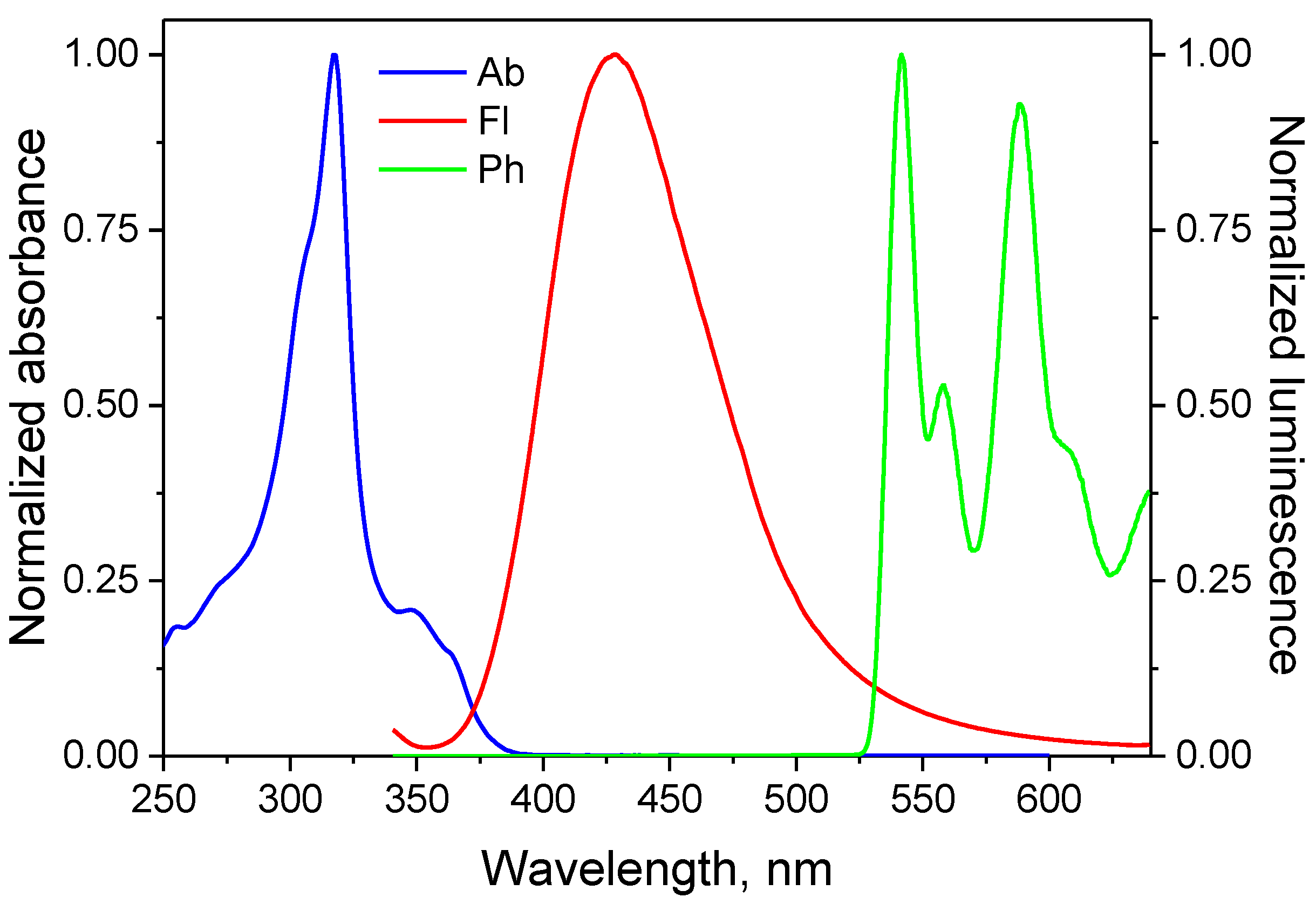
3.2. Spectroscopic Properties
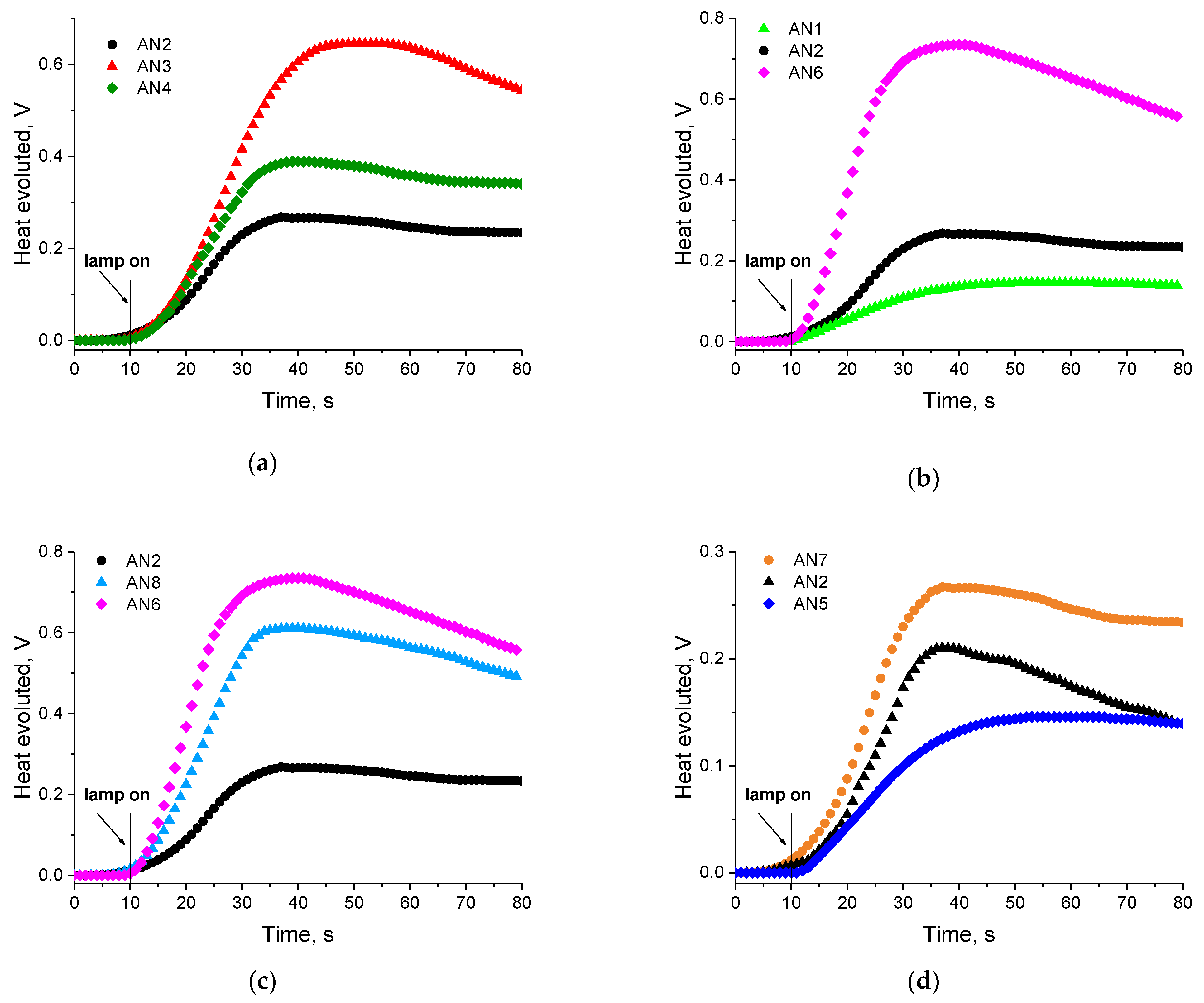
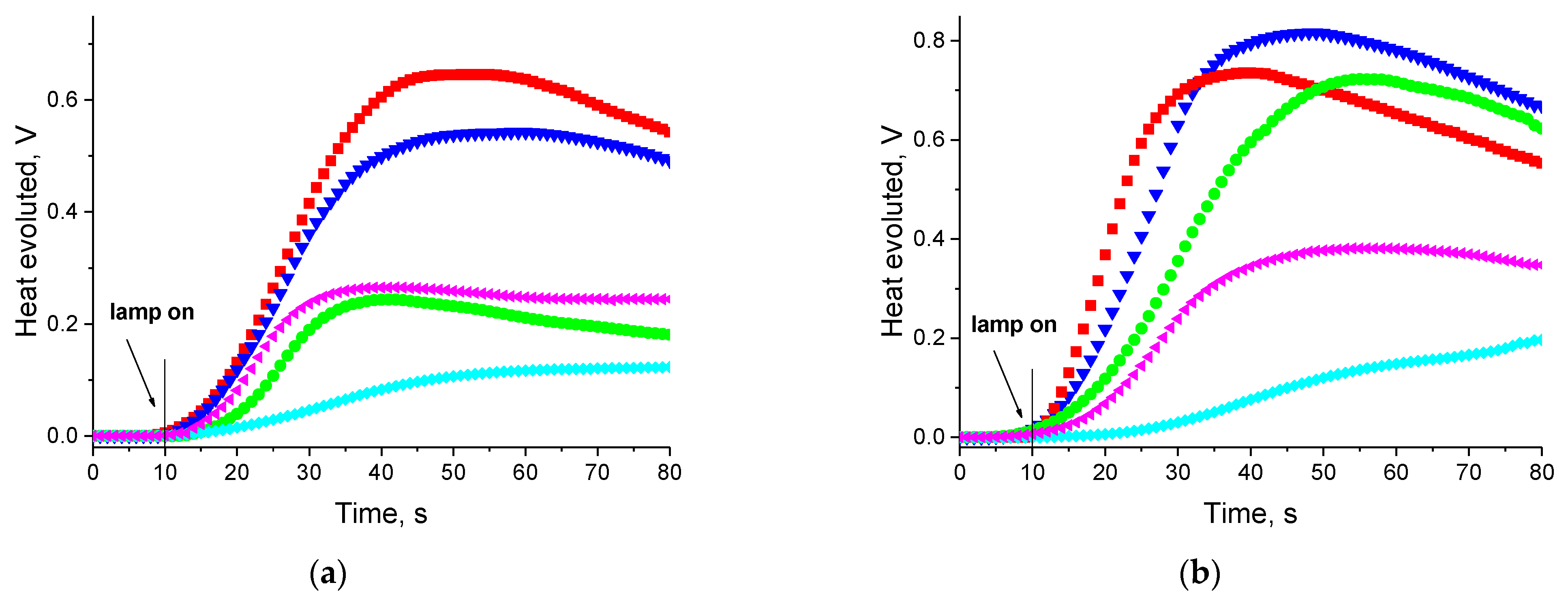
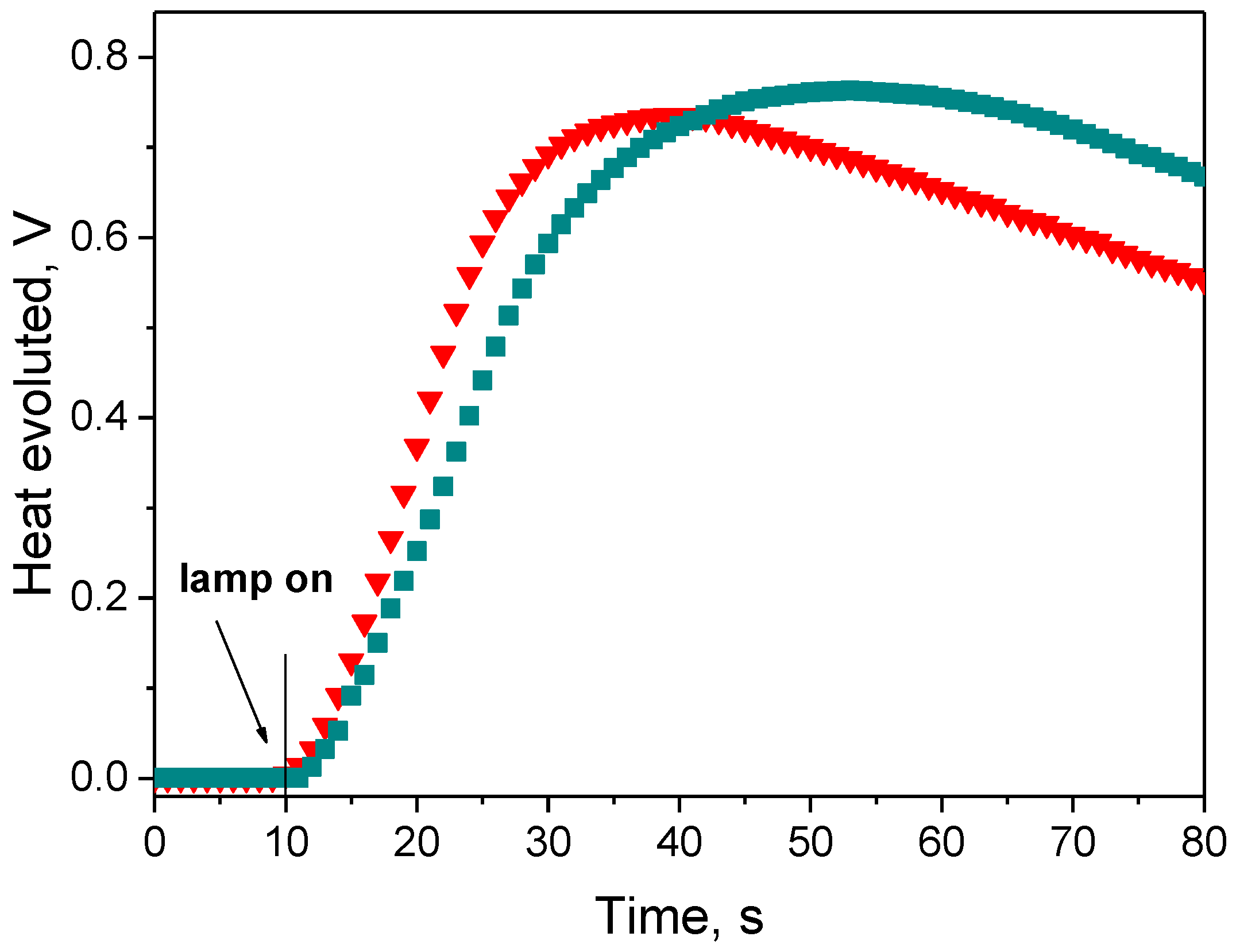
3.3. Kinetic Study of Multifunctional Acrylate Polymerization
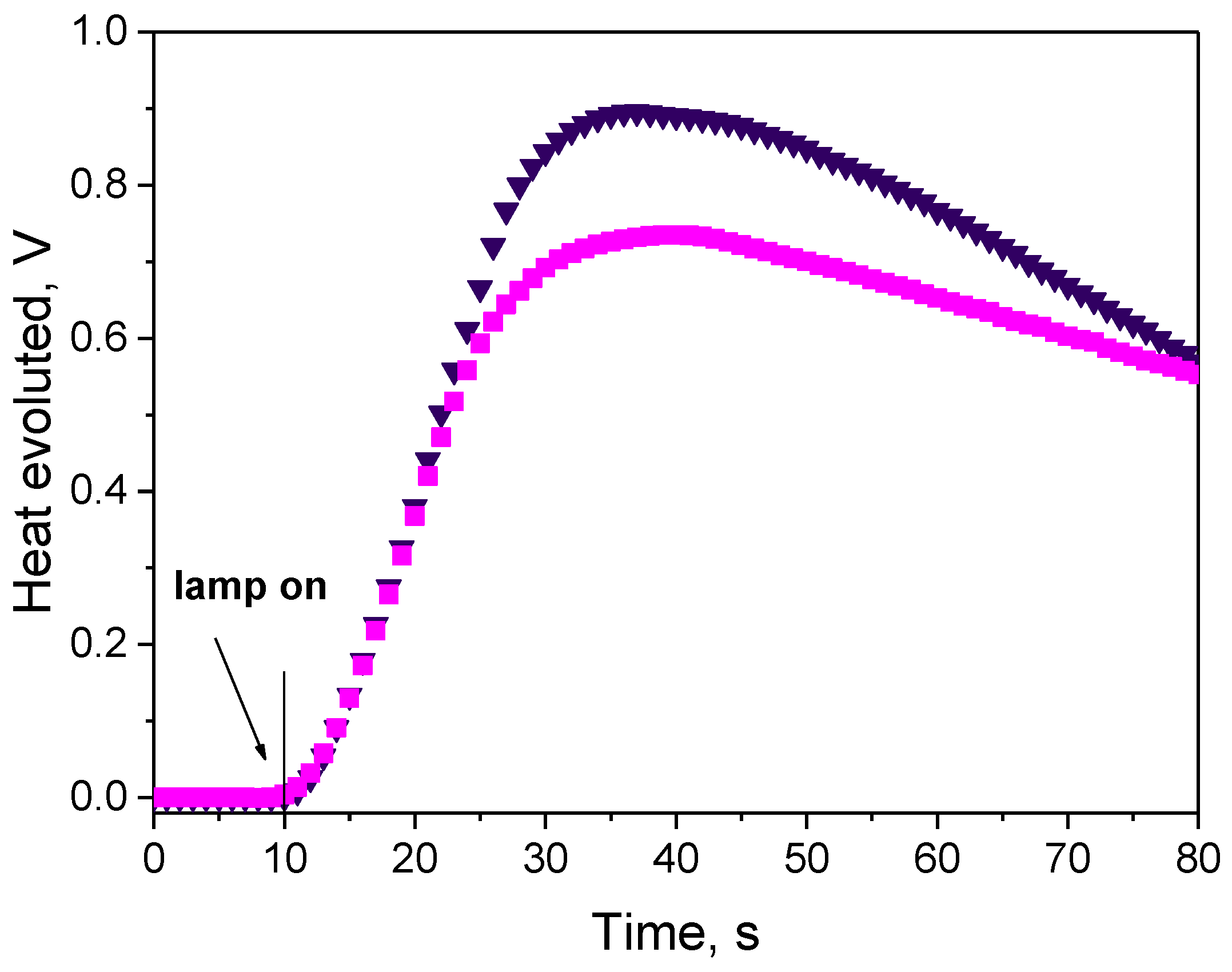
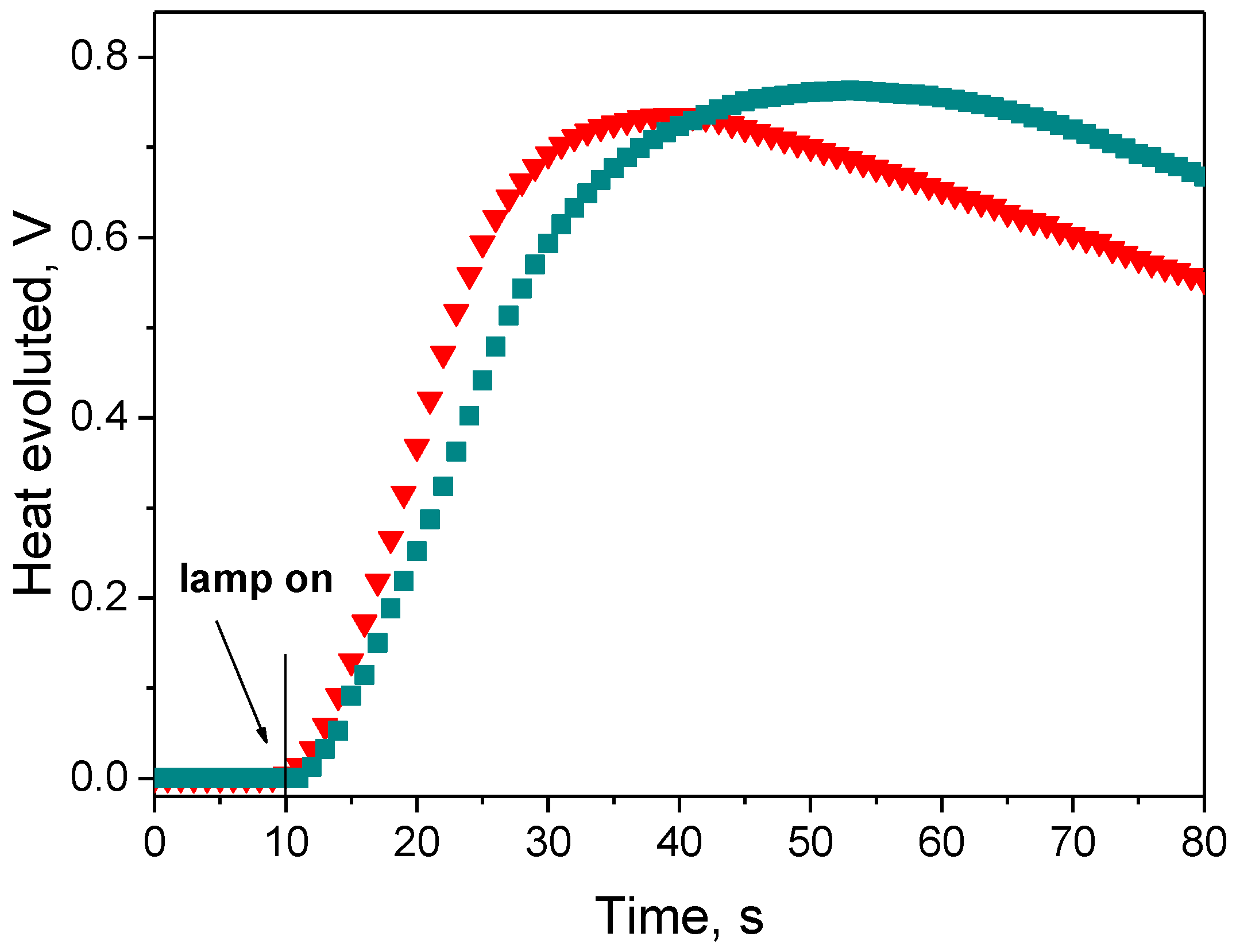
3.3.1. Effect of Photoinitiator Structure
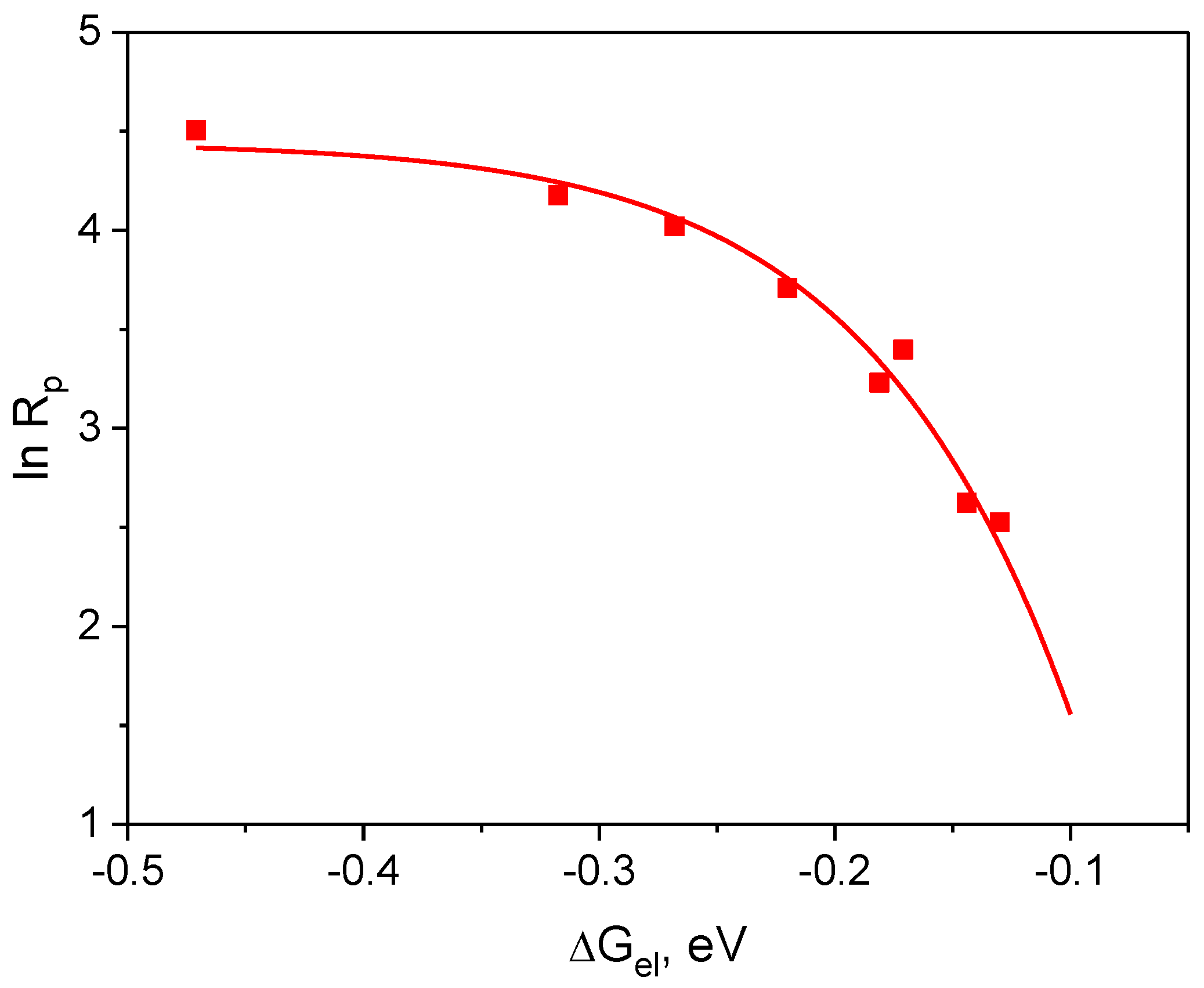
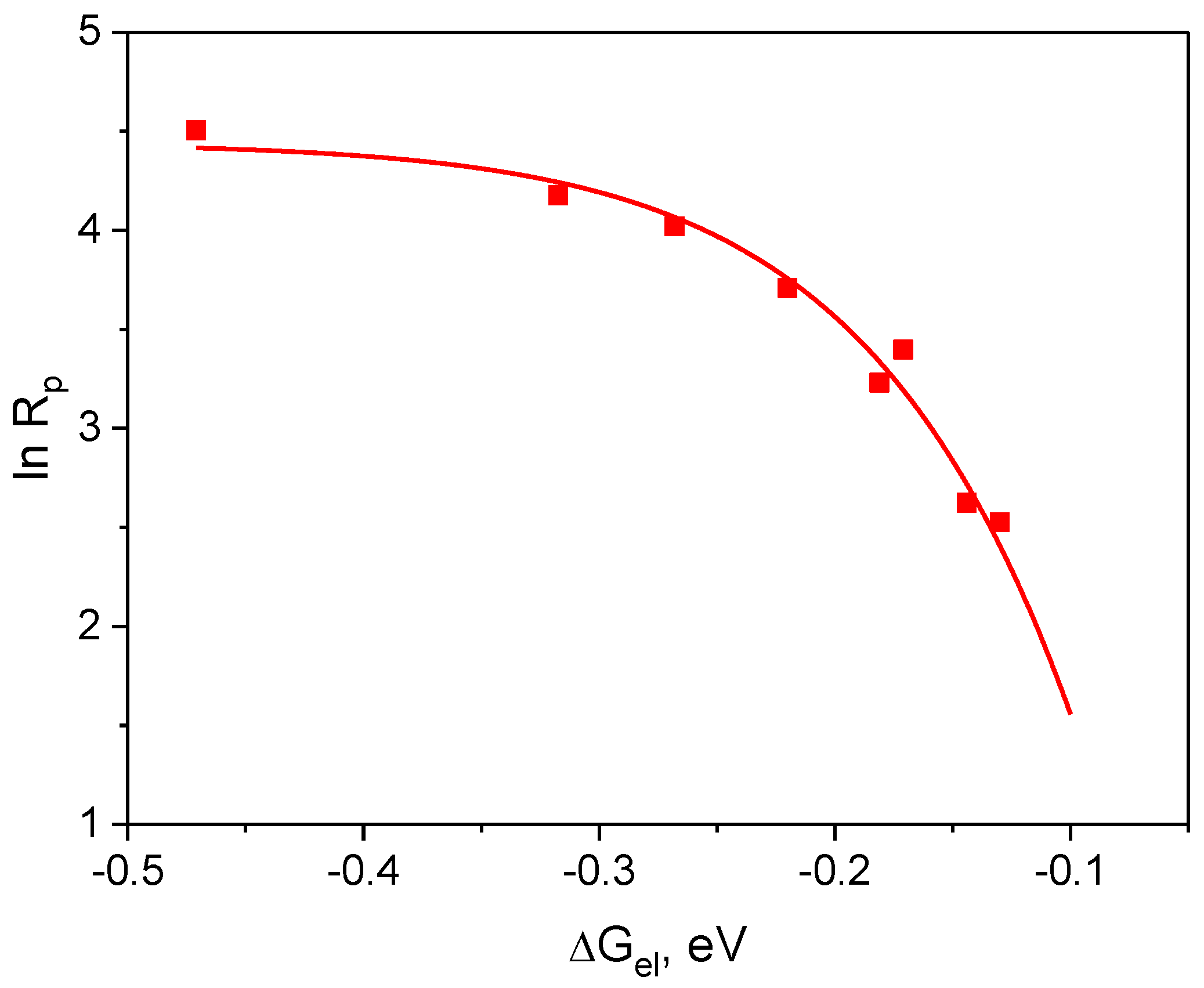
3.3.2. Influence of Thermodynamics Parameter
3.3.3. Photoinitiation by Hydrogen Atom Transfer Mechanism
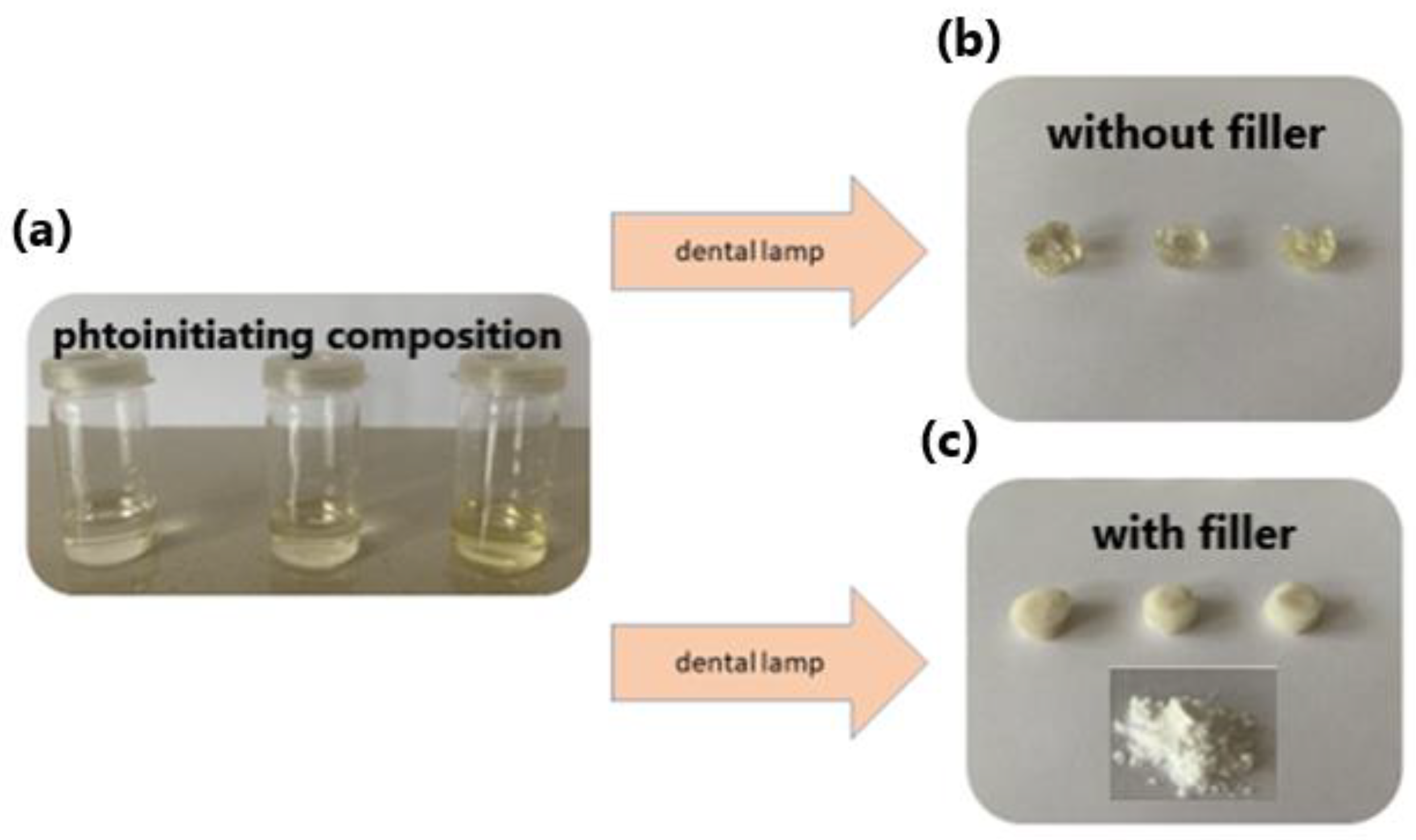
3.3.4. Dental Test Composition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dietliker, K. Chemistry and Technology of UV and EB Formulation for Coatings, Inks and Paints; SITA Technology LED: London, UK, 1991; Volume III. [Google Scholar]
- Allen, N.S. Photochemistry and Photophysics of Polymer Materials; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar] [CrossRef]
- Yagci, Y.; Jockusch, S.; Turro, N.J. Photoinitiated Polymerization: Advances, Challenges, and Opportunities. Macromolecules 2010, 43, 6245–6260. [Google Scholar] [CrossRef]
- Fouassier, J.P.; Allonas, X. Basics of Photopolymerization Reactions; Researching Post: Trivandrum, India, 2010. [Google Scholar]
- Fouassier, J.P.; Allonas, X. Dyes and Chromophores in Polymer Science; ISTE Ltd.: London, UK; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015. [Google Scholar]
- Fouassier, J.-P.; Lalevée, J. Photoinitiators for Polymer Synthesis-Scope, Reactivity, and Efficiency; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012. [Google Scholar]
- Fouassier, J.-P.; Morlet-Savary, F.; Lalevée, J.; Allonas, X.; Ley, C. Dyes as Photoinitiators or Photosensitizers of Polymerization Reactions. Materials 2010, 3, 5130–5142. [Google Scholar] [CrossRef]
- Hageman, H. Photoinitiators for free radical polymerization. Prog. Org. Coat. 1985, 13, 123–150. [Google Scholar] [CrossRef]
- Fouassier, J.; Jacques, P.; Lougnot, D.; Pilot, T. Lasers, photoinitiators and monomers: A fashionable formulation. Polym. Photochem. 1984, 5, 57–76. [Google Scholar] [CrossRef]
- Wrzyszczynski, A.; Bartoszewicz, J.; Hug, G.L.; Marciniak, B.; Pączkowski, J. Photochemical studies of a photodissociative initiator based on a benzophenone derivative possessing a thioether moiety. J. Photochem. Photobiol. A Chem. 2003, 155, 253–259. [Google Scholar] [CrossRef]
- Abraham, W.; Glänzel, A.; Stösser, R.; Grummt, U.-W.; Köppel, H. Desilylation by ether cleavage. J. Photochem. Photobiol. A Chem. 1990, 51, 359–370. [Google Scholar] [CrossRef]
- Tsunooka, M.; Tanaka, S.; Tanaka, M. Photocrosslinking of poly (2,3-epoxypropyl methacrylate) films with organic sulfur compounds. Makromol. Chem. Rapid Commun. 1983, 4, 539–541. [Google Scholar] [CrossRef]
- Walton, J.C. Functionalised Oximes: Emergent Precursors for Carbon-, Nitrogen- and Oxygen-Centred Radicals. Molecules 2016, 21, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, H. Photoinitiators for free radical polymerization. Prog. Polym. Sci. 1992, 17, 953–1044. [Google Scholar] [CrossRef]
- Gupta, S.N.; Thijs, L.; Neckers, D.C. Synthesis and polymerization of p-(p′-vinylbenzoyl)-peroxybenzoic acid tert-butylester: A monomer containing a photodissociable radical source. J. Polym. Sci. Polym. Chem. Ed. 1981, 19, 855–868. [Google Scholar] [CrossRef]
- Kucybała, Z.; Wrzyszczyński, A. Photolysis of N-[(4-benzoyl)benzenesulfonyl]benzenesulfonamide. J. Photochem. Photobiol. A Chem. 2002, 153, 109–112. [Google Scholar] [CrossRef]
- Neckers, D.; Hassoon, S.; Klimtchuk, E. Photochemistry and photophysics of hydroxyfluorones and xanthenes. J. Photochem. Photobiol. A Chem. 1996, 95, 33–39. [Google Scholar] [CrossRef]
- Encinas, M.V.; Garrido, J.; Lissi, E.A. Polymerization photoinitiated by carbonyl compounds. VIII. Solvent and photoinitiator concentration effects. J. Polym. Sci. Part A Polym. Chem. 1989, 27, 139–145. [Google Scholar] [CrossRef]
- Pyszka, I.; Kucybała, Z.; Pączkowski, J. Reinvestigation of the Mechanism of the Free Radical Polymerization Photoinitiation Process by Camphorquinone-Coinitiator Systems: New Results. Macromol. Chem. Phys. 2004, 205, 2371–2375. [Google Scholar] [CrossRef]
- Pączkowski, J. Electron-transfer photoinitiators of free radical polymerization. The effect of the co-iniciator structure on photoinitiation ability. In Photochemistry and UV Curing: New Trends; Fouassier, J.-P., Ed.; Research Signpost: Kerala, India, 2006; p. 101. [Google Scholar]
- Pączkowski, J.; Neckers, D.C. Photoinduced Electron Transfer Initiating Systems for Free-Radical Polymerization. In Electron Transfer in Chemistry; Wiley: Hoboken, NJ, USA, 2008; Volume 5, pp. 516–585. [Google Scholar]
- Jakubiak, J.; Allonas, X.; Fouassier, J.; Sionkowska, A.; Andrzejewska, E.; Linden, L.; Rabek, J. Camphorquinone-amines photoinitating systems for the initiation of free radical polymerization. Polymer 2003, 44, 5219–5226. [Google Scholar] [CrossRef]
- Jędrzejewska, B.; Wejnerowska, G. Highly Effective Sensitizers Based on Merocyanine Dyes for Visible Light Initiated Radical Polymerization. Polymer 2020, 12, 1242. [Google Scholar] [CrossRef] [PubMed]
- Ścigalski, F.; Jędrzejewska, B. Structural effect of oxazolone derivatives on the initiating abilities of dye-borate photoredox systems in radical polymerization under visible light. RSC Adv. 2020, 10, 21487–21494. [Google Scholar] [CrossRef]
- Jędrzejewska, B.; Osmialowski, B. Difluoroboranyl derivatives as efficient panchromatic photoinitiators in radical polymerization reactions. Polym. Bull. 2017, 75, 3267–3281. [Google Scholar] [CrossRef] [Green Version]
- Kabatc, J. The influence of a radical structure on the kinetics of photopolymerization. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 1575–1589. [Google Scholar] [CrossRef] [Green Version]
- Strzelczyk, R.; Podsiadły, R. Naphthoylenebenzimidazolone dyes as one-component photoinitiators. Color. Technol. 2017, 133, 178–183. [Google Scholar] [CrossRef]
- Al Mousawi, A.; Dietlin, C.; Graff, B.; Morlet-Savary, F.; Toufaily, J.; Hamieh, T.; Fouassier, J.P.; Chachaj-Brekiesz, A.; Ortyl, J.; Lalevée, J. Meta-Terphenyl Derivative/Iodonium Salt/9H-Carbazole-9-ethanol Photoinitiating Systems for Free Radical Promoted Cationic Polymerization upon Visible Lights. Macromol. Chem. Phys. 2016, 217, 1955–1965. [Google Scholar] [CrossRef]
- Pyszka, I.; Kucybała, Z.; Jędrzejewska, B. Effective Singlet Oxygen Sensitizers Based on the Phenazine Skeleton as Efficient Light Absorbers in Dye Photoinitiating Systems for Radical Polymerization of Acrylates. Materials 2021, 14, 3085. [Google Scholar] [CrossRef]
- Podsiadły, R.; Sokołowska, J.; Kolińska, J.; Grzelakowska, A. Synthesis and photochemical reaction of benzo[a]quinoxalino[2,3-c]phenazine dyes. Color. Technol. 2017, 133, 498–505. [Google Scholar] [CrossRef]
- Balcerak, A.; Kabatc, J. The photooxidative sensitization of bis(p-substituted diphenyl)iodonium salts in the radical polymerization of acrylates. RSC Adv. 2019, 9, 28490–28499. [Google Scholar] [CrossRef] [Green Version]
- Fouassier, J.; Allonas, X.; Burget, D. Photopolymerization reactions under visible lights: Principle, mechanisms and examples of applications. Prog. Org. Coat. 2003, 47, 16–36. [Google Scholar] [CrossRef]
- Corcione, C.E.; Frigione, M. Factors influencing photo curing kinetics of novel UV-cured siloxane-modified acrylic coatings: Oxygen inhibition and composition. Thermochim. Acta 2012, 534, 21–27. [Google Scholar] [CrossRef]
- Corcione, C.E.; Striani, R.; Frigione, M. UV-cured siloxane-modified methacrylic system containing hydroxyapatite as potential protective coating for carbonate stones. Prog. Org. Coat. 2013, 76, 1236–1242. [Google Scholar] [CrossRef]
- Striani, R.; Corcione, C.E.; Muia, G.D.; Frigione, M. Durability of a sunlight-curable organic–inorganic hybrid protective coating for porous stones in natural and artificial weathering conditions. Prog. Org. Coat. 2016, 101, 1–14. [Google Scholar] [CrossRef]
- Sun, K.; Xu, Y.; Dumur, F.; Morlet-Savary, F.; Chen, H.; Dietlin, C.; Graff, B.; Lalevée, J.; Xiao, P. In silico rational design by molecular modeling of new ketones as photoinitiators in three-component photoinitiating systems: Application in 3D printing. Polym. Chem. 2020, 11, 2230–2242. [Google Scholar] [CrossRef]
- Imran, O.Q.; Kim, N.K.; Bodkin, L.N.; Dwulet, G.E.; Feng, X.; Kawabata, K.; Elimelech, M.; Gin, D.L.; Osuji, C.O. Nanoscale Thickness Control of Nanoporous Films Derived from Directionally Photopolymerized Mesophases. Adv. Mater. Interfaces 2021, 8, 2001977. [Google Scholar] [CrossRef]
- Li, H.; Zhu, X.; Zheng, F.; Xia, P. UV-Curable Adhesive Composition for Compact Discs. CN Patent 101857776B, 16 January 2013. [Google Scholar]
- Mavila, S.; Sinha, J.; Hu, Y.; Podgórski, M.; Shah, P.K.; Bowman, C.N. High Refractive Index Photopolymers by Thiol–Yne “Click” Polymerization. ACS Appl. Mater. Interfaces 2021, 13, 15647–15658. [Google Scholar] [CrossRef]
- Ni, M.; Luo, W.; Wang, D.; Zhang, Y.; Peng, H.; Zhou, X.; Xie, X. Orthogonal Reconstruction of Upconversion and Holographic Images for Anticounterfeiting Based on Energy Transfer. ACS Appl. Mater. Interfaces 2021, 13, 19159–19167. [Google Scholar] [CrossRef] [PubMed]
- Sabel, T.; Orlic, S.; Pfeiffer, K.; Ostrzinski, U.; Grützner, G. Free-surface photopolymerizable recording material for volume holography. Opt. Mater. Express 2013, 3, 329. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, Y.; Li, L.; Yan, B.; Bao, J.; Zhang, A. Acrylate-based photosensitive resin for stereolithographic three-dimensional printing. J. Appl. Polym. Sci. 2019, 136, 47487. [Google Scholar] [CrossRef]
- Chen, H.; Noirbent, G.; Liu, S.; Brunel, D.; Graff, B.; Gigmes, D.; Zhang, Y.; Sun, K.; Morlet-Savary, F.; Xiao, P.; et al. Bis-chalcone derivatives derived from natural products as near-UV/visible light sensitive photoinitiators for 3D/4D printing. Mater. Chem. Front. 2021, 5, 901–916. [Google Scholar] [CrossRef]
- Vivero-Lopez, M.; Xu, X.; Muras, A.; Otero, A.; Concheiro, A.; Gaisford, S.; Basit, A.W.; Alvarez-Lorenzo, C.; Goyanes, A. Anti-biofilm multi drug-loaded 3D printed hearing aids. Mater. Sci. Eng. C 2021, 119, 111606. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Lindén, L.Å.; Rabek, J.F.; Ekstrand, J. Photocuring kinetic studies of new dental restorative resins based on poly(ethylene glycol) diacrylate and tris[2-(acryloyloxy)-ethyl]isocyanurate. Angew. Makromol. Chem. 1998, 257, 47–52. [Google Scholar] [CrossRef]
- Park, Y.-J.; Chae, K.-H.; Rawls, H. Development of a new photoinitiation system for dental light-cure composite resins. Dent. Mater. 1999, 15, 120–127. [Google Scholar] [CrossRef]
- Jandt, K.; Mills, R.; Blackwell, G.; Ashworth, S. Depth of cure and compressive strength of dental composites cured with blue light emitting diodes (LEDs). Dent. Mater. 2000, 16, 41–47. [Google Scholar] [CrossRef]
- Pérez-Mondragón, A.A.; Cuevas-Suárez, C.E.; González-López, J.A.; Trejo-Carbajal, N.; Herrera-González, A.M. Evaluation of new coinitiators of camphorquinone useful in the radical photopolymerization of dental monomers. J. Photochem. Photobiol. A Chem. 2020, 403, 112844. [Google Scholar] [CrossRef]
- Cadenaro, M.; Maravic, T.; Comba, A.; Mazzoni, A.; Fanfoni, L.; Hilton, T.; Ferracane, J.; Breschi, L. The role of polymerization in adhesive dentistry. Dent. Mater. 2019, 35, e1–e22. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, A.; Sokolowski, J.; Bociong, K. The Photoinitiators Used in Resin Based Dental Composite—A Review and Future Perspectives. Polymers 2021, 13, 470. [Google Scholar] [CrossRef]
- Barszczewska-Rybarek, I.M. A Guide through the Dental Dimethacrylate Polymer Network Structural Characterization and Interpretation of Physico-Mechanical Properties. Materials 2019, 12, 4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maffezzoli, A.; Pietra, A.; Rengo, S.; Nicolais, L.; Valletta, G. Photopolymerization of dental composite matrices. Biomaterials 1994, 15, 1221–1228. [Google Scholar] [CrossRef]
- 5Linden, L.A. Dental Polymers (Overview). In Polymeric Materials Encyclopedia; Salamone, J.C., Ed.; CRC Press: Boca Raton, FL, USA, 1996; Volume 12, p. 1849. [Google Scholar]
- De Bragança, G.F.; Mazão, J.D.; Versluis, A.; Soares, C.J. Effect of luting materials, presence of tooth preparation, and functional loading on stress distribution on ceramic laminate veneers: A finite element analysis. J. Prosthet. Dent. 2021, 125, 778–787. [Google Scholar] [CrossRef]
- Elshereksi, N.; Muchtar, A.; Azhari, C. Effects of nanobarium titanate on physical and mechanical properties of poly(methyl methacrylate) denture base nanocomposites. Polym. Polym. Compos. 2021, 29, 484–496. [Google Scholar] [CrossRef]
- Kantrong, N.; Mongkontunpimon, W.; Supameteeworakul, S.; Wongkhantee, S. Differential induction of surface chemical compositional change on tooth structure by glass ionomer restorative materials. Odontology 2021, 109, 124–138. [Google Scholar] [CrossRef]
- Hoshika, S.; Ting, S.; Ahmed, Z.; Chen, F.; Toida, Y.; Sakaguchi, N.; Van Meerbeek, B.; Sano, H.; Sidhu, S.K. Effect of conditioning and 1 year aging on the bond strength and interfacial morphology of glass-ionomer cement bonded to dentin. Dent. Mater. 2021, 37, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Dai, Y.; Zhang, X.; Xiao, Y. Synthesis and photophysical properties of three ladder-type chromophores with large and rigid conjugation structures. Dye. Pigment. 2013, 102, 1–5. [Google Scholar] [CrossRef]
- Amer, A.M.; El-Bahnasawi, A.A.; Mahran, M.R.H.; Lapib, M. On the Synthesis of Pyrazino[2,3-b]phenazine and 1H-Imidazo[4,5-b]phenazine Derivatives. Mon. Chem. Chem. Mon. 1999, 130, 1217–1225. [Google Scholar] [CrossRef]
- Boido, A.; Vazzana, I.; Sparatore, F. N-[(N-halogenoacyl)amino]acenapthlene-quinoxalines as potential antitumoral agents. Il Farm. 1994, 49, 97–104. [Google Scholar]
- Klessinger, M.; Michl, J. Excited States and Photochemistry of Organic Molecules; Wiley-VCH: New York, NY, USA, 1995. [Google Scholar]
- Laschuk, N.; Obua, A.; Ebralidze, I.I.; Fruehwald, H.; Poisson, J.; Egan, J.G.; Gaspari, F.; Naumkin, F.Y.; Easton, E.B.; Zenkina, O.V. Spacer Conjugation and Surface Support Effects in Monolayer Electrochromic Materials. ACS Appl. Electron. Mater. 2019, 1, 1705–1717. [Google Scholar] [CrossRef]
- Jablonski, A. Efficiency of Anti-Stokes Fluorescence in Dyes. Nat. Cell Biol. 1933, 131, 839–840. [Google Scholar] [CrossRef]
- Murov, S.L.; Carmichael, I.; Hug, G.L. Handbook of Photochemistry; Marcel Dekker: New York, NY, USA, 1993. [Google Scholar]
- Yang, J.-S.; Liau, K.-L.; Wang, C.-M.; Hwang, C.-Y. Substituent-Dependent Photoinduced Intramolecular Charge Transfer inN-Aryl-Substitutedtrans-4-Aminostilbenes. J. Am. Chem. Soc. 2004, 126, 12325–12335. [Google Scholar] [CrossRef] [PubMed]
- Lalevée, J.; Fouassier, J.-P. Photopolymerisation Initiating Systems; Royal Society of Chemistry: Croydon, UK, 2018. [Google Scholar]
- Paczkowski, J.; Pietrzak, M.; Kucybała, Z. Generalization of the Kinetic Scheme for Photoinduced Polymerization via an Intermolecular Electron Transfer Process. 2. Application of the Marcus Theory. Macromolecules 1996, 29, 5057–5064. [Google Scholar] [CrossRef]
- Paczkowski, J.; Kucybała, Z. Generalization of the Kinetic Scheme for a Dye-Photosensitized Free-Radical Polymerization Initiating System via an Intermolecular Electron-Transfer Process. Application of Marcus Theory. Macromolecules 1995, 28, 269–273. [Google Scholar] [CrossRef]
- Rehm, D.; Weller, A. Kinetics of Fluorescence Quenching by Electron and H-Atom Transfer. Isr. J. Chem. 1970, 8, 259–271. [Google Scholar] [CrossRef]
- A Marcus, R. Chemical and Electrochemical Electron-Transfer Theory. Annu. Rev. Phys. Chem. 1964, 15, 155–196. [Google Scholar] [CrossRef]
- Marcus, R.A. On the Theory of Electron-Transfer Reactions. VI. Unified Treatment for Homogeneous and Electrode Reactions. J. Chem. Phys. 1965, 43, 679–701. [Google Scholar] [CrossRef] [Green Version]
- Pyszka, I.; Kucybała, Z. The effect of co-initiator structure on photoinitiating efficiency of photoredox couples composed of quinoline[2,3-b]-1H-imidazo[1,2-a]pyridinium bromide and phenoxyacetic acid or N,N-dimethylaniline derivatives. Polym. Bull. 2008, 61, 553–562. [Google Scholar] [CrossRef]
- Jędrzejewska, B.; Pietrzak, M.; Pączkowski, J. Tetramethylammonium phenyltrialkylborates as co-initiators with novel two-cationic styrylbenzimidazolium dyes in highly efficient, visible light polymerization of acrylate. J. Photochem. Photobiol. A Chem. 2010, 214, 276–283. [Google Scholar] [CrossRef]
- Brimage, D.R.G.; Davidson, R.S.; Steiner, P.R. Use of heterocyclic compounds as photosensitisers for the decarboxylation of carboxylic acids. J. Chem. Soc. Perkin Trans. 1973, 1, 526–529. [Google Scholar] [CrossRef]
- Pyszka, I. Benzo-i dibenzopochodne fenazyny jako barwnikowe fotoinicjatory polimeryzacji rodnikowej triakrylanu trimetylolopropanu. Efekt podstawnika w parze fotoredoks. Przem. Chem. 2019, 1, 120–124. [Google Scholar] [CrossRef]
- Kucybała, Z.; Pietrzak, M.; Osmańska, K.; Pączkowski, J. Mechanistic and Kinetic Study of Free Radical Polymerization Photoinitiated by Xanthene Dye-n-Phenylglycine Derivatives Photoredox Pairs. Pol. J. Chem. 2005, 79, 851–866. [Google Scholar]
- Weller, A. Photoinduced Electron Transfer in Solution: Exciplex and Radical Ion Pair Formation Free Enthalpies and their Solvent Dependence. Z. Phys. Chem. 1982, 133, 93–98. [Google Scholar] [CrossRef]
- Eberson, L. Electron Transfer in Organic Chemistry; Springer: New York, NY, USA, 1987. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
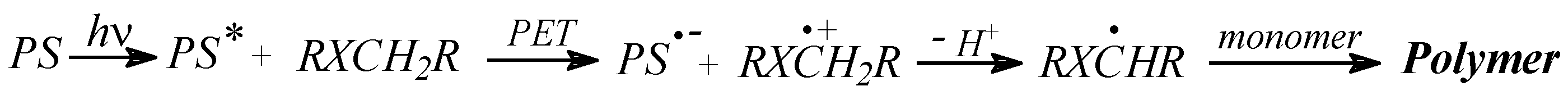{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | εmax, dm3 mol−1 cm−1 | ΔνSt, cm−1 | |||
|---|---|---|---|---|---|
| AN1 | 316 | 27,200 | 385.4 | 530.6 | 5699 |
| 547.2 | |||||
| 574.2 | |||||
| 621.4 | |||||
| AN2 | 317 | 65,200 | 427.6 | 541.4 | 5432 |
| 347 | 14,600 | 587.8 | |||
| AN3 | 327 | 82,400 | 449 | 544.8 | 8309 |
| 592.2 | |||||
| AN4 | 321 | 92,000 | 395.6 | 542.4 | 5875 |
| 589.4 | |||||
| AN5 | 319 | 110,100 | 362.6 | 539.6 | 3769 |
| 586.0 | |||||
| AN6 | 335 | 145,800 | 529.8 | 543.4 | 10976 |
| 586.8 | |||||
| AN7 | 347 | 170,000 | 452.8 | 546.8 | 5076 |
| 363 | 189,900 | 585.2 | |||
| AN8 | 339 | 146,200 | 473.4 | 522.4 | 8375 |
| 561.4 | |||||
| CQ 1 | 472 | 40 | - | - | - |
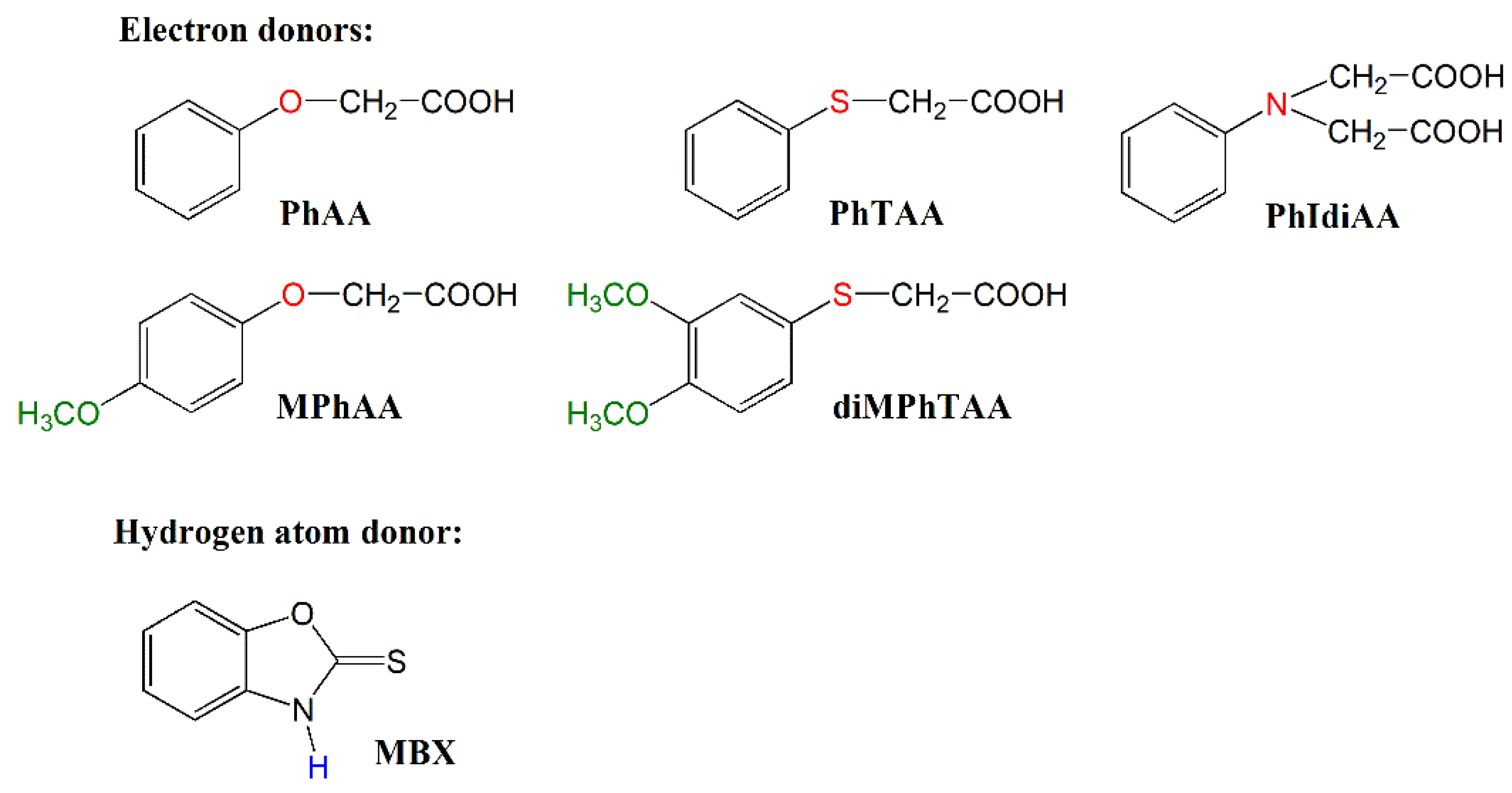
| Dye | Rp, μmol/s | |||||
|---|---|---|---|---|---|---|
| PhAA | MPhAA | PhTAA | diMPhTAA | PhIdiAA | MBX | |
| AN1 | 1.4 | 9.2 | 6.5 | 14.0 | 12.5 | 12.8 |
| AN2 | 3.9 | 22.6 | 17.9 | 35.9 | 29.9 | 33.1 |
| AN3 | 9.0 | 44.4 | 39.7 | 68.1 | 55.7 | 57.1 |
| AN4 | 5.3 | 42.5 | 21.9 | 52.3 | 40.8 | 46.1 |
| AN5 | 3.2 | 21.3 | 13.2 | 29.2 | 25.3 | 28.7 |
| AN6 | 10.2 | 80.2 | 61.5 | 116.1 | 90.4 | 91.2 |
| AN7 | 1.3 | 10.0 | 7.2 | 14.0 | 13.8 | 13.1 |
| AN8 | 7.3 | 47.1 | 38.1 | 72.8 | 65.1 | 65.4 |
| CQ | 15.8 | 82.3 | 71.4 | 129.7 | 107.8 | 93.6 |
| Electron Donor | Eox [V] |
|---|---|
| PhAA | 1.450 |
| MPhAA | 1.170 |
| PhTAA | 1.076 |
| diMPhTAA | 0.779 |
| PhIdiAA | 0.718 |
| Dye | Ered, V | 1 ΔGel, eV | |
|---|---|---|---|
| AN1 | −1.482 | 2.33 | −0.130 |
| AN2 | −1.401 | 2.29 | −0.171 |
| AN3 | −1.284 | 2.27 | −0.268 |
| AN4 | −1.422 | 2.36 | −0.220 |
| AN5 | −1.401 | 2.30 | −0.181 |
| AN6 | −1.091 | 2.28 | −0.471 |
| AN7 | −1.398 | 2.26 | −0.144 |
| AN8 | −1.335 | 2.37 | −0.317 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pyszka, I.; Jędrzejewska, B. Acenaphthoquinoxaline Derivatives as Dental Photoinitiators of Acrylates Polymerization. Materials 2021, 14, 4881. https://doi.org/10.3390/ma14174881
Pyszka I, Jędrzejewska B. Acenaphthoquinoxaline Derivatives as Dental Photoinitiators of Acrylates Polymerization. Materials. 2021; 14(17):4881. https://doi.org/10.3390/ma14174881
Chicago/Turabian StylePyszka, Ilona, and Beata Jędrzejewska. 2021. "Acenaphthoquinoxaline Derivatives as Dental Photoinitiators of Acrylates Polymerization" Materials 14, no. 17: 4881. https://doi.org/10.3390/ma14174881
APA StylePyszka, I., & Jędrzejewska, B. (2021). Acenaphthoquinoxaline Derivatives as Dental Photoinitiators of Acrylates Polymerization. Materials, 14(17), 4881. https://doi.org/10.3390/ma14174881