
New Insights on the Vibrational Dynamics of 2-Methoxy-, 4-Methoxy- and 4-Ethoxy-Benzaldehyde from INS Spectra and Periodic DFT Calculations

 ,
,  ,
,  , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
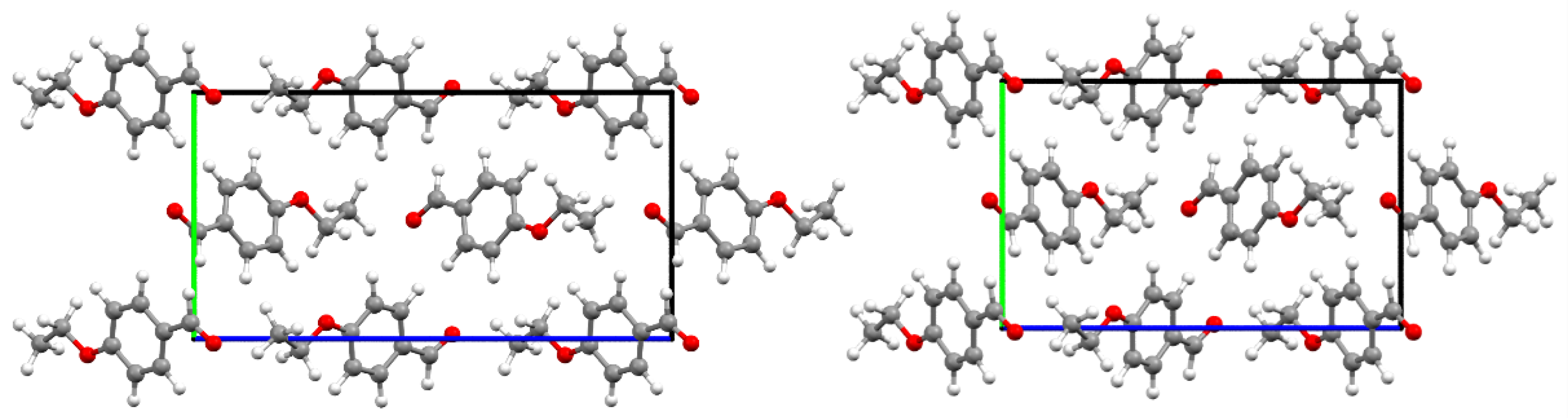
3. Results
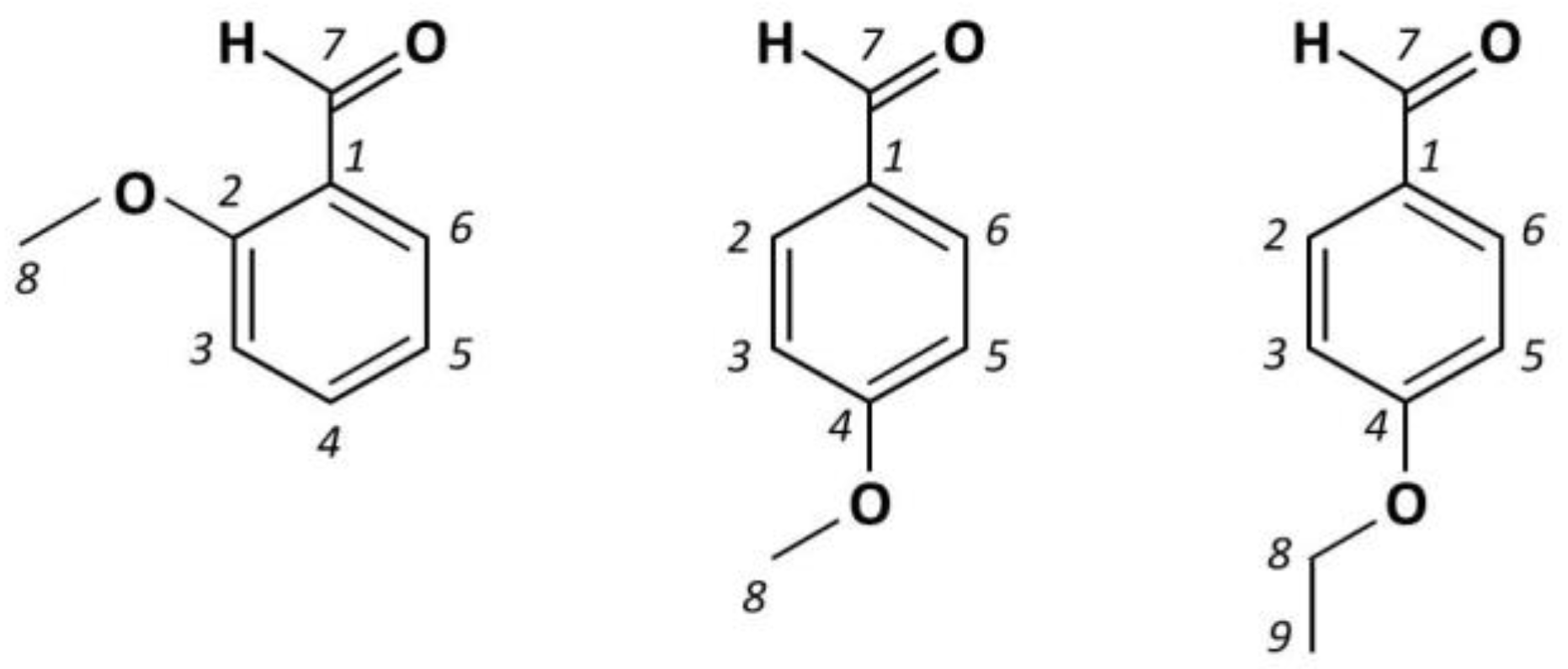
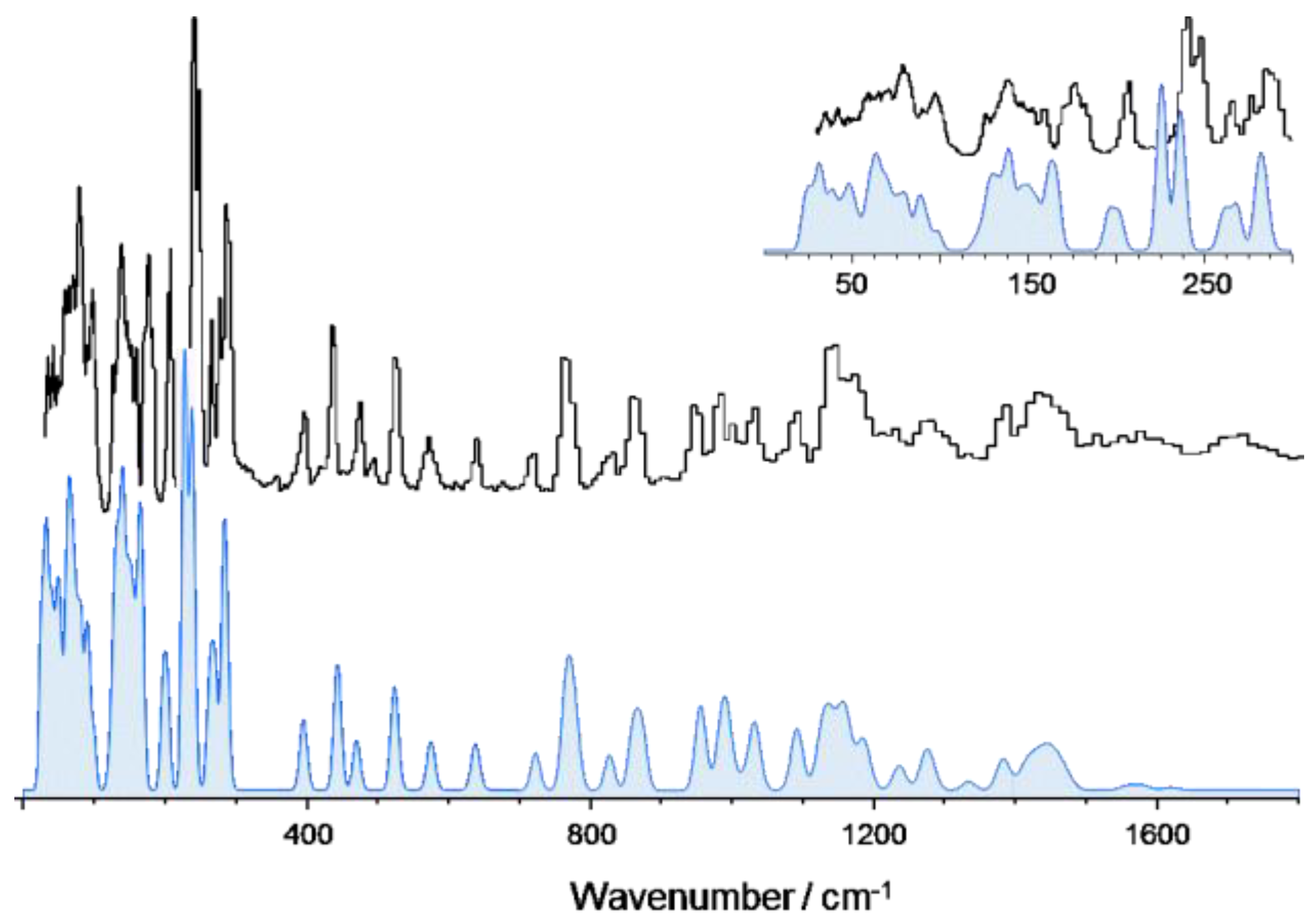
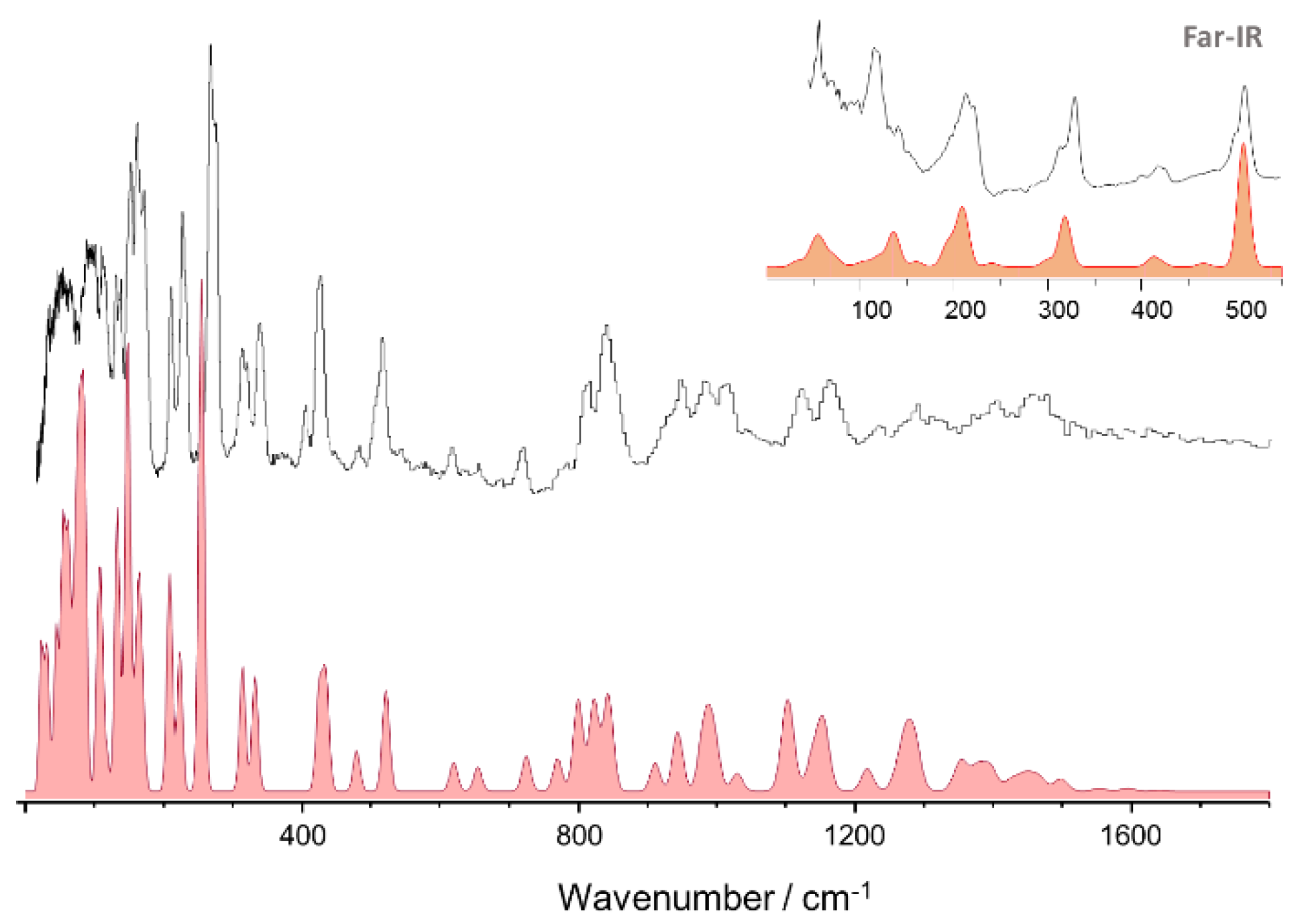
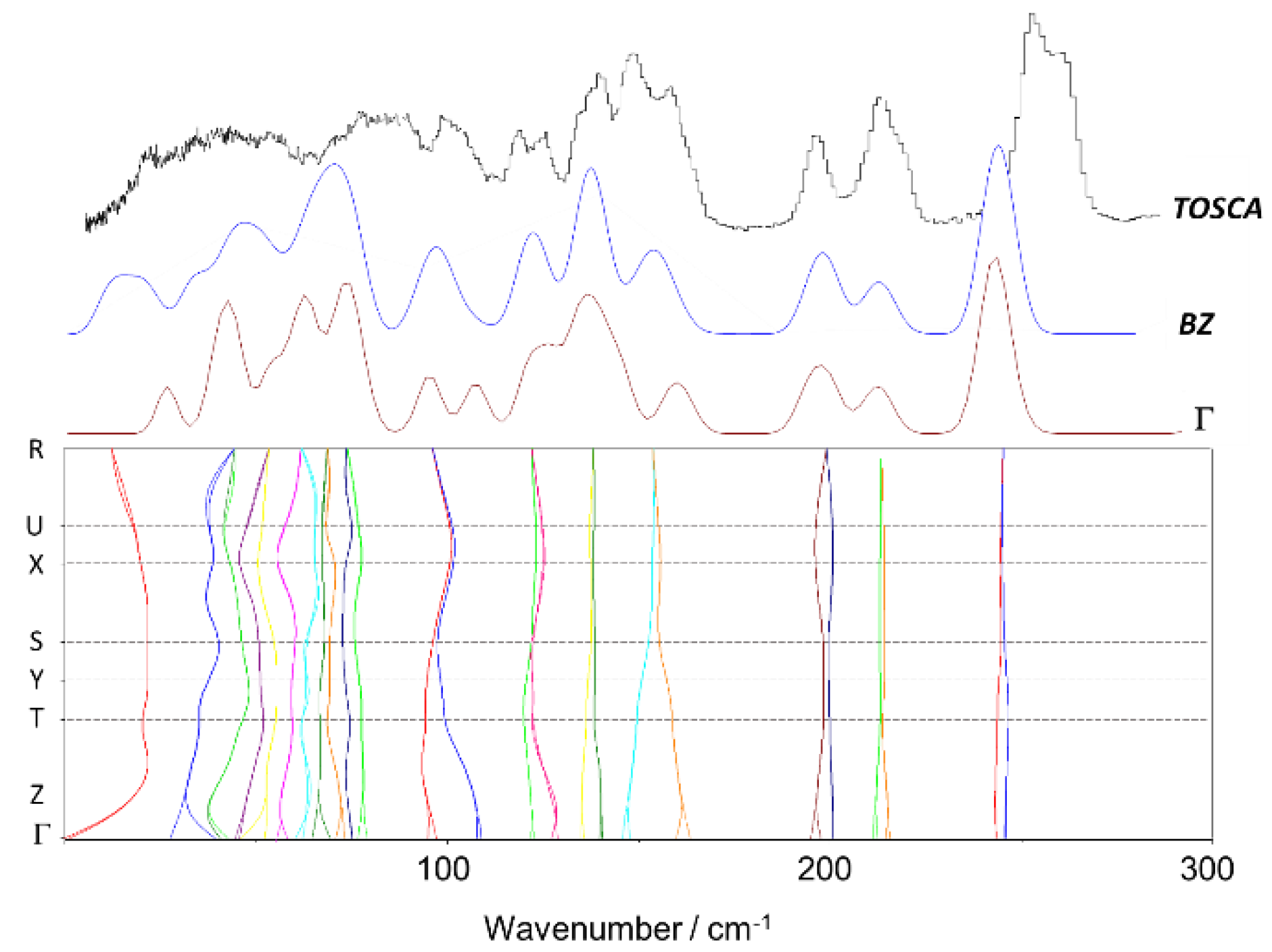
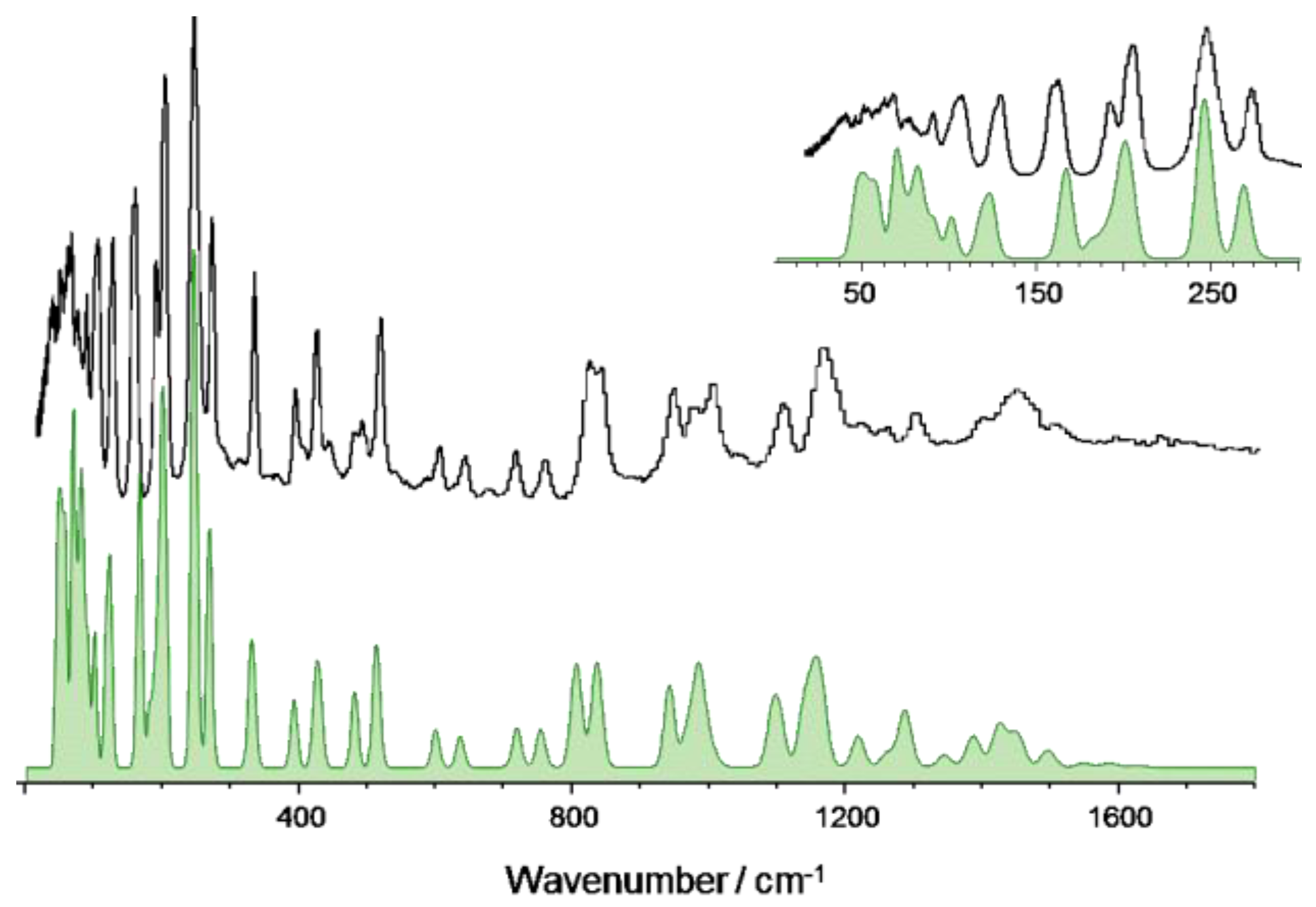
3.1. 2-Methoxybenzaldehyde (2MeOB)
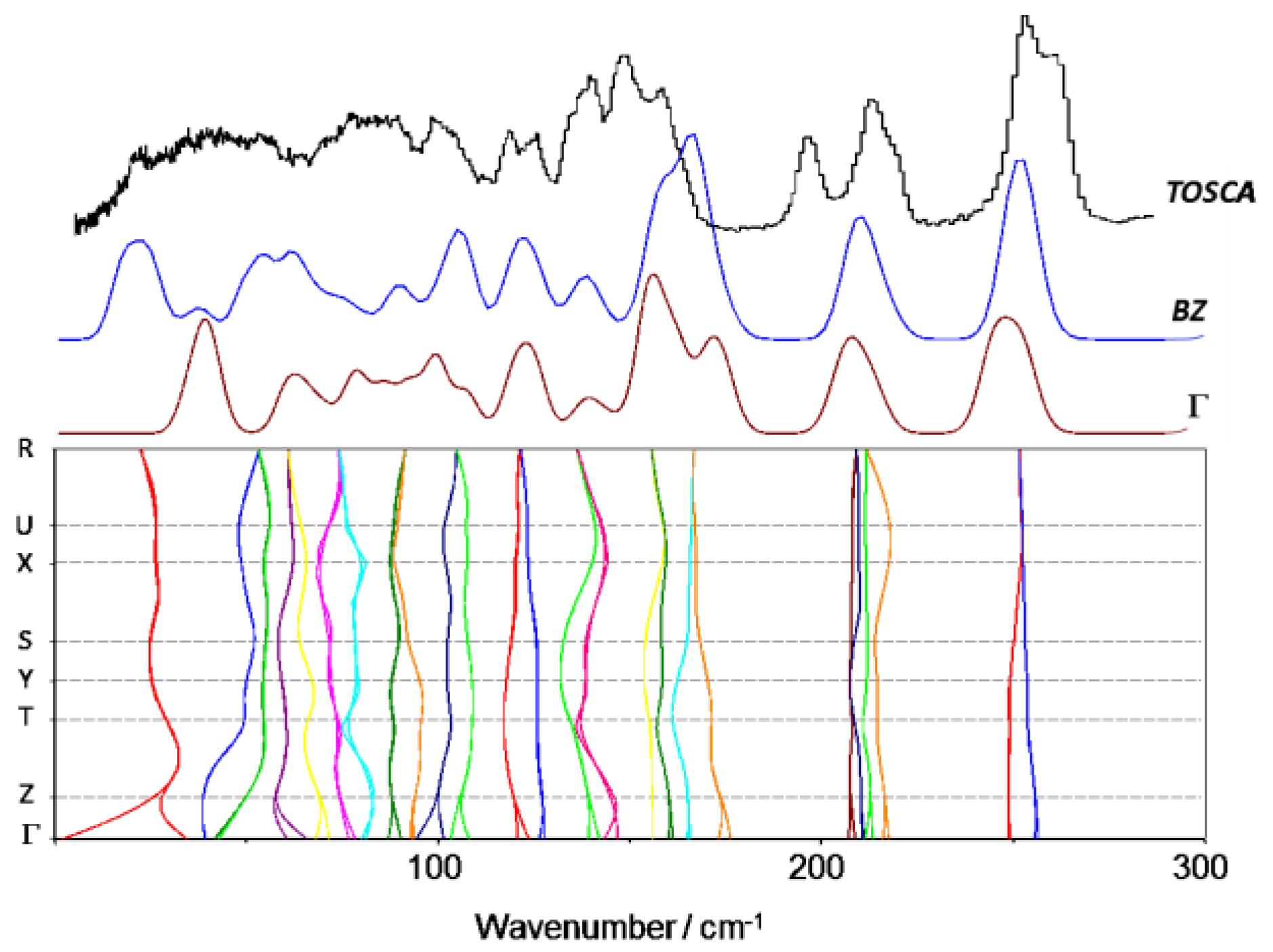
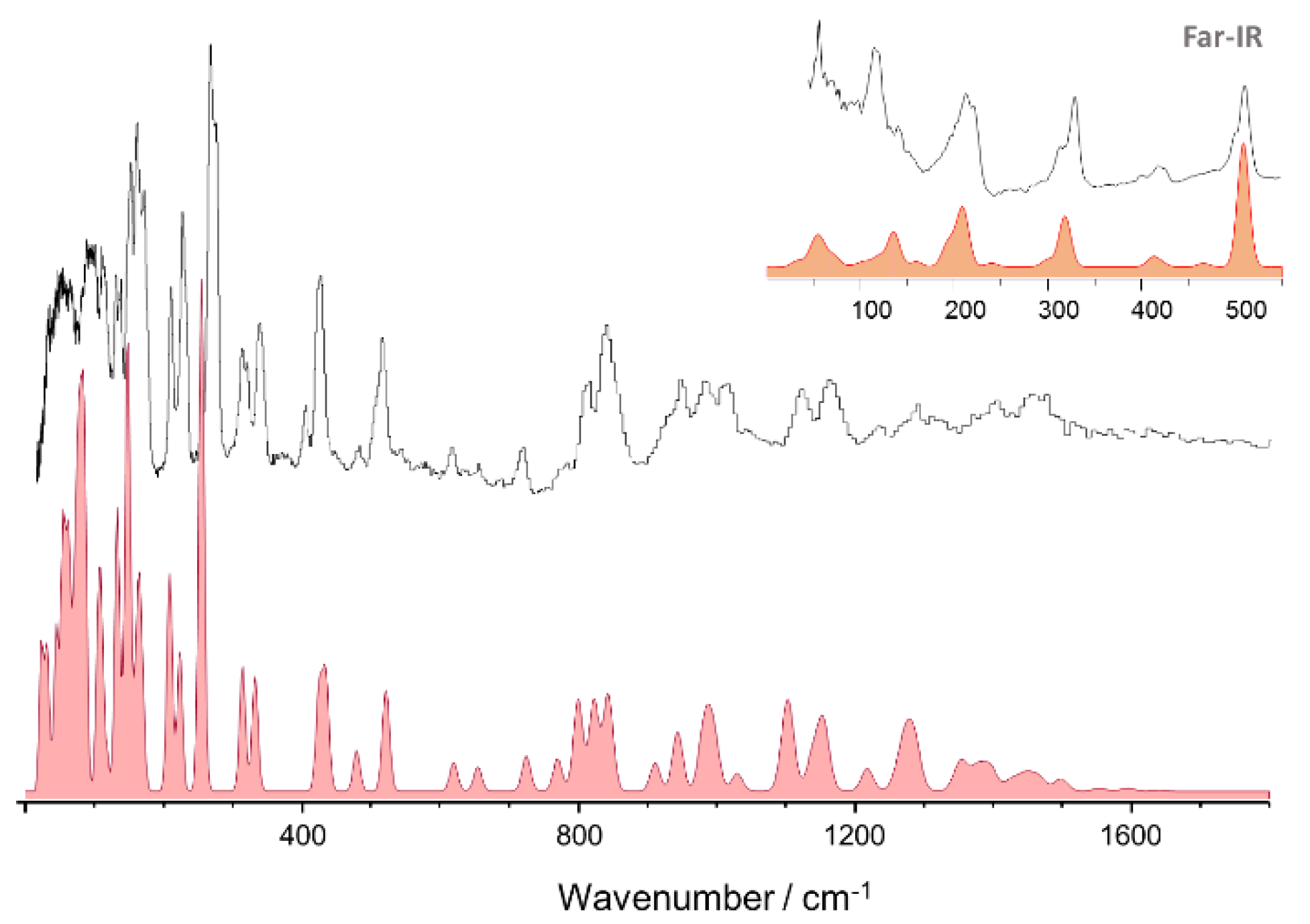
3.2. 4-Methoxybenzaldehyde (4MeOB)
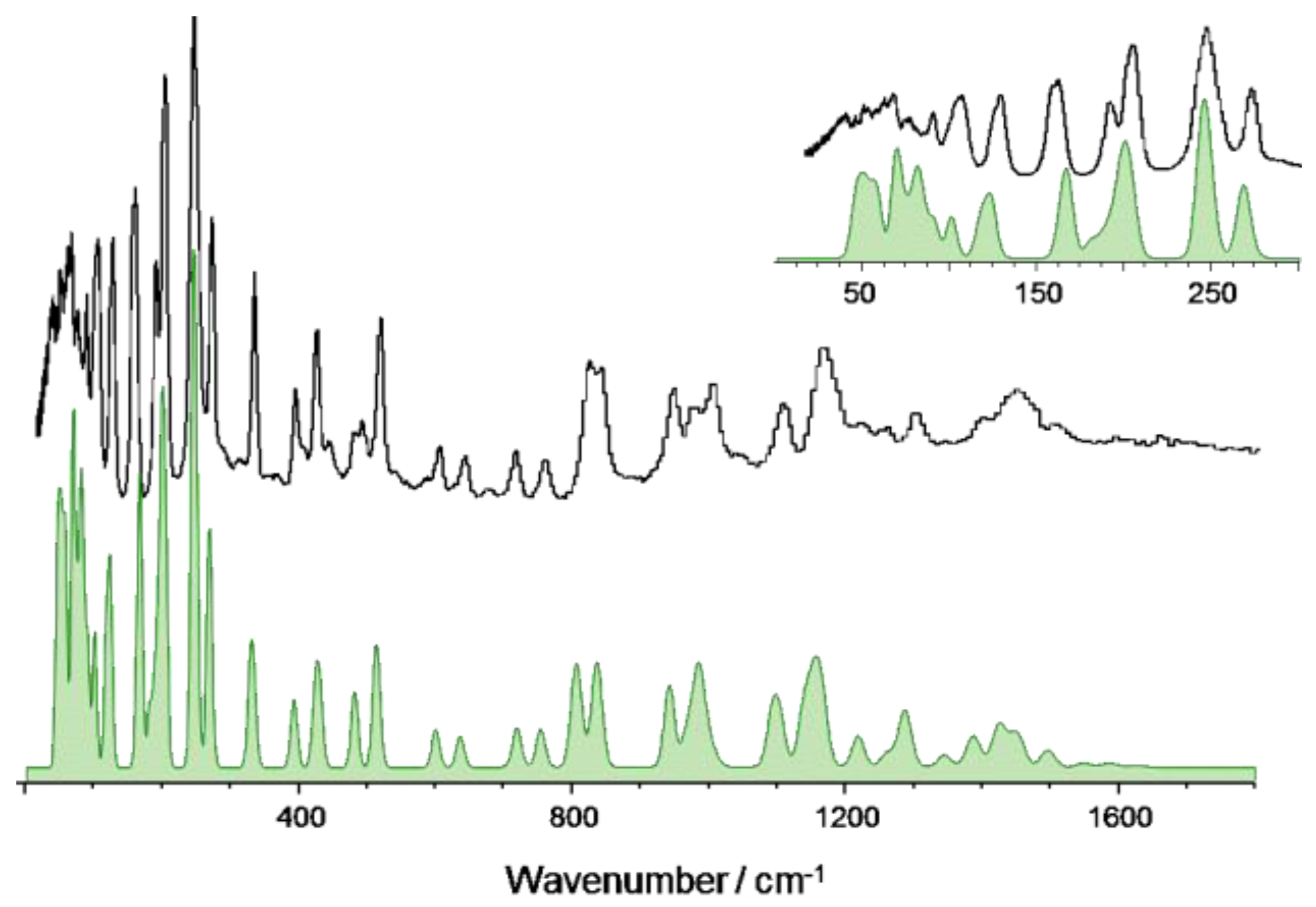
3.3. 4-Ethoxybenzaldehyde (4EtOB)
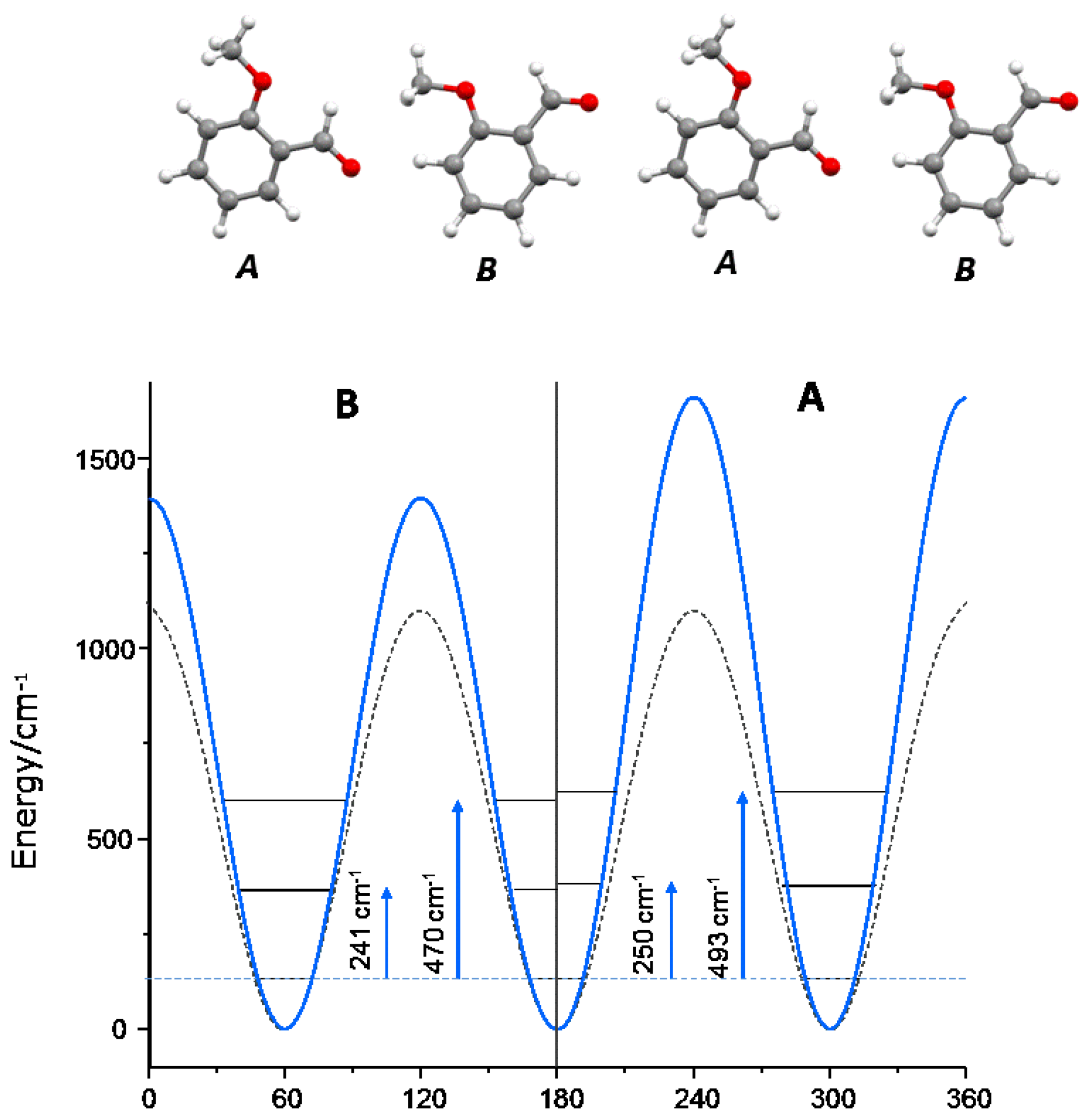
3.4. Dynamics of Internal Rotors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Araujo, C.F.; Freire, C.S.R.; Nolasco, M.M.; Ribeiro-Claro, P.; Rudic, S.; Silvestre, A.J.; Vaz, P.D. Hydrogen Bond Dynamics of Cellulose through Inelastic Neutron Scattering Spectroscopy. Biomacromolecules 2018, 19, 1305–1313. [Google Scholar] [CrossRef]
- Araujo, C.F.; Nolasco, M.M.; Ribeiro-Claro, P.J.A.; Rudić, S.; Silvestre, A.J.D.; Vaz, P.D.; Sousa, A.F. Inside PEF: Chain conformation and dynamics in crystalline and amorphous domains. Macromolecules 2018, 51, 3515–3526. [Google Scholar] [CrossRef]
- Parker, S.F.; Butler, I.R. Synthesis, computational studies, inelastic neutron scattering, infrared and raman spectroscopy of ruthenocene. Eur. J. Inorg. Chem. 2019, 2019, 1142–1146. [Google Scholar] [CrossRef] [Green Version]
- Nolasco, M.M.; Araujo, C.F.; Vaz, P.D.; Amado, A.M.; Ribeiro-Claro, P. Vibrational dynamics of crystalline 4-phenylbenzaldehyde from INS spectra and periodic DFT calculations. Molecules 2020, 25, 1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilski, P.; Drużbicki, K.; Jenczyk, J.; Mielcarek, J.; Wąsicki, J. Molecular and vibrational dynamics in the cholesterol-lowering agent lovastatin: Solid-state NMR, inelastic neutron scattering, and periodic DFT Study. J. Phys. Chem. B 2017, 121, 2776–2787. [Google Scholar] [CrossRef] [PubMed]
- Pawlukojć, A.; Hetmańczyk, Ł. INS, DFT and temperature dependent IR studies on dynamical properties of acetylcholine chloride. Vib. Spectrosc. 2016, 82, 37–43. [Google Scholar] [CrossRef]
- Drużbicki, K.; Mikuli, E.; Pałka, N.; Zalewski, S.; Ossowska-Chruściel, M.D. Polymorphism of resorcinol explored by complementary vibrational spectroscopy (FT-RS, THz-TDS, INS) and first-principles solid-state computations (Plane-Wave DFT). J. Phys. Chem. B 2015, 119, 1681–1695. [Google Scholar] [CrossRef]
- Zhong, L.; Parker, S.F. Structure and vibrational spectroscopy of methanesulfonic acid. R. Soc. Open Sci. 2018, 5, 181363. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro-Claro, P.J.; Vaz, P.D.; Nolasco, M.M.; Araujo, C.F.; Gil, F.; Amado, A.M. Vibrational dynamics of 4-fluorobenzaldehyde from periodic DFT calculations. Chem. Phys. Lett. X 2019, 2, 100006. [Google Scholar] [CrossRef]
- Harrelson, T.F.; Cheng, Y.Q.; Li, J.; Jacobs, I.E.; Ramirez-Cuesta, A.; Faller, R.; Moulé, A.J. Identifying Atomic Scale Structure in Undoped/Doped Semicrystalline P3HT Using Inelastic Neutron Scattering. Macromolecules 2017, 50, 2424–2435. [Google Scholar] [CrossRef] [Green Version]
- Nolasco, M.M.; Araujo, C.F.; Thiyagarajan, S.; Rudić, S.; Vaz, P.D.; Silvestre, A.J.D.; Ribeiro-Claro, P.J.A.; Sousa, A.F. Asymmetric monomer, amorphous polymer structure–property relationships in 2,4-FDCA and 2,4-PEF. Macromolecules 2020, 53, 1380–1387. [Google Scholar] [CrossRef]
- Vilela, C.; Freire, C.S.R.; Araújo, C.; Rudić, S.; Silvestre, A.J.D.; Vaz, P.D.; Ribeiro-Claro, P.J.A.; Nolasco, M.M. Understanding the structure and dynamics of nanocellulose-based composites with neutral and ionic poly(methacrylate) derivatives using inelastic neutron scattering and DFT calculations. Molecules 2020, 25, 1689. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro-Claro, P.J.A.; Drew, M.G.B.; Félix, V. C-H⋯O bonded dimers in 2-methoxy-benzaldehyde studied by X-ray crystallography, vibrational spectroscopy, and ab initio calculations. Chem. Phys. Lett. 2002, 356, 318–324. [Google Scholar] [CrossRef]
- Kirchner, M.T.; Bläser, D.; Boese, R.; Thakur, T.S.; Desiraju, G.R. Weak C-H⋯O hydrogen bonds in anisaldehyde, salicyl-aldehyde and cinnamaldehyde. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2011, 67, 387–390. [Google Scholar] [CrossRef] [Green Version]
- Parker, S.F.; Lennon, D.; Albers, P.W. Vibrational Spectroscopy with Neutrons: A Review of New Directions. Appl. Spectrosc. 2011, 65, 1325–1341. [Google Scholar] [CrossRef]
- Parker, S.F.; Fernandez-Alonso, F.; Ramirez-Cuesta, A.J.; Tomkinson, J.; Rudic, S.; Pinna, R.S.; Gorini, G.; Fernandez Castanon, J. Recent and future developments on TOSCA at ISIS. J. Phys. Conf. Ser. 2014, 554, 012003. [Google Scholar] [CrossRef]
- Arnold, O.; Bilheux, J.C.; Borreguero, J.M.; Buts, A.; Campbell, S.I.; Chapon, L.; Doucet, M.; Draper, N.; Leal, R.F.; Gigg, M.A.; et al. Mantid-Data analysis and visualization package for neutron scattering and mu SR experiments. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrometers Detect. Assoc. Equip. 2014, 764, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Refson, K.; Tulip, P.R.; Clark, S. Variational density-functional perturbation theory for dielectrics and lattice dynamics. Phys. Rev. B 2006, 73, 73. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro-Claro, P.; Vaz, P.D.; Nolasco, M.M.; Amado, A.M. Understanding the vibrational spectra of crystalline isoniazid: Raman, IR and INS spectroscopy and solid-state DFT study. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 204, 452–459. [Google Scholar] [CrossRef]
- Tkatchenko, A.; Scheffler, M. Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef] [Green Version]
- Jmol Development Team. Jmol. 2016. Available online: http://jmol.sourceforge.net/ (accessed on 8 August 2021).
- Dymkowski, K.; Parker, S.F.; Fernandez-Alonso, F.; Mukhopadhyay, S. AbINS: The modern software for INS interpretation. Phys. B Condens. Matter 2018, 551, 443–448. [Google Scholar] [CrossRef]
- Groner, P. Program Barrier. 2019. Available online: http://www.ifpan.edu.pl/~kisiel/prospe.htm (accessed on 8 August 2021).
- Drużbicki, K.; Mielcarek, J.; Kiwilsza, A.; Toupet, L.; Collet, E.; Pajzderska, A.; Wąsicki, J. Computationally assisted (solid-state density functional theory) structural (X-ray) and vibrational spectroscopy (FT-IR, FT-RS, TDs-THz) characterization of the cardiovascular drug lacidipine. Cryst. Growth Des. 2015, 15, 2817–2830. [Google Scholar] [CrossRef]
- Vennila, P.; Govindaraju, M.; Venkatesh, G.; Kamal, C. Molecular structure, vibrational spectral assignments (FT-IR and FT-RAMAN), NMR, NBO, HOMO-LUMO and NLO properties of O-methoxybenzaldehyde based on DFT calculations. J. Mol. Struct. 2016, 1111, 151–156. [Google Scholar] [CrossRef]
- Ribeiro-Claro, P.J.A.; De Batista Carvalho, L.A.E.; Amado, A.M. Evidence of dimerization through C-H⋯O interactions in liquid 4-methoxybenzaldehyde from raman spectra and Ab Initio calculations. J. Raman Spectrosc. 1997, 28, 867–872. [Google Scholar] [CrossRef]
- Gunasekaran, S.; Seshadri, S.; Muthu, S.; Kumaresan, S.; Arunbalaji, R. Vibrational spectroscopy investigation using ab initio and density functional theory on p-anisaldehyde. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2008, 70, 550–556. [Google Scholar] [CrossRef]
- Altun, A.; Swesi, O.; Alhatab, B. Structural and spectroscopic (UV–Vis, IR, Raman, and NMR) characteristics of anisaldehydes that are flavoring food additives: A density functional study in comparison with experiments. J. Mol. Struct. 2017, 1128, 590–605. [Google Scholar] [CrossRef]
- Venkoji, P. Vibrational spectra of monosubstituted benzaldehydes—o-ethoxy-, p-ethoxy-, p-carboxy- and p-cyanobenzaldehydes. Spectrochim. Acta Part A Mol. Spectrosc. 1986, 42, 1301–1305. [Google Scholar] [CrossRef]
- Ferres, L.; Mouhib, H.; Stahl, W.; Nguyen, H.V.L. Methyl internal rotation in the microwave spectrum of o-methyl anisole. ChemPhysChem 2017, 18, 1855–1859. [Google Scholar] [CrossRef]
- Ferres, L.; Stahl, W.; Kleiner, I.; Nguyen, H.V.L. The effect of internal rotation in p-methyl anisole studied by microwave spectroscopy. J. Mol. Spectrosc. 2018, 343, 44–49. [Google Scholar] [CrossRef]
- Melandri, S.; Maris, A.; Favero, P.G.; Favero, L.B.; Caminati, W.; Meyer, R. Millimeter-wave absorption free jet spectrum, barriers to internal rotation, and torsional relaxation in p-anisaldehyde. J. Mol. Spectrosc. 1997, 185, 374–383. [Google Scholar] [CrossRef]
- Bohn, R.K.; Farag, M.S.; Ott, C.M.; Radhakrishnan, J.; Sorenson, S.A.; True, N.S. Rotational spectra of para-anisaldehyde—Assignment of the planar conformers and observation of torsionally excited states. J. Mol. Struct. 1992, 268, 107–121. [Google Scholar] [CrossRef]
- Zachariou, A.; Hawkins, A.; Collier, P.; Howe, R.F.; Lennon, D.; Parker, S.F. The methyl torsion in unsaturated compounds. ACS Omega 2020, 5, 2755–2765. [Google Scholar] [CrossRef] [Green Version]
- Adilina, I.B.; Aulia, F.; Fitriady, M.A.; Oemry, F.; Widjaya, R.R.; Parker, S.F. Computational and spectroscopic studies of carbon disulfide. Molecules 2020, 25, 1901. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | X-ray [13] | CASTEP |
|---|---|---|
| Bond length/pm | ||
| C7=O1 | 121.1 | 123.2 |
| C2–O2 | 135.8 | 135.8 |
| O2–C8 | 142.8 | 143.5 |
| Bond angle/° | ||
| C1–C7=O1 | 124.1 | 124.2 |
| C2–O2–C8 | 118.6 | 117.8 |
| Dihedral angle/° | ||
| C6C1–C7O1 | −5.4 | −3.3 |
| C1C2–O2C8 | −178.7 | −177.8 |
| C-H…O distance/pm | ||
| C3(A)…O1(B) | 347.4 | 336.3 |
| Calculated/cm–1 (CASTEP) 1 | Experimental/cm–1 (TOSCA) | Approximate Description |
|---|---|---|
| 3105 | 3070 | νCH |
| 1440 | 1435 | βCH3 |
| 1382 | 1384 | βCH(=O) |
| 1277 | 1279 | βCH |
| 1235 | 1232 | βCH |
| 1184 | 1176 | βCH |
| 1135–1145 | 1145 | βCH+rock CH3 |
| 1091 | 1088 | βCH |
| 1031 | 1031 | βCH |
| 1015 | 1002 | νO–CH3 |
| 991 | 983 | γCH(=O) |
| 956 | 951 | γCH |
| 867 | 865 | γCH |
| 828 | 830 | αα ring |
| 770 | 766 | γCH |
| 724 | 718 | δ ring |
| 638 | 640 | α ring |
| 575 | 575 | α ring |
| 526 | 527 | δ ring |
| 472 | 475 | α ring |
| 445 | 438 | δ ring |
| 396 | 399 | β ring–OCH3 |
| 285 | 290 | γ ring–OCH3 |
| 270 | 280 | β C–O–C (A) |
| 263 | 267 | β C–O–C (B) |
| 238 | 250 | τ O–CH3 (A) |
| 228 | 241 | τ O–CH3 (B) |
| 200 | 208 | β ring–CHO |
| 168 | 178 | γ ring–CHO (A) |
| 162 | 172 | γ ring–CHO (B) |
| 156 | 160 | τ C–OCH3 (A) |
| 146 | 148 | τ C–OCH3 (B) |
| 140 | 139 | τ C–CHO (B) |
| 132 | 126 | τ C–CHO (A) |
| 91 | 98 | Libration |
| 67 | 81 | Libration |
| 50 | 70 | Translation |
| 40 | 62 | Translation |
| Parameter | X-ray [14] | CASTEP |
|---|---|---|
| Bond length/pm | ||
| C7=O1 | 121.4 | 123.0 |
| C2–O2 | 135.7 | 135.3 |
| O2–C8 | 142.5 | 144.0 |
| Bond angle/° | ||
| C1–C7=O1 | 126.5 | 126.2 |
| C4–O2-C8 | 118.0 | 117.6 |
| Dihedral angle/° | ||
| C6C1–C7O1 | 4.3 | 3.1 |
| C3C4–O2C8 | −2.9 | −2.8 |
| C-H…O distance/pm | ||
| C3(H)…O2 | 360.3 | 336.2 |
| C5(H)…O1 | 344.7 | 335.7 |
| C8(HA)…O1 | 358.2 | 350.9 |
| C8(HB)…O1 | 343.4 | 337.0 |
| Calculated/cm–1 (CASTEP) 1 | Experimental/cm–1 (TOSCA) | Approximate Description |
|---|---|---|
| 3110 | 3096 | νCH |
| 1496 | 1508 | βCH |
| 1446 | 1451 | βCH3 |
| 1387 | 1396 | βCH(=O) |
| 1287 | 1304 | βCH |
| 1156 | 1170 | βCH |
| 1097 | 1110 | βCH |
| 986 | 1004 | γCH(=O) |
| 970 | 977 | γCH |
| 941 | 948 | γCH |
| 837 | 840 | γCH |
| 806 | 827 | γCH |
| 753 | 760 | νCC |
| 719 | 717 | δ ring |
| 636 | 644 | α ring |
| 600 | 605 | α ring |
| 513 | 518 | δ ring |
| 481 | 479 | β ring–OCH3 |
| 425 | 426 | δ ring |
| 392 | 395 | α ring |
| 329 | 335 | γ ring–OCH3 |
| 269 | 273 | β C–O–C |
| 245 | 247 | τ O–CH3 |
| 200 | 205 | γ ring–CHO |
| 183 | 190 | β ring–CHO |
| 165 | 160 | τ C–OCH3 |
| 121 | 128 | τ C–CHO |
| 100 | 105 | Libration |
| 88 | 90 | Libration |
| 80 | 75 | Libration |
| 69 | 67 | Libration |
| 54 | 51 | Translation |
| 49 | 39 | Translation |
| Parameter | Fixed Cell | Relaxed Cell |
|---|---|---|
| Cell dimensions/pm | ||
| a | 560 | 616.2 |
| b | 903.4 | 892.6 |
| c | 1750 | 1442.6 |
| Bond length/pm | ||
| C7=O1 | 122.8 | 122.8 |
| C4–O2 | 135.4 | 135.2 |
| O2–C8 | 145.1 | 145.2 |
| Bond angle/° | ||
| C1–C7=O1 | 126.3 | 126.2 |
| C4–O2–C8 | 118.1 | 117.8 |
| Dihedral angle/° | ||
| C6C1–C7O1 | 1.8 | 4.5 |
| C3C4–O2C8 | −2.3 | −4.1 |
| C-H…O distance/pm | ||
| C5(H)…O1 | 341.3 | 339.3 |
| C9(H)…O1 | 342.8 | 352.1 |
| C8(HB)…O2 | 400.0 | 392.0 |
| Calculated/cm–1 (CASTEP) 1 | Experimental/cm–1 (TOSCA) | Approximate Description |
|---|---|---|
| 3141 | 3124 | νCH |
| 1440 | 1470 | βCH3 |
| 1381 | 1401 | βCH(=O) |
| 1346 | 1375 | δCH2 wag |
| 1266 | 1293 | δCH2 twist |
| 1208 | 1236 | νC–CHO |
| 1140 | 1165 | βCH |
| 1094 | 1123 | βCH |
| 1021 | 1044 | νO–CH2CH3 |
| 979 | 1016 | γCH(=O) |
| 933 | 985 | γ CH |
| 900 | 950 | γ CH |
| 832 | 840 | γ CH |
| 792 | 812 | γ CH |
| 760 | 780 | νCC |
| 714 | 720 | δ ring |
| 647 | 655 | α ring |
| 611 | 618 | α ring |
| 511 | 519 | δ ring |
| 471 | 482 | β ring–OCH2CH3 |
| 421 | 426 | δ ring |
| 322 | 339 | γ ring–OCH2CH3 |
| 304 | 315 | β O–C–C |
| 245 | 268/274 | τ O–CH3 |
| 214 | 227/233 | β C–O–C |
| 199 | 211 | γ ring–CHO |
| 155 | 161/170 | β ring–CHO |
| 138 | 152 | τ C–OCH2CH3 |
| 123 | 131/137 | τ O–CH2CH3 |
| 98 | 115 | τ C–CHO |
| 71 | 96 | Libration |
| 49 | 61 | Libration |
| 35 | 46 | Translation |
| 17 | 34 | Translation |
| 2MeOB (B/D) | (A/C) | 4MeOB | 4EtOB | |
|---|---|---|---|---|
| 0–1 | 241 | 250 | 247 | 267 |
| 0–2 | 470 | 493 | 480 | 508 |
| V3 | 470 | 493 | 480 | 508 |
| V6 | −16 | −73 | 3 | 147 |
| V3 isolated (G09) | 1107 | 1055 | 1079 | |
| V3 gas phase | 804 [35] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro-Claro, P.J.A.; Vaz, P.D.; Nolasco, M.M.; Gil, F.P.S.C.; de Carvalho, L.A.E.B.; Marques, M.P.M.; Amado, A.M. New Insights on the Vibrational Dynamics of 2-Methoxy-, 4-Methoxy- and 4-Ethoxy-Benzaldehyde from INS Spectra and Periodic DFT Calculations. Materials 2021, 14, 4561. https://doi.org/10.3390/ma14164561
Ribeiro-Claro PJA, Vaz PD, Nolasco MM, Gil FPSC, de Carvalho LAEB, Marques MPM, Amado AM. New Insights on the Vibrational Dynamics of 2-Methoxy-, 4-Methoxy- and 4-Ethoxy-Benzaldehyde from INS Spectra and Periodic DFT Calculations. Materials. 2021; 14(16):4561. https://doi.org/10.3390/ma14164561
Chicago/Turabian StyleRibeiro-Claro, Paulo J. A., Pedro D. Vaz, Mariela M. Nolasco, Francisco P. S. C. Gil, Luís A. E. Batista de Carvalho, Maria Paula M. Marques, and Ana M. Amado. 2021. "New Insights on the Vibrational Dynamics of 2-Methoxy-, 4-Methoxy- and 4-Ethoxy-Benzaldehyde from INS Spectra and Periodic DFT Calculations" Materials 14, no. 16: 4561. https://doi.org/10.3390/ma14164561
APA StyleRibeiro-Claro, P. J. A., Vaz, P. D., Nolasco, M. M., Gil, F. P. S. C., de Carvalho, L. A. E. B., Marques, M. P. M., & Amado, A. M. (2021). New Insights on the Vibrational Dynamics of 2-Methoxy-, 4-Methoxy- and 4-Ethoxy-Benzaldehyde from INS Spectra and Periodic DFT Calculations. Materials, 14(16), 4561. https://doi.org/10.3390/ma14164561