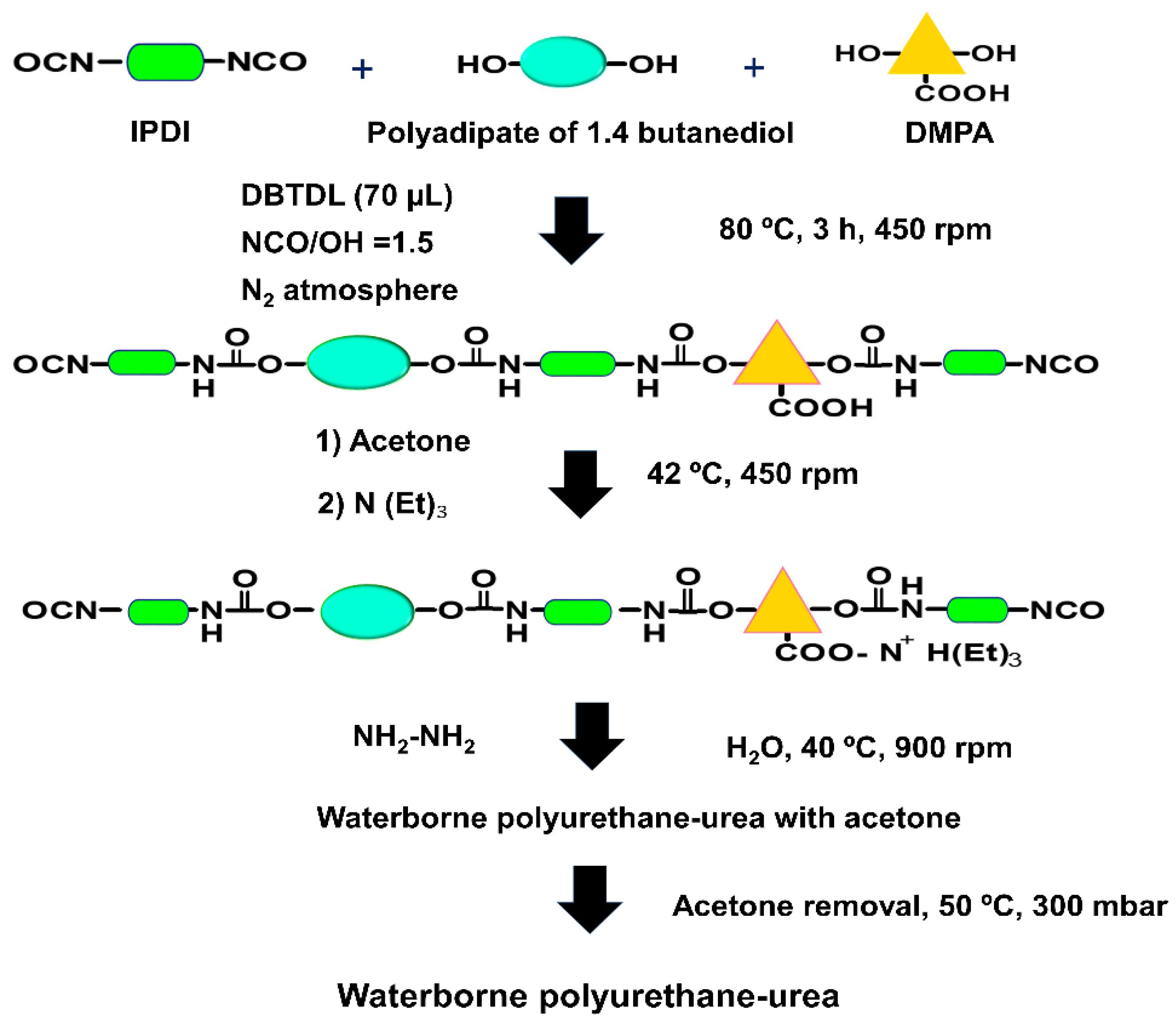
3.1. Characterization of the GO Derivatives
The amounts of the functional groups on the graphene oxide derivative surfaces (GO, A-GO, r-GO) obtained by XPS are shown in
Table 1. The GO surface shows the lowest amount of carbon and the highest amount of oxygen, indicating higher degree of surface oxidation, and both r-GO and A-GO surfaces have similar amounts of carbon and oxygen, but the amount of nitrogen is somewhat higher in A-GO.
Figure S1 of the Supplementary Materials file shows the curve fittings of the high resolution C1s spectra of the GO derivatives; the ones of O1s and N1s of A-GO are shown, as representative examples, in
Figure S2 of the Supplementary Materials file. The percentages of the carbon species on the GO, A-GO, and r-GO surfaces were assessed from the C1s spectra and they are shown in
Table 2; the assignment of the carbon species was carried out according to references [
26,
27]. The GO surface contains the lowest percentage of C-C and the highest number of C-O groups. The percentages of C-O groups are similar in the r-GO and A-GO surfaces, and the r-GO surface shows the highest percentage of C=O groups. Therefore, all GO derivative surfaces show the same kind of surface carbon-oxygen functional groups but in different amounts, and, additionally, the A-GO surface contains 88 at.% C-N (sp
3)—binding energy = 399.5 eV) and 12 at.% -NH
2 (sp
2) species—binding energy = 401.1 eV)—
Figure S2 of the Supplementary Materials.
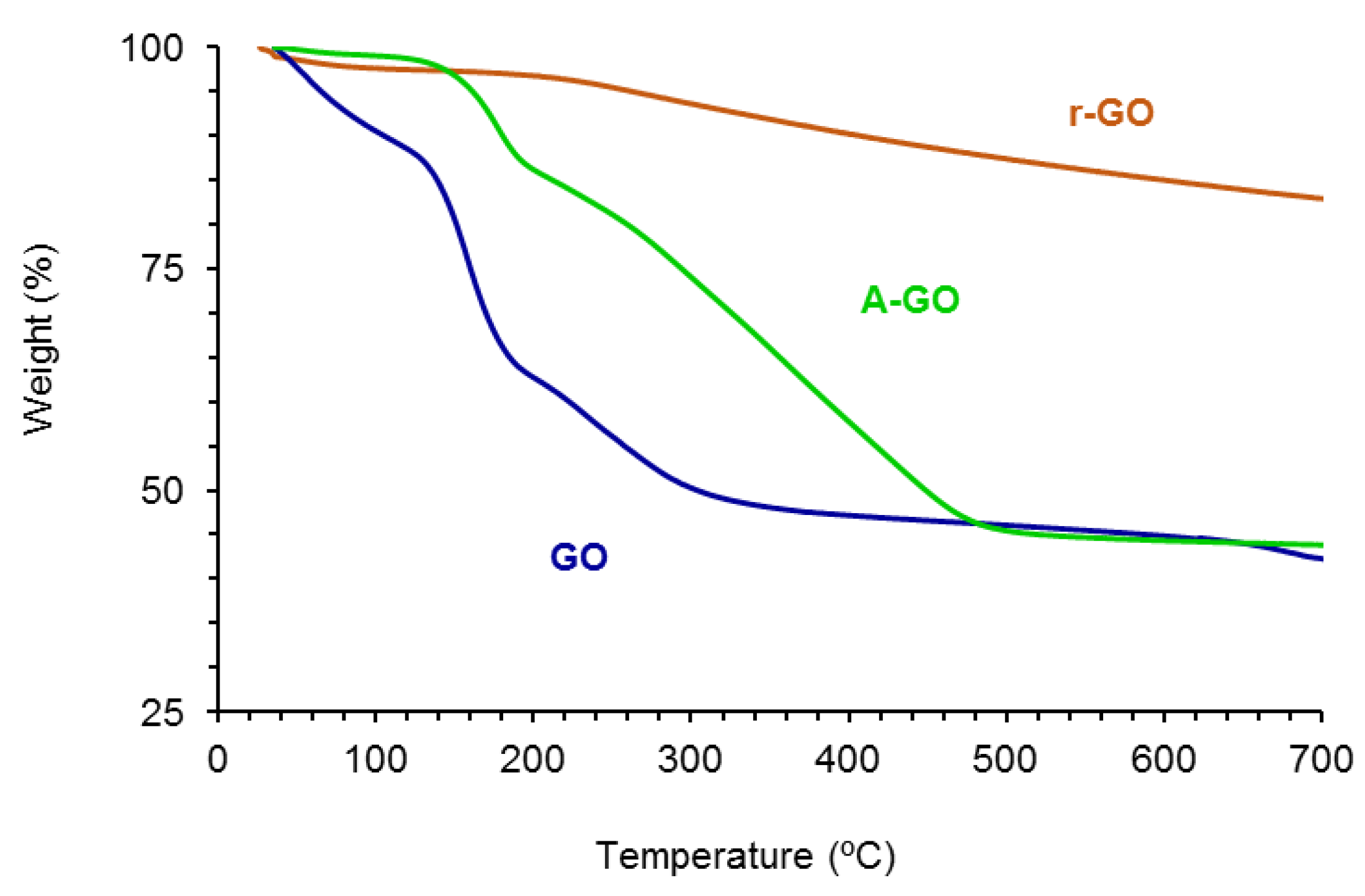
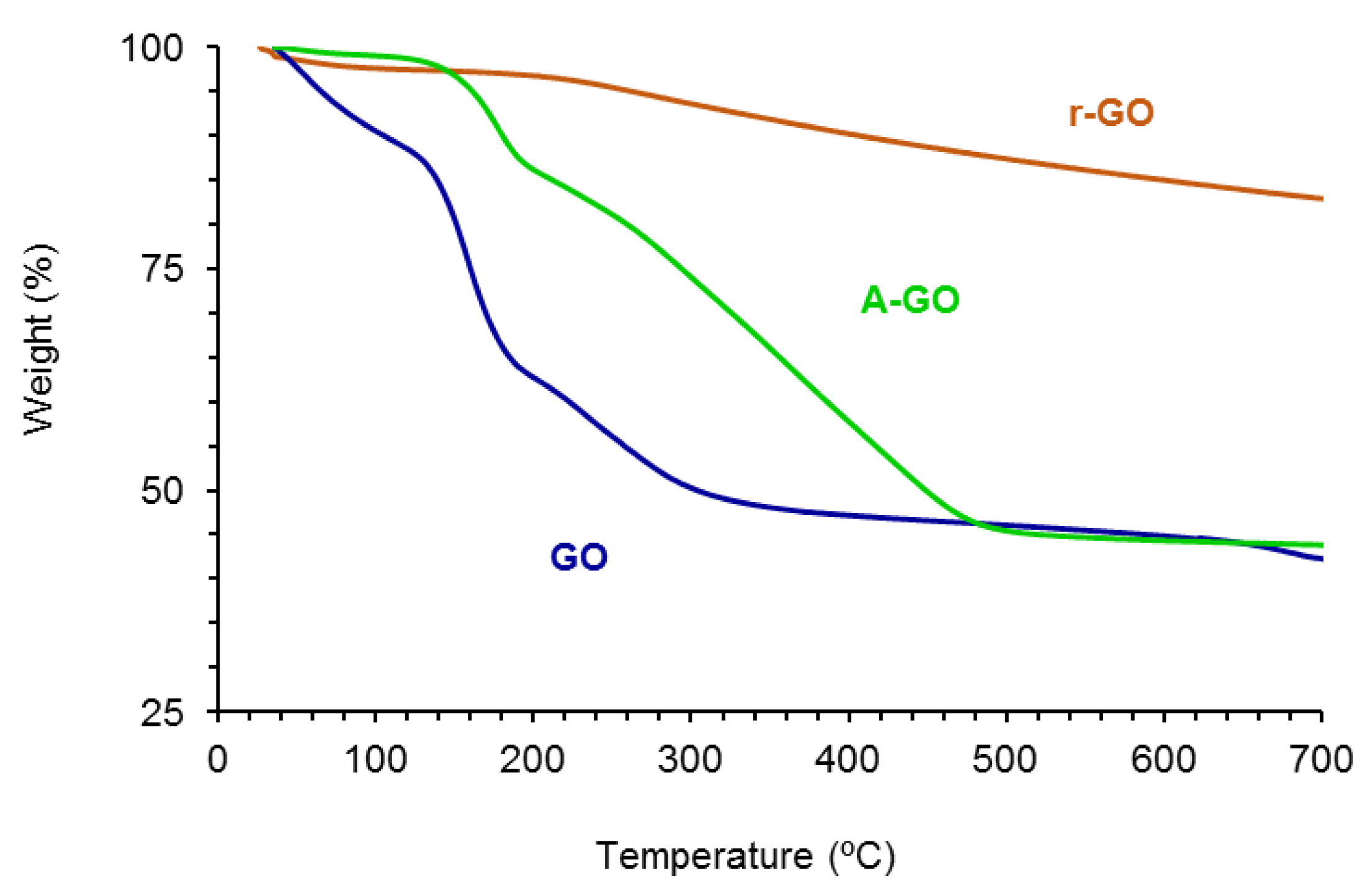
The amounts of functional groups of GO, A-GO and r-GO were assessed by TGA.
Figure 4 shows the variation of the weight as a function of the temperature of the GO derivatives. The amounts of functional groups are high and somewhat similar in GO (61 wt.%) and A-GO (56 wt.%), but r-GO shows only 18 wt.% functional groups. The nature of the functional groups in the GO derivatives was assessed from the derivative thermal gravimetric analysis plots (
Figure S3 of the Supplementary Materials file). GO shows the thermal decompositions due to absorbed moisture (49 °C), -C-OH (158, 231–257 °C), and –O-C=O (676–688 °C) groups—
Table 3, whereas A-GO shows the thermal decompositions of absorbed moisture (50 °C), -C-N (177 °C), -C-OH (263 °C), and C=O (361, 438 °C) groups. On the other hand, r-GO shows two thermal decompositions due to absorbed moisture (54 °C) and -C-OH groups (258 °C). The thermal decomposition of the C-OH groups appears at about 257–258 °C in all graphene oxide derivatives, but in GO some additional thermal decompositions at 158 and 231 °C of the C-OH groups also appear, indicating different interactions with the graphene sheets. Therefore, GO shows the highest percentage of -C-OH groups and A-GO shows the highest percentage of C=O groups; furthermore, A-GO contains 18 wt.% C-N groups due to the treatment of GO with dodecyl amine, in agreement with the findings of Wang et al. [
28].
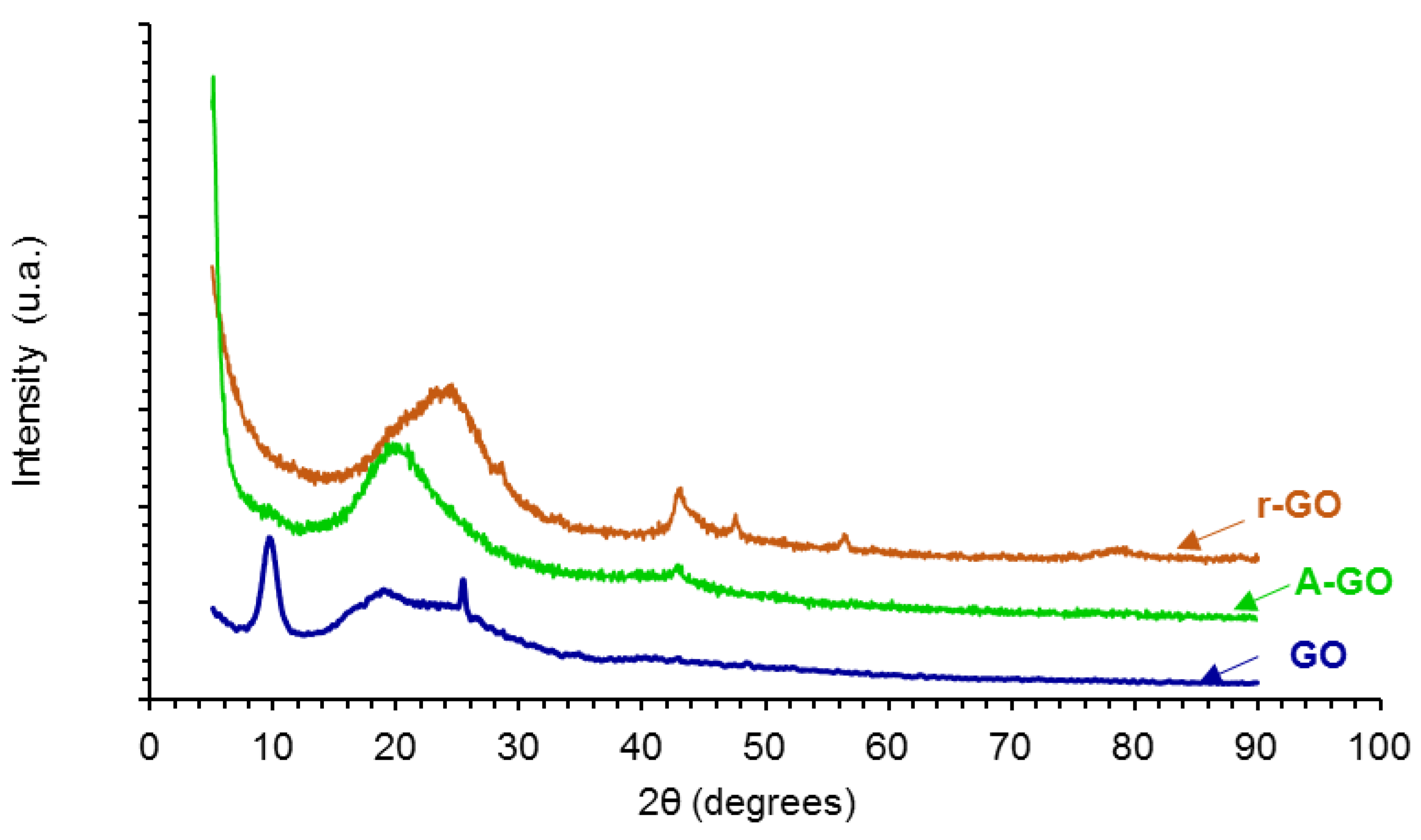
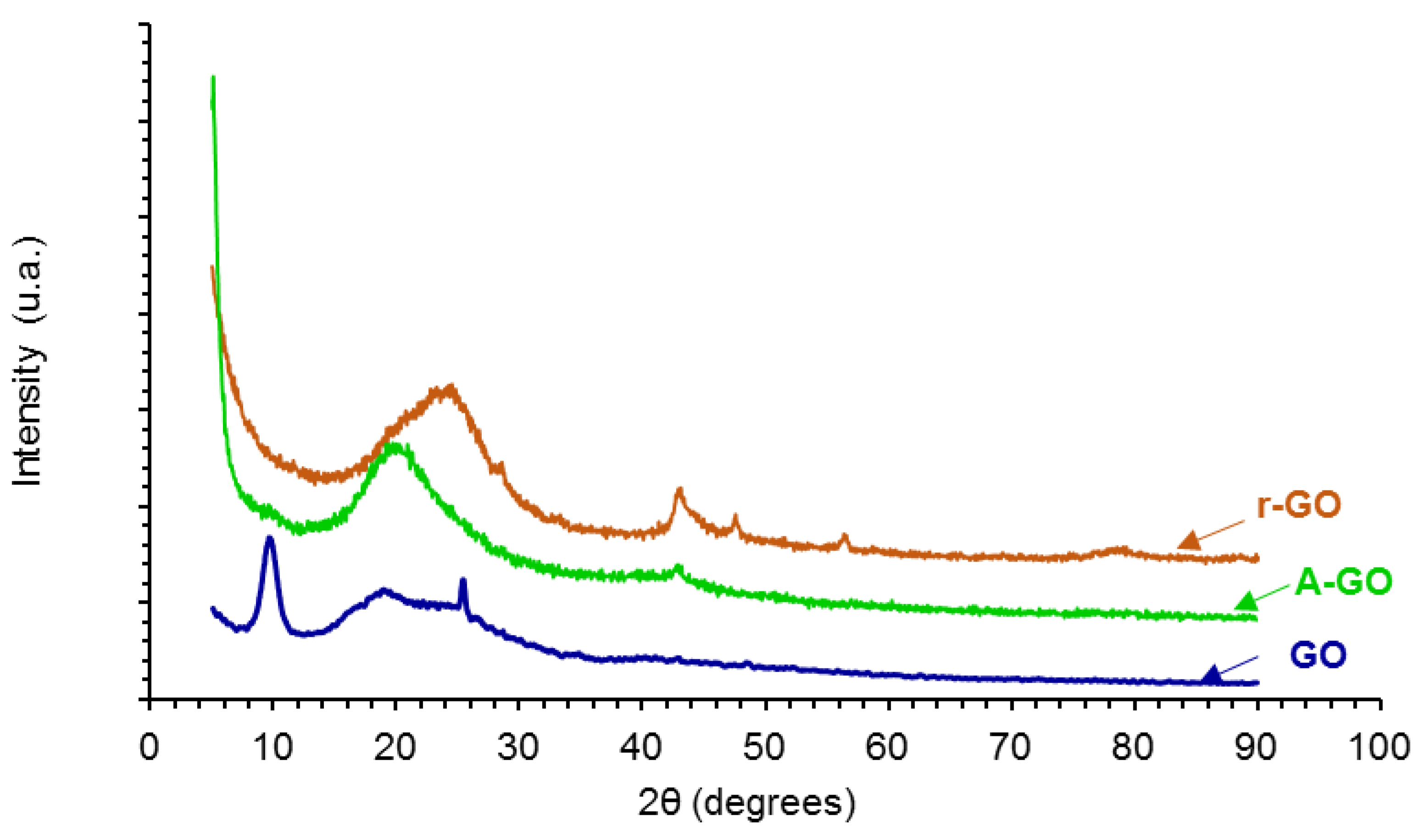
The X-ray diffractograms of the graphene oxide derivatives (GO, A-GO, r-GO) are shown in
Figure 5. GO shows three main diffraction peaks at 2Ɵ values of 9.55, 18.30, and 25.35°, the one at 9.55° is characteristic of GO [
29]. The peak at 2Ɵ value of 9.55° is associated to the (101) diffraction plane due to the formation of oxygen-containing groups in GO which causes an expansion of the C–C interplanar spacing of the graphite, and the two peaks at 2Ɵ values of 18.30 and 25.35° are associated to the oxidation of graphite [
29,
30]. On the other hand, the X-ray diffractogram of r-GO shows the varnishing of the peak at 2Ɵ value of 9.55° caused by the dismantle of the regular stacking of the GO sheets and the aggregation of the graphene sheets, the broad weak peak at 2Ɵ value of 20.75° is retained, indicating parallel stacking of the r-GO sheets [
29]. Furthermore, the reduction of the GO removes the most oxygen containing groups in the interstitial graphitic spaces and restores the C–C lattice of the graphite and, therefore, the diffractogram of r-GO shows some peaks of graphite at 2Ɵ values of 42.9°—(101) plane -, 47.55°—(102) plane -, 56.55°—(004) plane -, and 77.1°—(110) plane [
31]. The amine-functionalization of the GO also shows the varnishing of the peak at 2Ɵ value of 9.55° caused by the dismantle of the regular stacking of the GO sheets and the broad weak peak at 2Ɵ value of 19.15° is retained, indicating parallel stacking of the A-GO sheets; in addition, some aggregation of the graphene sheets in A-GO is evidenced by the diffraction peak at 2Ɵ value of 42.9° due to the (101) plane of the graphite [
30].
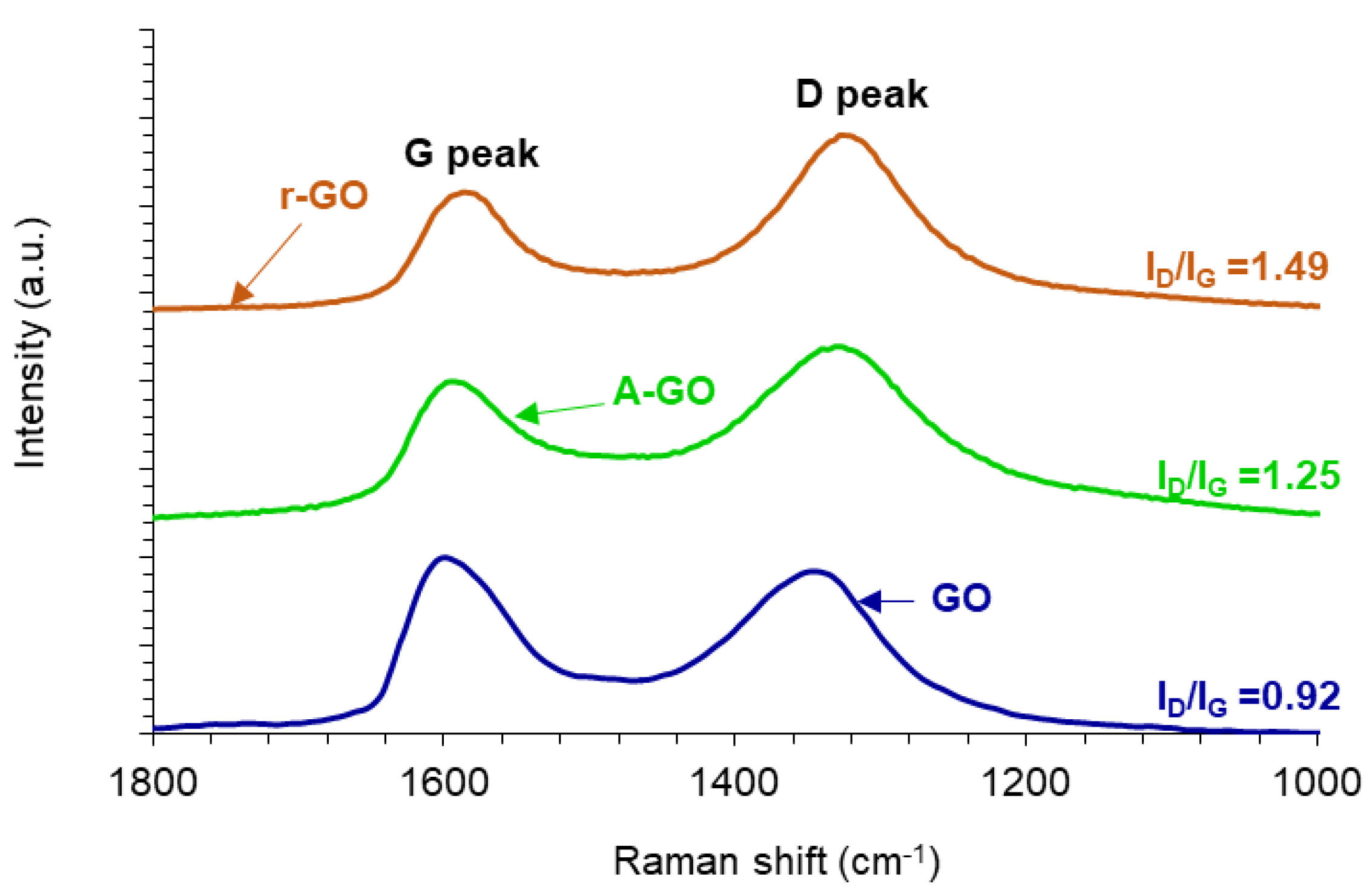
The structural changes produced during the reduction and amine-functionalization of GO are evidenced in the Raman spectra (
Figure 6). All Raman spectra show the D band at 1342 cm
−1 related to the disordered structure of GO, and the G band at 1598 cm
−1 related to C-C bond of sp
2 carbon. The ratio of the intensities of the D and G bands (I
D/I
G) is related to the order of graphene nano-sheets, i.e., the increase of the I
D/I
G ratio indicates a decrease of the average size of the C-C sp
2 nano-sheets. The I
D/I
G ratios in the Raman spectra of r-GO and A-GO are higher than in GO due to the presence of unrepaired defects after the removal of oxygen-containing functional groups. Thus, GO has the highest average size of the C-C sp
2 nano-sheets whereas r-GO has the lowest. It is worthy to note that these I
D/I
G ratios are consistent with the values in the literature [
32,
33].
The increase in the ordered structure of the graphene nano-sheets in A-GO and r-GO is also evidenced in the TEM micrographs (
Figure 7). GO shows some individual graphene nano-sheets, but the most common are 4–10 stacked nano-sheets of less than 20 nm thick and 2–5 µm long. A-GO shows organized and highly stacked graphene nano-sheets whereas r-GO depicts large ultrathin and transparent silk curtain wave-like structure with a large number of wrinkles. Therefore, r-GO and, mainly, A-GO show larger number of stacked graphene nano-sheets than GO. This anticipates more net interactions between the polymers and GO than with r-GO and, particularly, A-GO.
In summary, the GO derivatives differ in surface chemistry and morphology. The GO surface contains the highest amount of oxygen functional groups, mainly C-O groups, and both r-GO and A-GO surfaces have lower and similar amounts of oxygen functional groups, but the amount of nitrogen is somewhat higher in A-GO. Furthermore, GO has a fewer number of stacked graphene nano-sheets, r-GO depicts large ultrathin wave-like stacked graphene nano-sheets, and A-GO shows the most organized and stacked graphene nano-sheets. Therefore, different extent of interactions of those GO derivatives with the waterborne poly(urethane urea) can be anticipated.
3.3. Characterization of the PUs
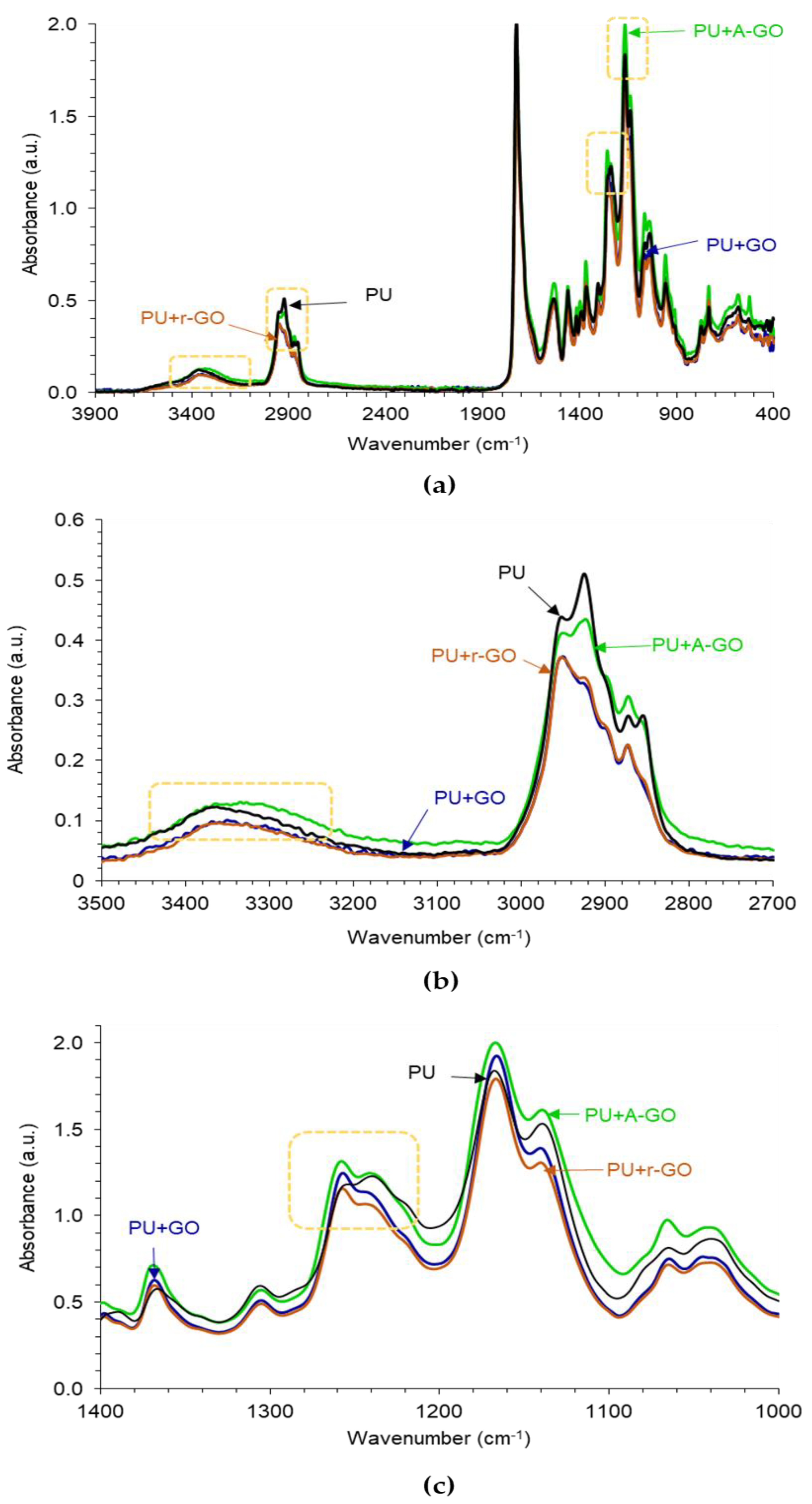
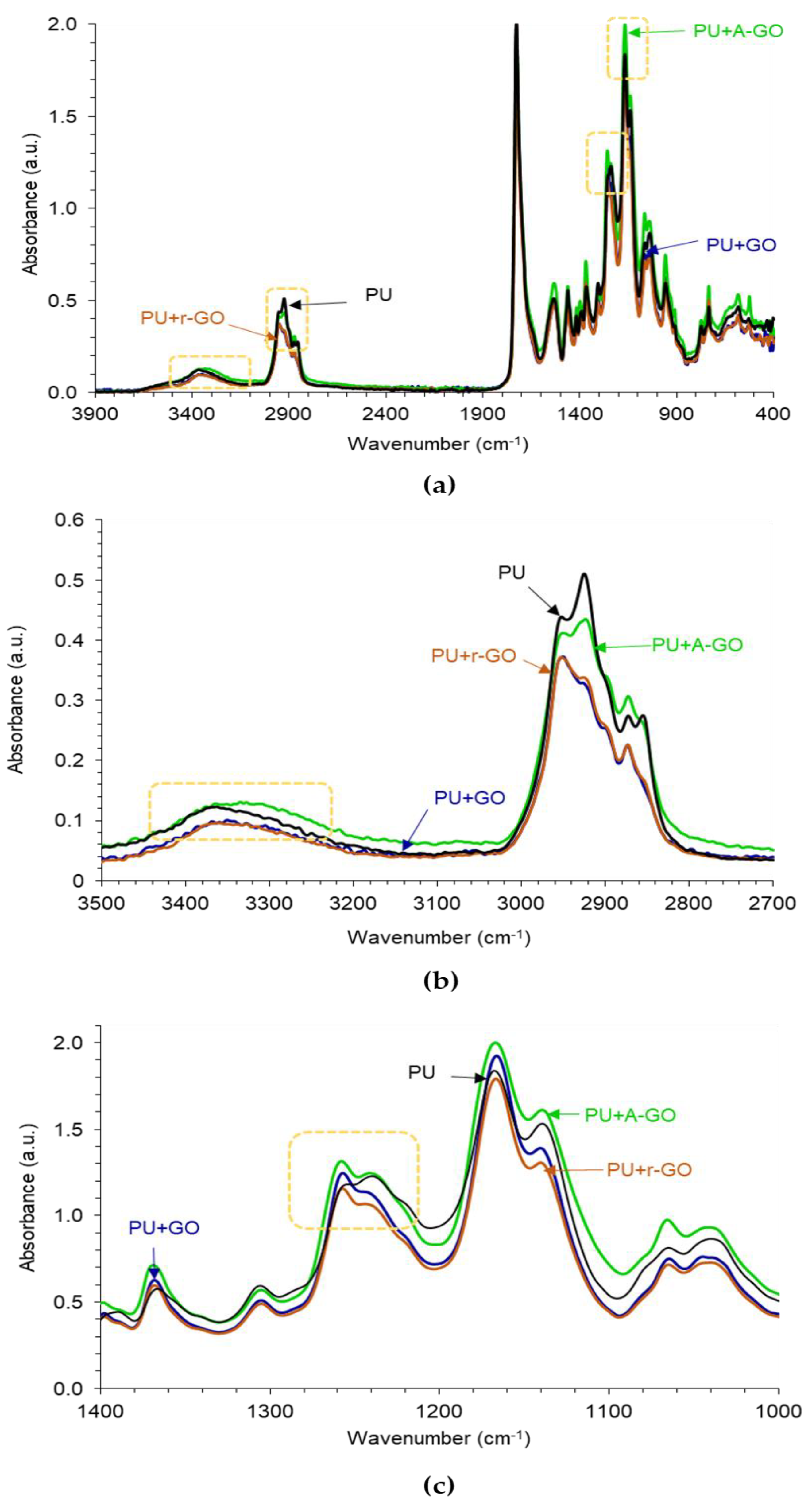
The ATR-IR spectra of the poly(urethane urea)s (PUs) without and with 0.04 wt.% GO derivatives are given in
Figure 9a. The typical absorption bands of the hard and the soft segments in the PUs can be distinguished in the ATR-IR spectra. The absorption bands due to the hard segments appear at 3364 cm
−1 (N-H stretching), 1727 cm
−1 (C=O stretching), and 1534 cm
−1 (C-N stretching). The bands due to the soft segments correspond to C-H groups at 2855, 2873, and 2925 cm
−1, CH
2 group at 1461 cm
−1, and several C-O bands of the polyester polyol at 1240, 1130, 1168, 1065, and 958 cm
−1. The addition of 0.04 wt.% GO derivative does not produce new bands in the ATR-IR spectra of the PUs, but the ratio of the intensities of the C-O-C (soft segments) with respect to the one of the C=O (hard segments) stretching bands changes. Thus, according to
Table 5, the I
C-O-C/I
C=O ratio is higher in PU+A-GO and, to less extent, in PU+GO with respect to PU, indicating higher soft segments content in PU+A-GO and anticipating differences in the degree of micro-phase separation among the PUs containing different GO derivatives.
Figure 9b shows that the addition of GO or A-GO displaces the wavenumber of the N-H stretching band from 3364 cm
−1 (PU without GO derivative) to 3351 or 3335 cm
−1 respectively, revealing the interactions between the OH and/or NH
2 functional groups on the GO and A-GO surfaces with the end NCO groups of the prepolymer that led to the formation of new urethane/urea hard segments. Furthermore, the intensity of the C-H stretching band at 2873 cm
−1 decreases in PU+GO and PU+r-GO with respect to the other PUs, due to a change in the structure of the soft segments (
Figure 9b). Similarly, the addition of GO and r-GO displaces the C-O band from 1240 cm
−1 (PU without GO derivative) to 1257 cm
−1 indicating again the structural change of the soft segments (
Figure 9c). Therefore, the addition of the GO derivative with different surface chemistry affects differently the structure of the hard and soft segments, i.e., the degree of micro-phase separation, of the PUs.
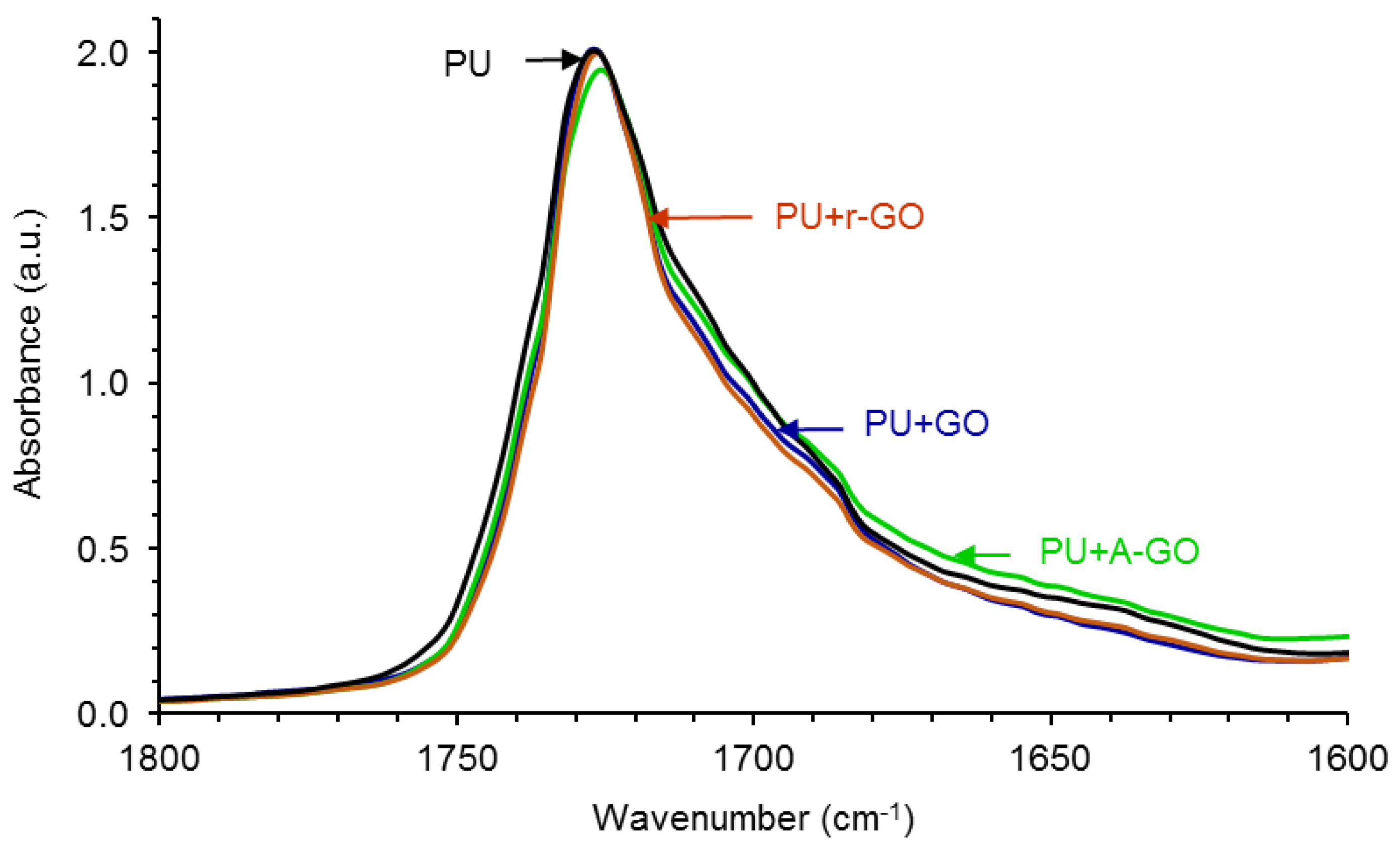
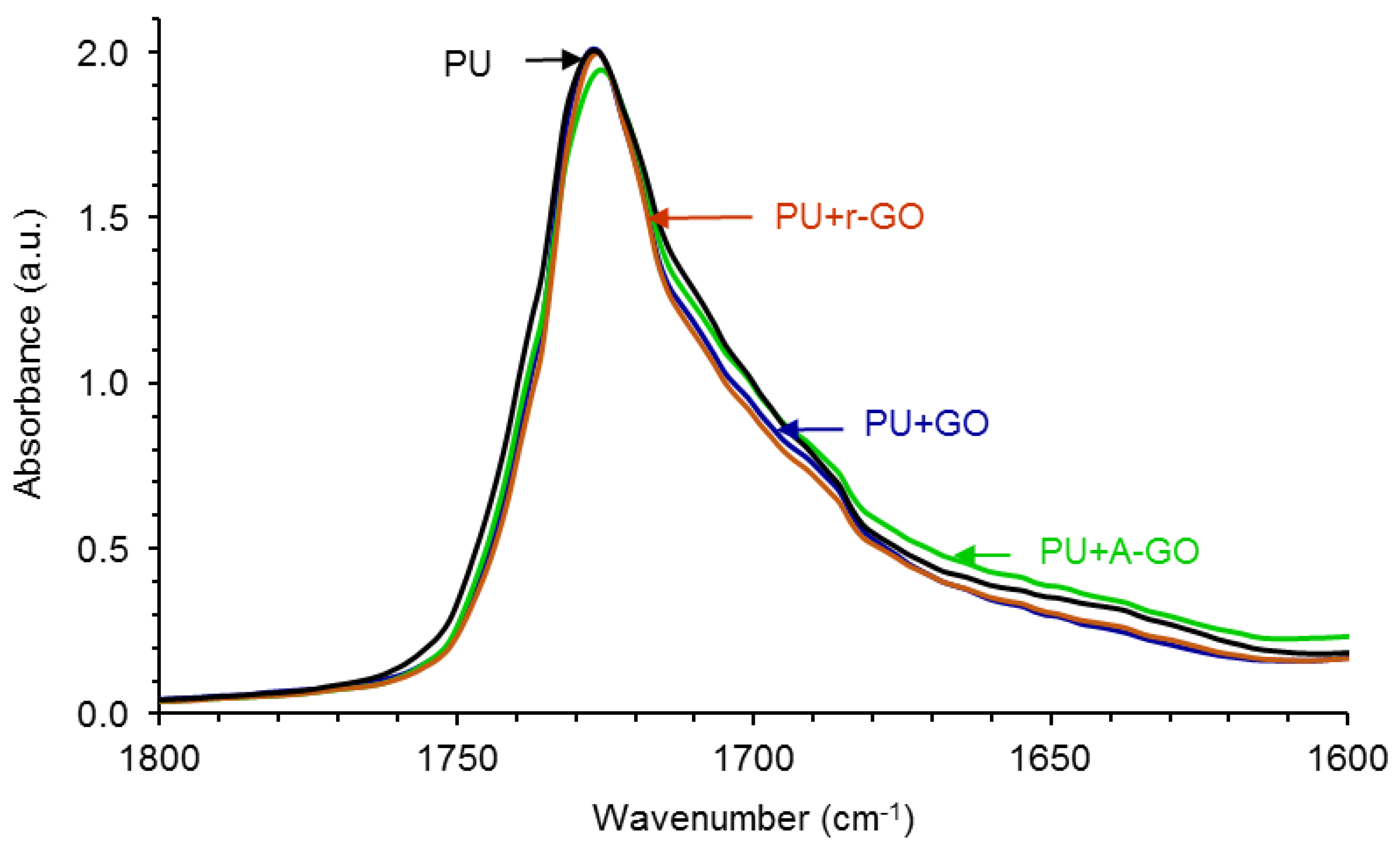
Figure 10 shows the carbonyl region (1800–1600 cm
−1) of the ATR-IR spectra of the PUs in which differences in the regions of the associated by hydrogen bond (H-bonded) urethane and urea groups are noticed. In order to quantify these differences, the curve fitting of the carbonyl region of the ATR-IR spectra of the PUs have been carried out.
Figure S4 of the Supplementary Materials file shows, as typical example, the curve fitting of the carbonyl region of the PU without GO derivative in which four contributions due to the free urethane, H-bonded urethane, free urea, and H-bonded urea species can be distinguished.
Table 6 shows that the free urethane groups (1727–1726 cm
−1) are dominant in all PUs and that the addition of the GO derivative decreases the percentage of the free urethane species and increases the percentage of the associated by hydrogen bond urethane species (1712–1708 cm
−1), more noticeably in PU+r-GO. Furthermore, the amount of the free urea species (1693–1688 cm
−1) is lower and the amount of the associated by hydrogen bond urea species (1677–1652 cm
−1) is higher in PU+r-GO. Therefore, the structure of the hard segments changes by adding the GO derivative in a different manner depending on the surface functional groups, this should cause differences in the degree of micro-phase separation in the PUs.
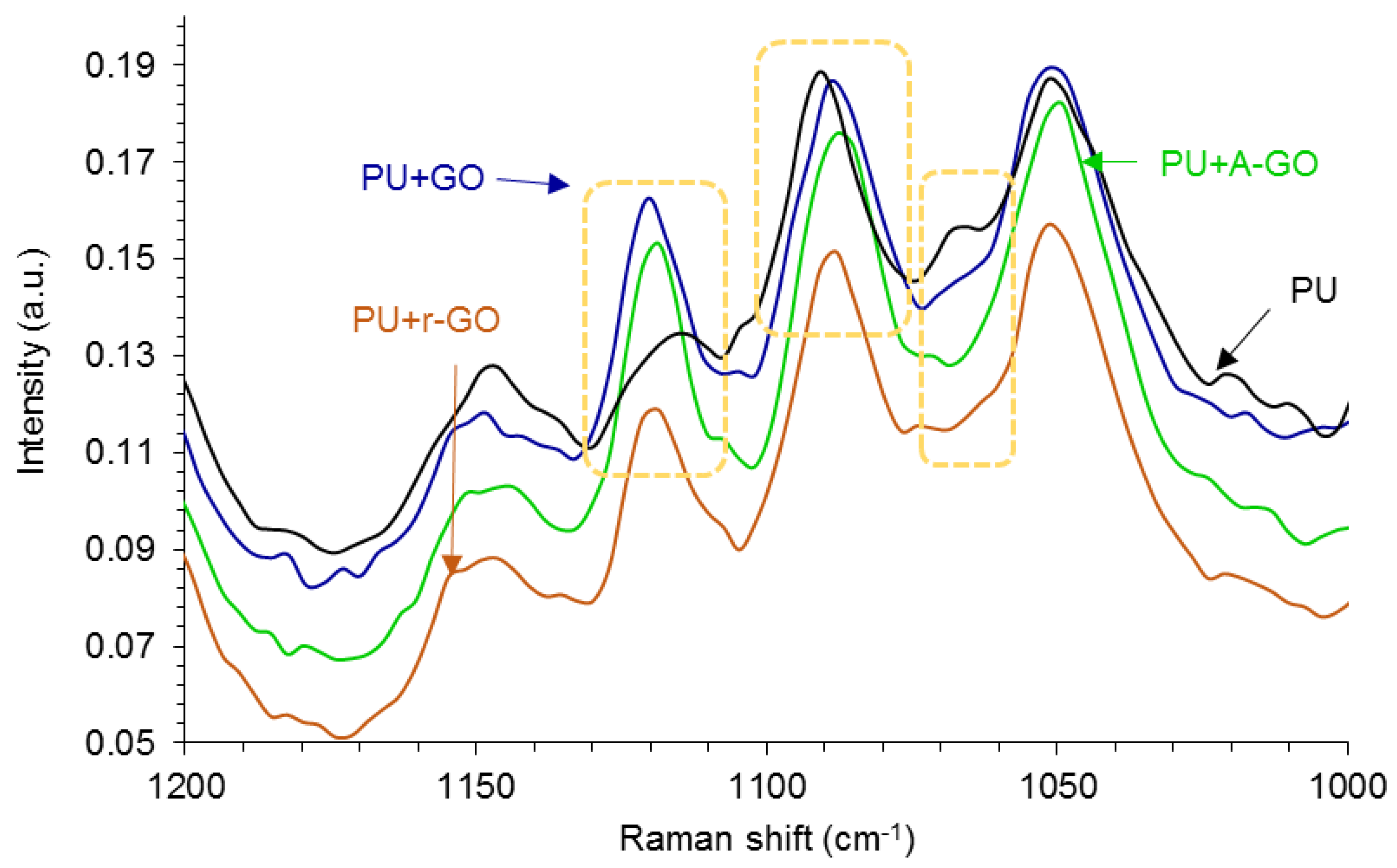
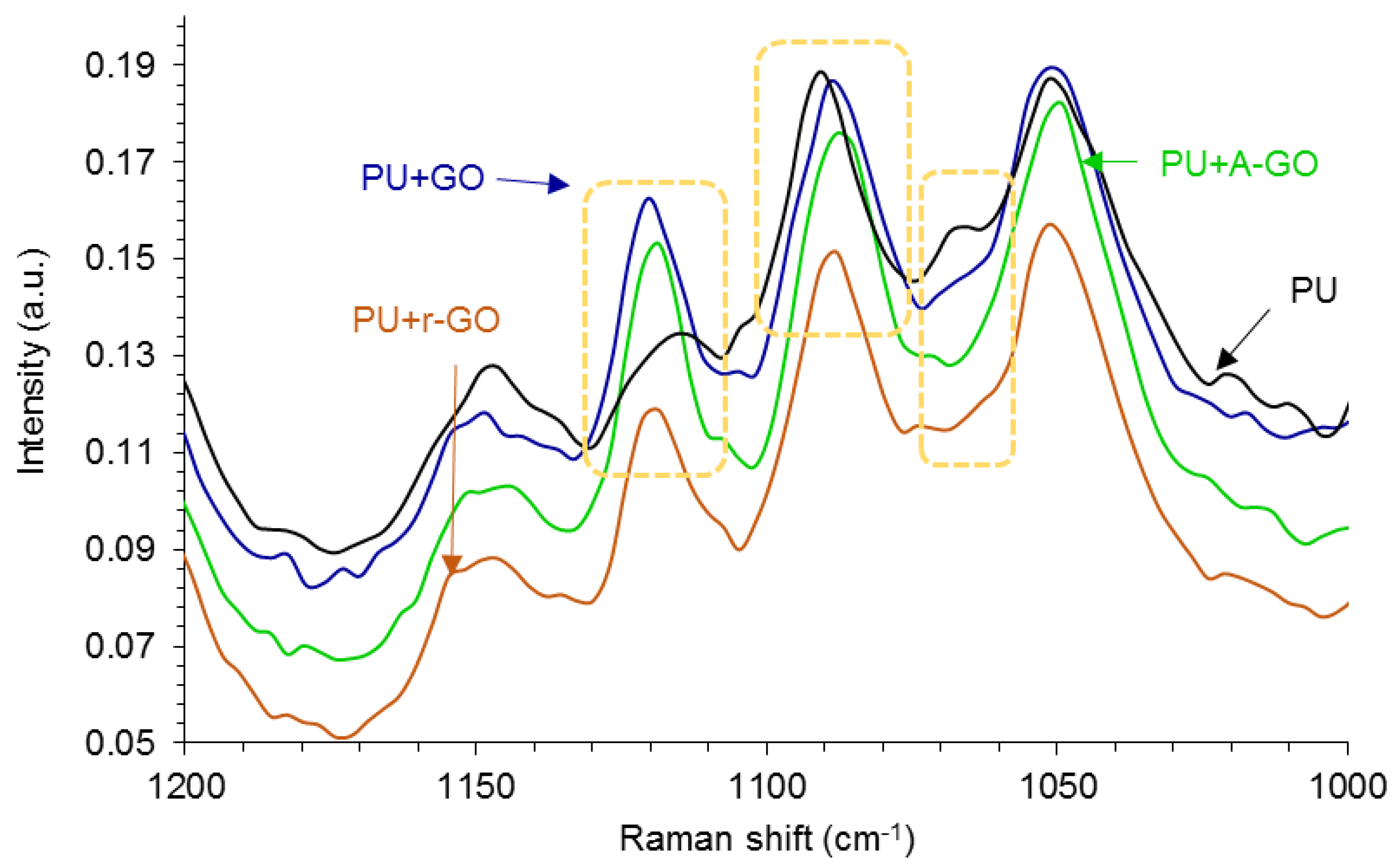
Raman spectroscopy is more sensitive than IR spectroscopy due to the interactions between nanoparticles and polyurethane chains.
Figure 11 shows some changes in the Raman shifts of the soft segments at 1124, 1090, and 1065 cm
−1 (due to the C-N, C-O-C, and C-C groups respectively) indicating the existence of interactions between the soft segments of the poly(urethane urea) and the GO derivative.
The DSC traces corresponding to the second heating runs of the PUs are shown in
Figure S5 of the Supplementary Materials file, and the thermal events in the DSC traces are given in
Table 7. The PU without GO derivative shows the glass transition temperatures (T
gs) of the soft segments at −53 °C and 5 °C, and the T
g of the hard segments at 193 °C. The addition of the GO derivative slightly increases the T
g of the soft segments and decreases the T
g of the hard segments (
Table 7), this indicates lower degree of micro-phase separation which is somewhat more marked in PU+r-GO. Moreover, one melting peak at 45 °C of the soft segments appears only in PU+A-GO, because of its higher percentage of soft segments evidenced by ATR-IR spectroscopy—this indicates different interactions than in the other PUs. As a result, the addition of the GO derivative with different surface functionality changes differently the degree of the micro-phase separation in the PU.
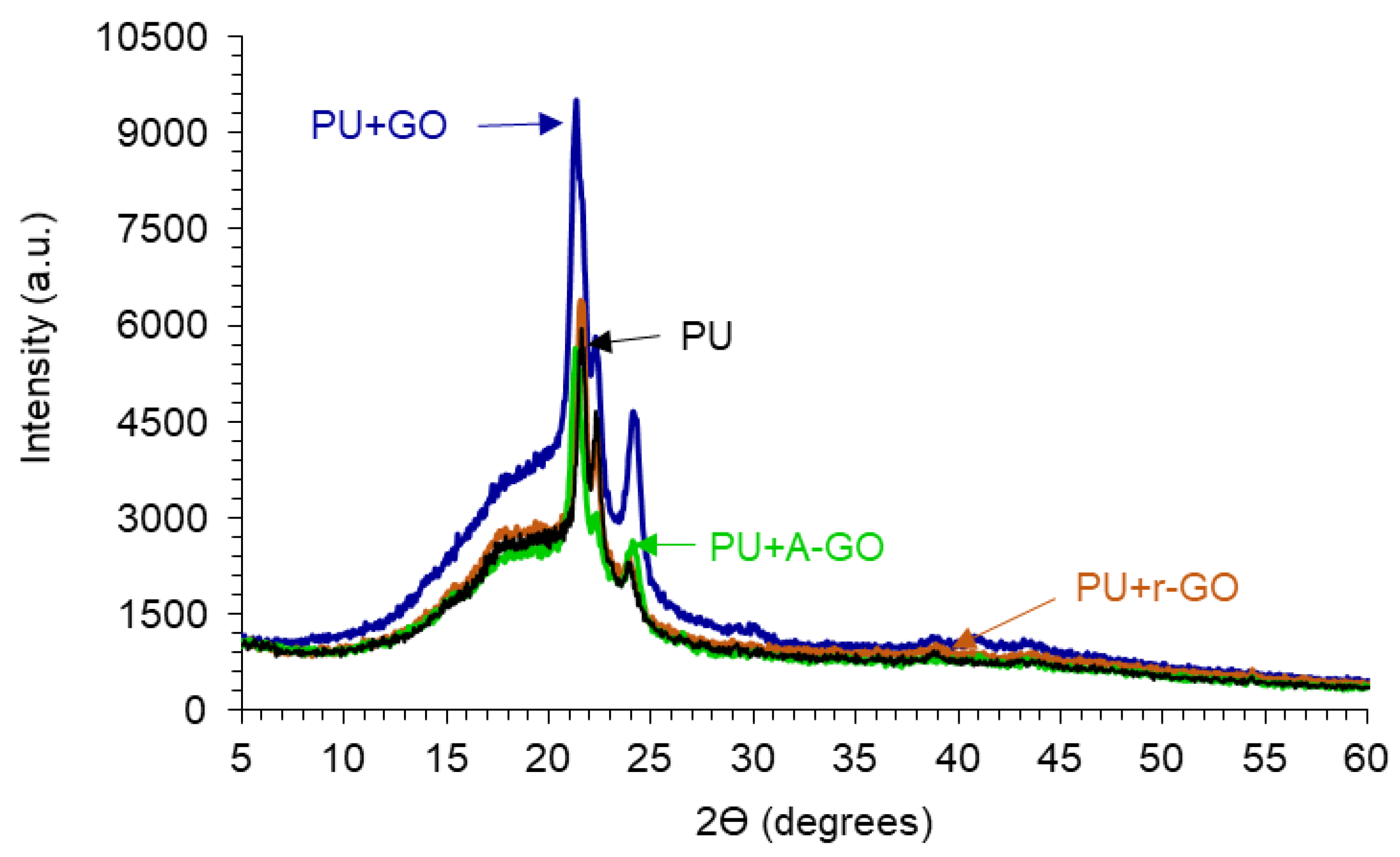
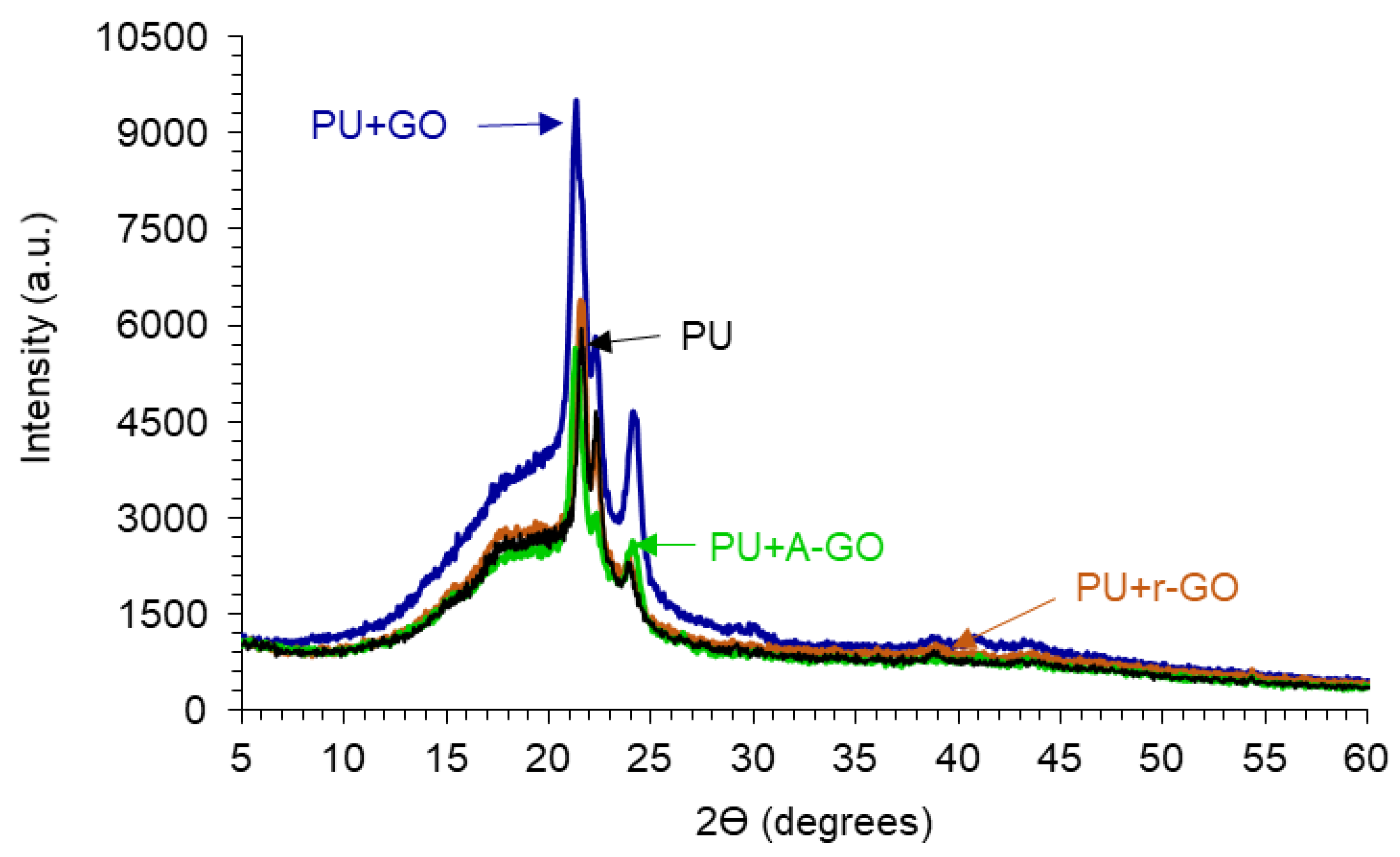
The crystallinity of the PUs is due to the interactions between the hard and the soft segments, as assessed by X-ray diffraction.
Figure 12 shows the X-ray diffractograms of the PUs in which three diffraction peaks at 2Ɵ values of 21.30–21.65, 22.20–22.35, and 23.75–24.10° can be distinguished, they are somewhat similar irrespective of the functional groups on the GO derivatives. The diffraction peaks at 2Ɵ values of 18.3–20.75° of the GO derivatives do not appear in the X-ray diffractograms of the PUs because of its adequate dispersion and low amount added. On the other hand, the intensities of the diffraction peaks are similar in all PUs, except in PU+GO in which more noticeable interactions between the hard and the soft segments are produced, likely caused by more net GO-poly(urethane urea) chains interactions.
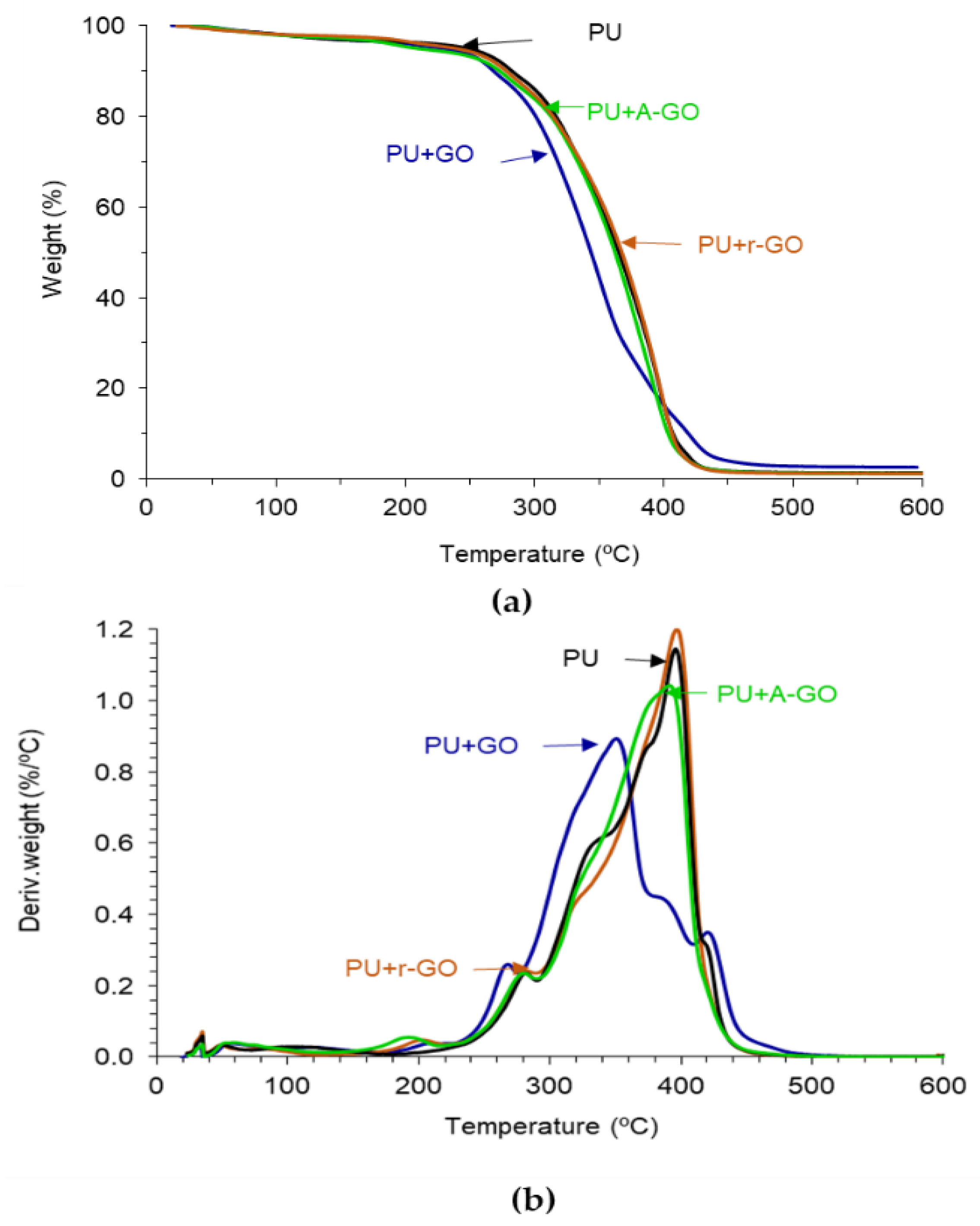
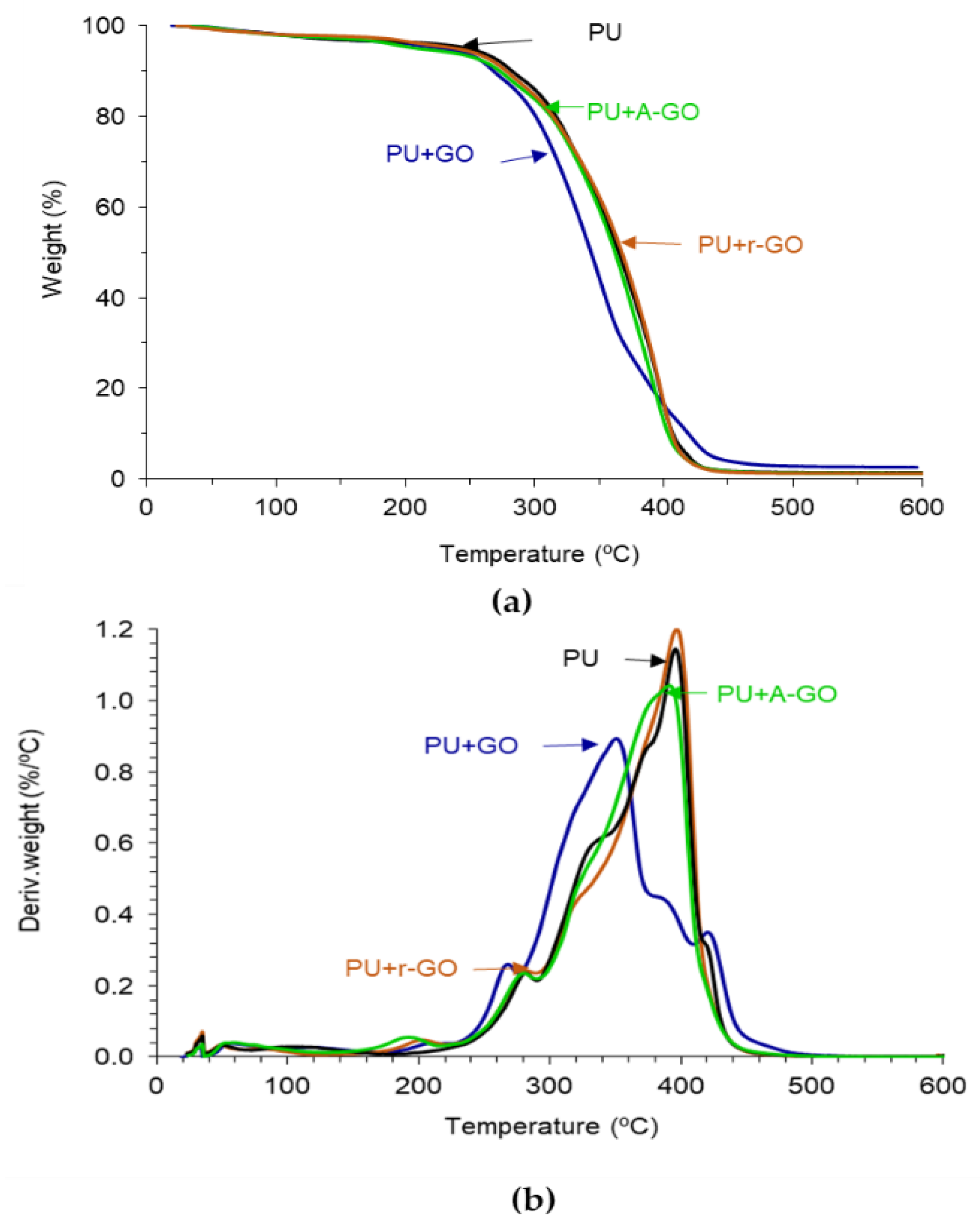
The thermal stability and structure of the PUs were assessed by TGA.
Figure 13a shows the variation of the weight as a function of the temperature of the PUs. PU+GO shows lower thermal stability than PU and the other PU+GO derivatives, likely due to more noticeable structural changes. The thermal stabilities of the PUs were quantified by the temperatures at which 5 (T
5%) and 50 (T
50%) wt.% are lost. According to
Table 8, the addition of all GO derivatives decreases the value of T
5%, due to the interactions between the hard and soft domains caused by the intercalation of the graphene nano-sheets. The lowest value of T
5% corresponds to PU+A-GO, as the soft segment content is higher than in the rest. On the other hand, the value of T
50% is similar in all PUs, except in PU+GO which is lower. Some literature has established the improved thermal stability of the polyurethane composites containing graphene derivatives [
17,
34,
35], but the opposite trend is found in this study, this can be due to the small amount added of the GO derivatives and to the dominant effect of the GO derivative morphology of the nano-sheets in changing the structure of the hard and soft domains. Thus, the thermal stability of the PU is differently affected by adding GO derivatives with different functional groups.
The structural changes in the PUs caused by adding 0.04 wt.% GO derivatives can be better distinguished in the derivative TGA plots (
Figure 13b). All PUs show one or two small thermal decompositions at 50–53 °C and 116–118 °C due to residual moisture (
Table 9). The PU without GO derivative shows five additional thermal decompositions due to the urethane (280 °C) and urea (329 °C) hard domains, and the soft domains (372, 396, and 422 °C). All PUs containing GO derivative show an additional thermal decomposition at 190–206 °C with weight losses of 2–3 wt.%, this can be ascribed to the existence of GO derivative-poly(urethane urea) interactions. This thermal decomposition is produced at lower temperatures in PU+A-GO, this indicates less interactions between A-GO and the polymer chains. On the other hand, the temperature and the weight loss of the most thermal decompositions of the PU do not change by adding A-GO and r-GO, but the thermal decompositions of the soft domains at 372 and 422 °C do not appear as they are embedded in one unique thermal decomposition of the soft segments at 390–398 °C (
Table 9). Still, the temperature of decomposition and the amount of the urea hard domains decrease in PU+GO due to stronger GO-polyurethane interactions (
Table 9). This is in agreement with the different structure of PU+GO evidenced by ATR-IR spectroscopy, X-ray diffraction, and DSC.
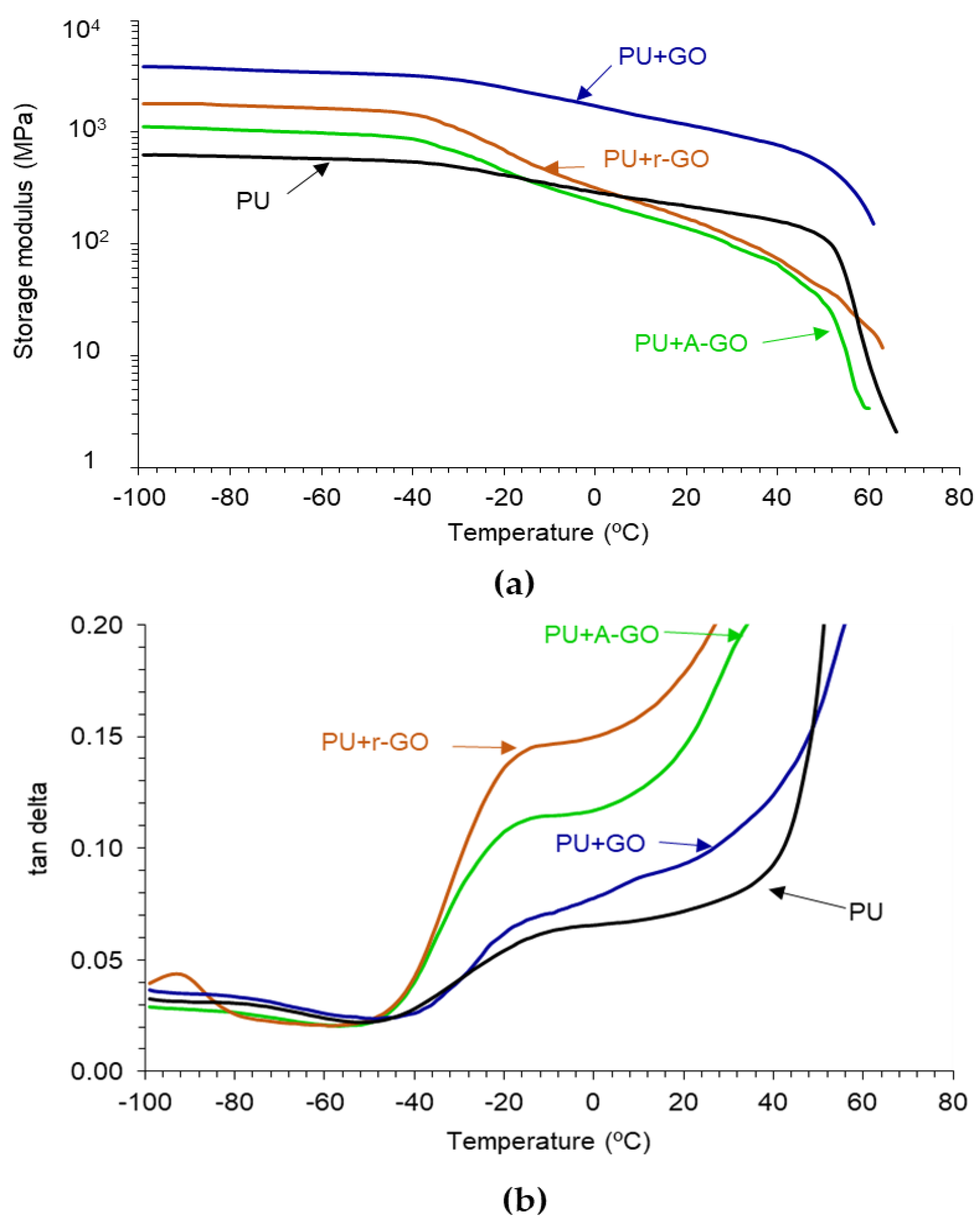
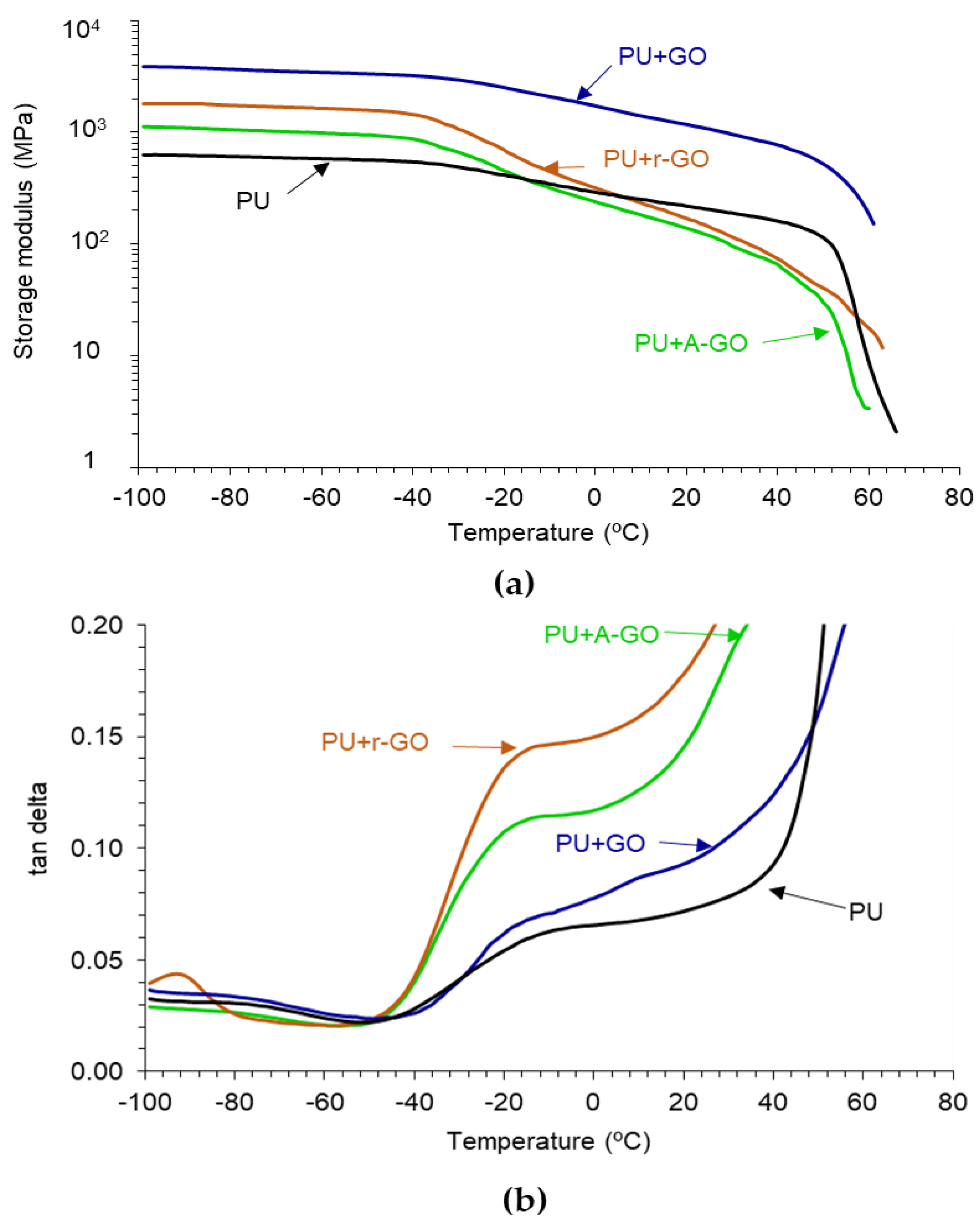
The viscoelastic properties of the PUs have been assessed by DMA.
Figure 14a indicates that all PUs show the glassy, glass transition, rubbery plateau, and melting regions. In the glassy region, the storage modulus of the PU increases by adding 0.04 wt.% GO derivatives, more noticeably in PU+GO and less markedly in PU+A-GO, which has higher percentage of soft segments. The glass transition and the rubbery plateau regions become steeper in the PUs with GO derivatives, except in PU+GO, and the melting is produced at a lower temperature. Therefore, the viscoelastic properties of the PUs are modified by adding GO derivatives and differently for PU+GO than for PU+A-GO and PU+r-GO. The viscoelastic behavior of PU+GO is somewhat similar to that of the PU without GO derivative, but the values of the storage moduli are higher due to the covalently bonded GO nano-sheets to the polymer chains. However, r-GO and, mainly, A-GO interact less effectively than GO with the polymer chains and disturb the interactions between the poly(urethane urea) chains leading to steeper glass transition and rubbery plateau regions.
Figure 14b shows the existence of two structural relaxations in the tan δ plots which can be associated to the glass transition temperatures of the PUs. The addition of the GO derivatives decreases the temperatures of the two structural relaxations (
Table 10), this indicates better interactions between the soft and the hard segments, i.e., lower degree of phase separation, more noticeably in PU+r-GO and PU+A-GO. Furthermore, the value of the maximum of tan delta in PU+r-GO and PU+A-GO are significantly higher than in PU and PU+GO due to the intercalation of the GO derivative between the polymer chains that disturbs the interactions between the soft and the hard segments, and to the higher soft segments content in PU+A-GO. Therefore, the viscoelastic properties of the PUs are determined differently by adding the GO derivatives with different functional groups.
The dispersion of the GO derivatives particles in the poly(urethane urea) matrix in the PUs was assessed by confocal laser microscopy (
Figure 15). All GO derivatives are well dispersed into the PU matrix, the r-GO and A-GO particles have similar size (4–8 µm) and they do not agglomerate. However, most GO particles cannot be distinguished in PU+GO but some agglomerates of nano-sheets can be noticed.
Because of the different polarity of the polyurethane and the GO derivatives with different surface chemistry, the wettability of the PU+GO derivative surface may change. The water contact angle on the PU without GO derivative surface is 47 degrees, and the addition of r-GO or GO does not change the wettability (47–48 degrees). Conversely, the addition of A-GO decreases the water contact angle value to 33 degrees, likely due to the high polarity of the nitrogen functional groups, this is in agreement with the lower pH obtained in PUD+A-GO.
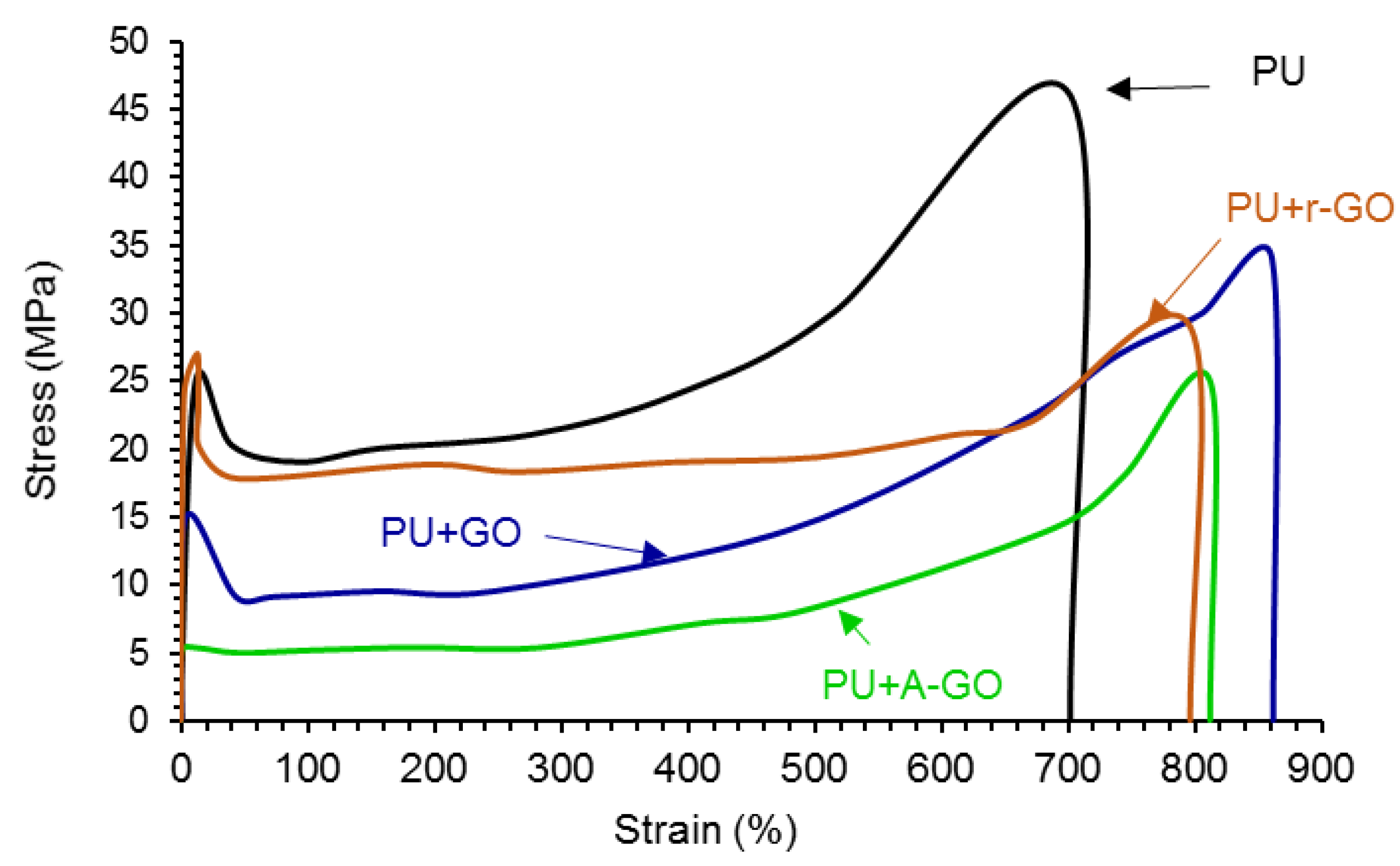
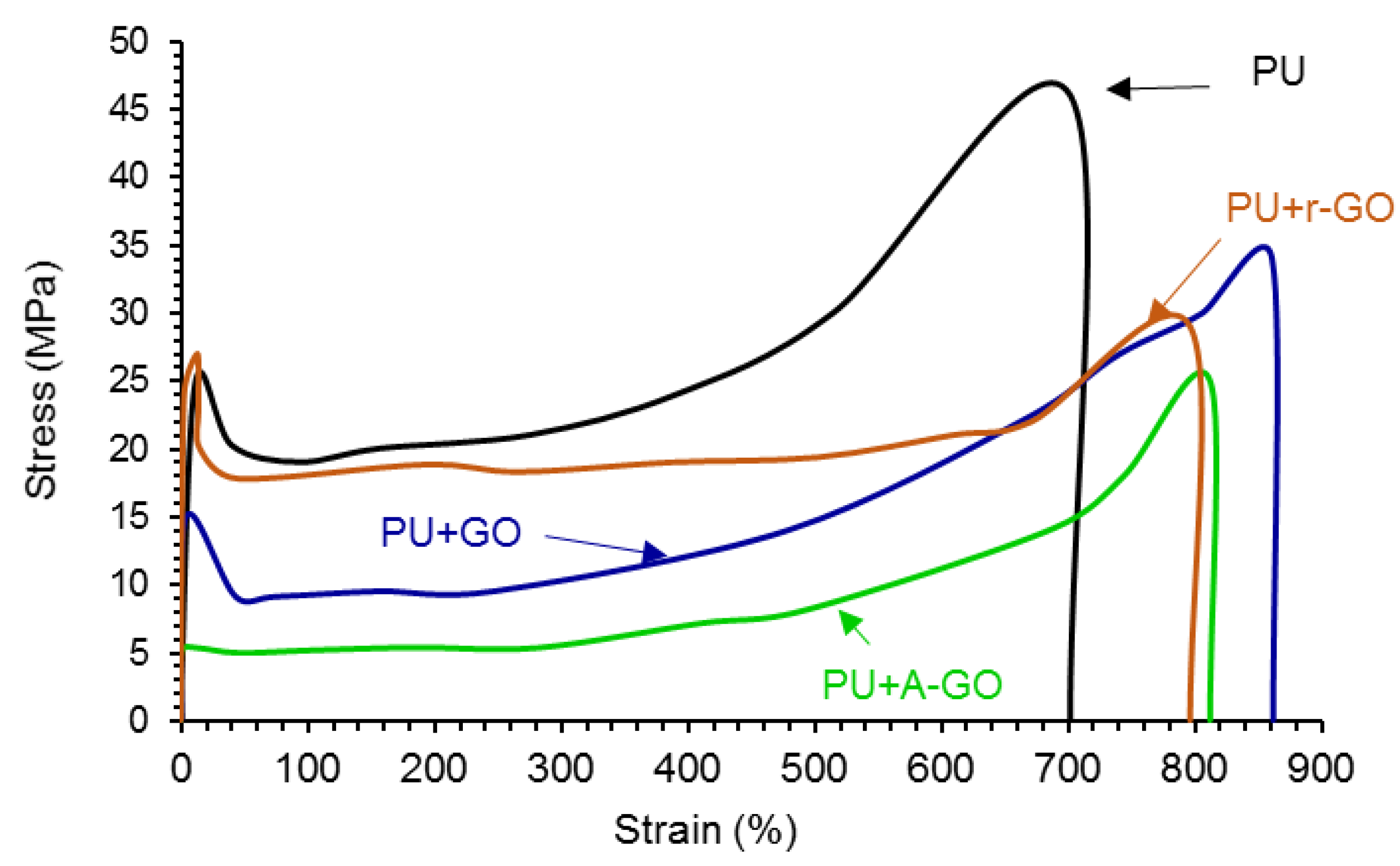
The addition of the GO derivatives also affects the mechanical properties of the PUs which were assessed by stress-strain tests. According to
Figure 16, all stress-strain curves show a noticeable yield point (less marked in PU+A-GO) followed by the plastic deformation until the PU breaks. The addition of the GO derivatives increases the elongation-at-break (
Table 11) and decreases the tensile strength and the yield stress of the PU. PU+GO shows higher elongation-at-break and tensile strength values than PU+r-GO and PU+A-GO. This trend agrees with the better viscoelastic properties and degree of micro-phase separation of PU+GO with respect to PU+r-GO and PU+A-GO. Furthermore, PU+r-GO shows similar yield stress to PU (
Table 11) because of larger and thicker nano-sheets of r-GO which may cause fewer net interactions with the polymer chains. Therefore, the addition of the GO derivatives seems to impart some toughening to the PU, to a different extent depending on the size of the nano-sheets and the functional groups of the GO derivatives.
The addition of the carbon nanoparticles strongly influences the morphologies of the waterborne polyurethanes as has been observed for GO-PU [
36], TiO
2/r-GO-PU [
37], and PU-graphene [
38] composites. Therefore, the morphology of the PU should change when the GO derivatives are added—this was analyzed by SEM micrographs of the fractured surfaces of the PUs. Whereas PU without GO exhibits relativity smooth fractured surface as expected for an elastomeric material (
Figure 17), all PUs containing the GO derivatives show rough fractured surfaces indicating some toughness (as anticipated by the increased elongation-at-break values obtained in the stress-strain tests). The appearance of the fractured surfaces differs depending on the GO derivative. While a rough micro-fractured surface and some small cracks and voids are seen in PU+r-GO, a rough wrinkled and bumped fractured surface with some A-GO nano-sheets and a few cracks are distinguished in PU+A-GO. The fractured surface of PU+GO is less rough, no cracks but small fractured zones are distinguished and the adhesion of the GO nano-sheets to the fractured PU matrix is good. Therefore, the addition of the different GO derivatives causes different morphologies of the PUs. Similar findings have been reported in our recent work dealing with PUs containing different graphene derivatives [
39].
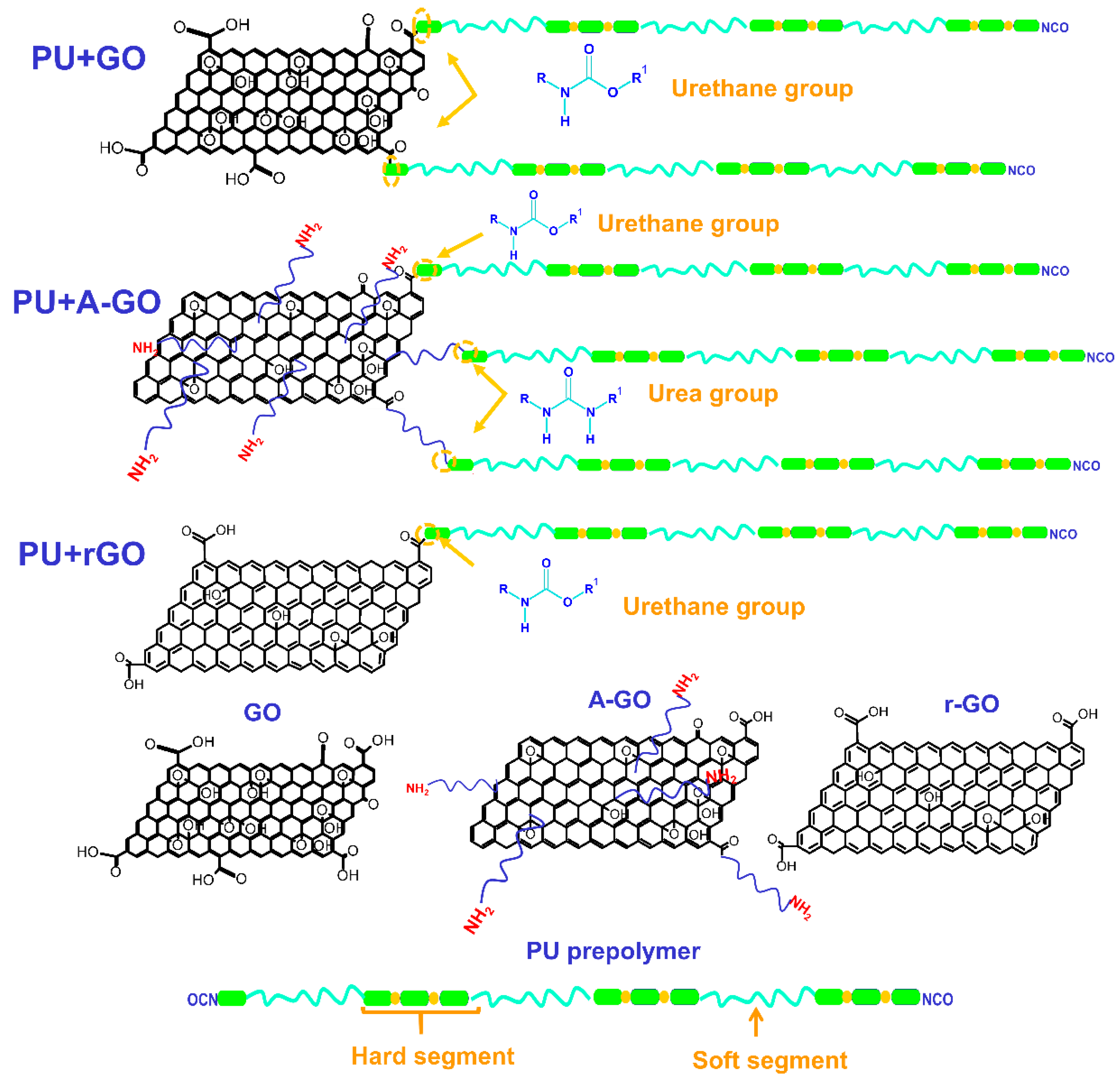
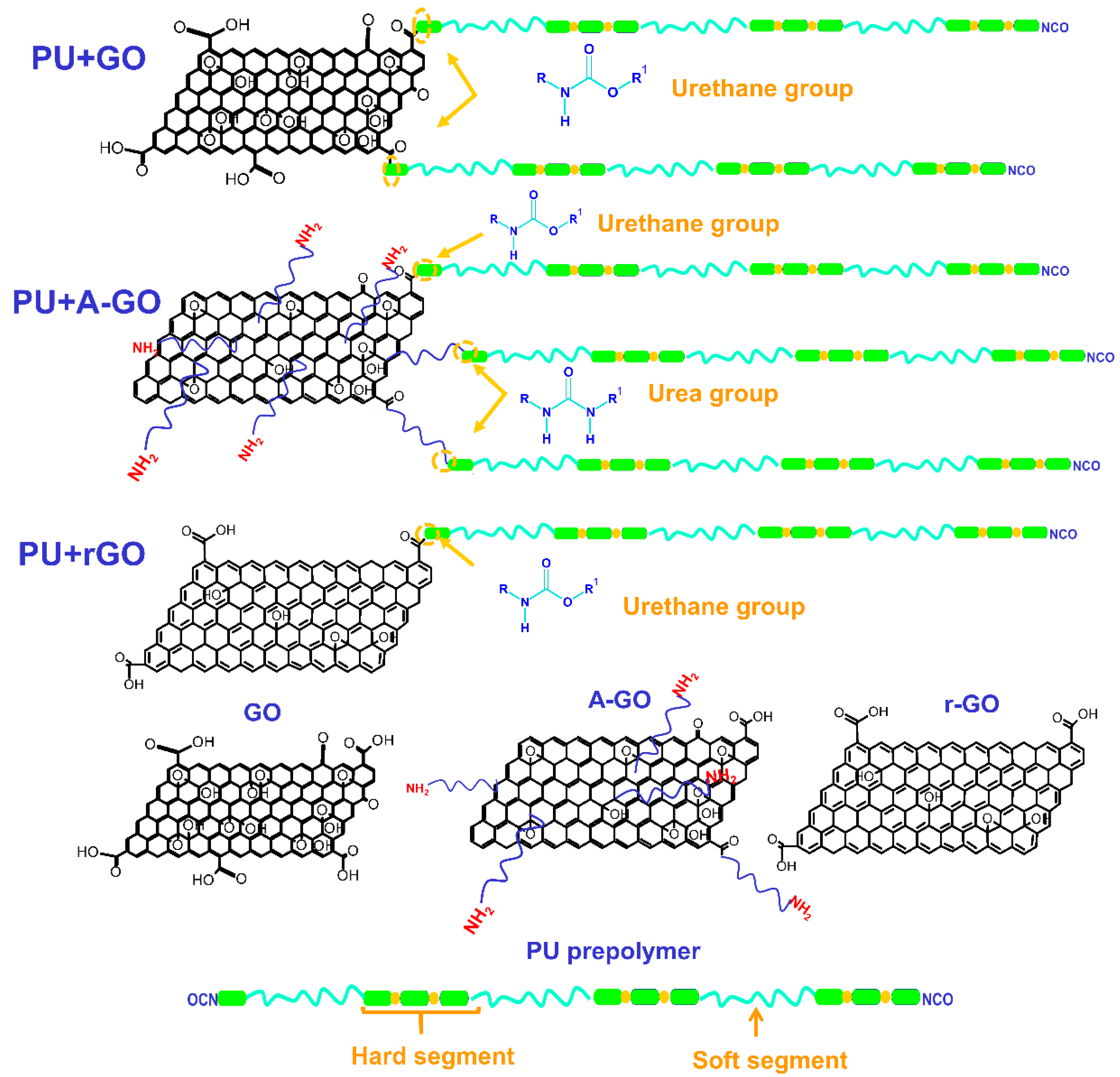
The functional groups (C-O, N-H) on the surface and edges of the GO derivatives covalently attach to the prepolymer chains during PUD synthesis. The interactions between the poly(urethane urea) chains with covalently bonded GO derivative nano-sheets changes the degree of micro-phase separation of the PU: these changes produce different thermal, viscoelastic, mechanical, and surface properties, i.e., different structure–property relationship are produced. During PUD synthesis, the oxygen functional groups on the GO and r-GO surface produce new urethane linkages by the reaction with the end NCO groups of the prepolymer chain (
Figure 18), to a greater extent in PU+GO than in PU+r-GO because of the higher amount of oxygen functional groups on the GO surface. Furthermore, the number of stacked graphene nano-sheets in r-GO is higher than in GO. Because the nano-sheets intercalate among the polymer chains, the degree of micro-phase separation will be different in PU+GO and PU+r-GO. GO has the highest number of functional groups and the thinnest graphene nano-sheets, so more net interactions with the polymer chains will be produced. Conversely, r-GO has the lowest content of functional groups and thicker stacked graphene nano-sheets than GO, so the improvement of the properties in PU+r-GO should be less pronounced.
A-GO surface had a similar amount of oxygen functional groups to r-GO and also some nitrogen functional groups. Because of the higher reactivity of the amines with the isocyanates, during PUD synthesis, the amine functional groups on the A-GO surface should react preferentially, producing new urea hard domains (
Figure 18); additionally, there is a chance that some oxygen functional groups on the A-GO surface may also react with the NCO groups. Furthermore, the number of stacked graphene nano-sheets is higher in A-GO than in GO, so the intercalation of the covalently A-GO nano-sheets will disturb differently the interactions between the poly(urethane urea) chains. As a consequence, the degree of micro-phase separation of PU+A-GO differs from the ones in PU+GO and PU+r-GO, this justifies the reduced increase in properties, i.e., PU+A-GO has the lowest thermal stability, the lowest temperature of the thermal decomposition of the A-GO/PU interactions, and the highest value of the maximum of tan delta, the highest surface polarity, and the less improved mechanical properties.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}