
Mechanism of Unusual Isosymmetric Order-Disorder Phase Transition in [Dimethylhydrazinium]Mn(HCOO)3 Hybrid Perovskite Probed by Vibrational Spectroscopy



Abstract
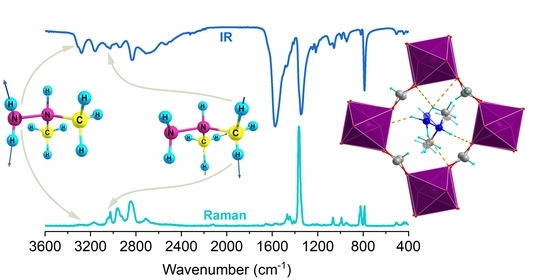
:
1. Introduction
2. Materials and Methods
2.1. Materials and Synthesis
2.2. Raman ad IR Spectroscopy
2.3. Quantum Chemical Calculations
3. Results
3.1. Crystal Structure and Geometry Optimization
3.2. Selection Rules and Factor Group Analysis
3.3. DFT Calculations
3.4. Room-Temperature IR and Raman Spectra and Assignment of Bands
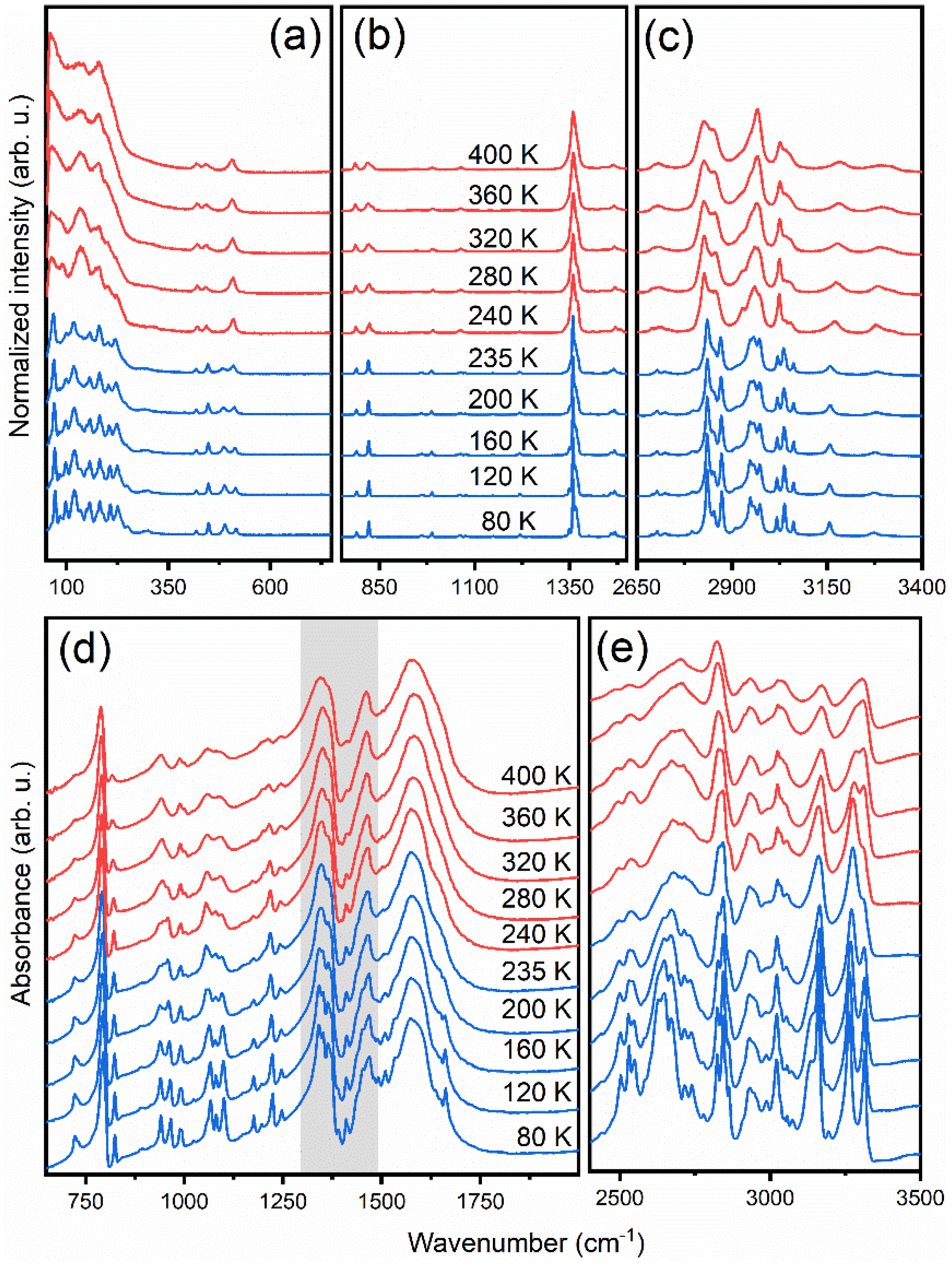
3.5. Temperature-Dependent IR and Raman Spectra
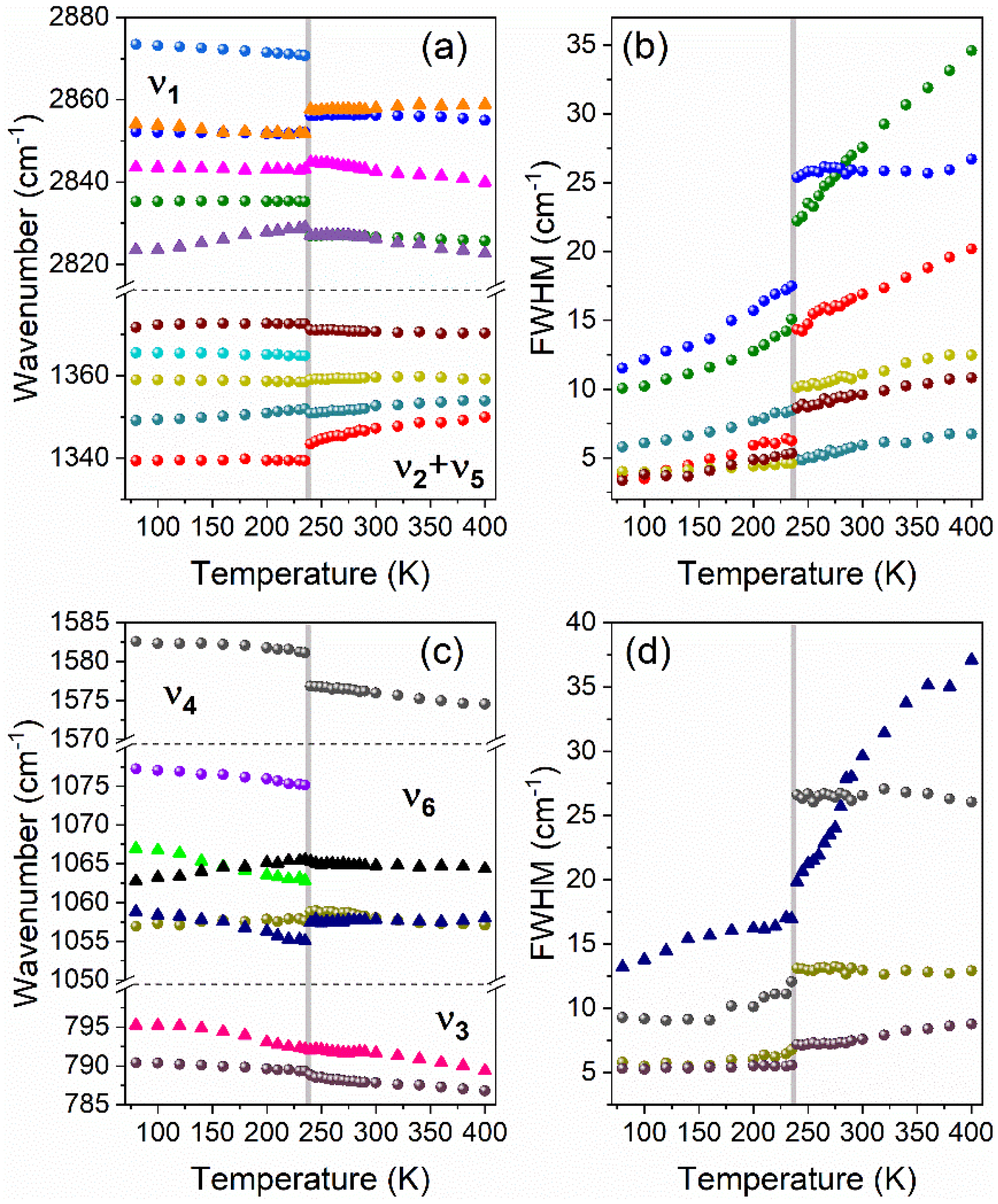
3.5.1. Internal Modes of Formate Ions
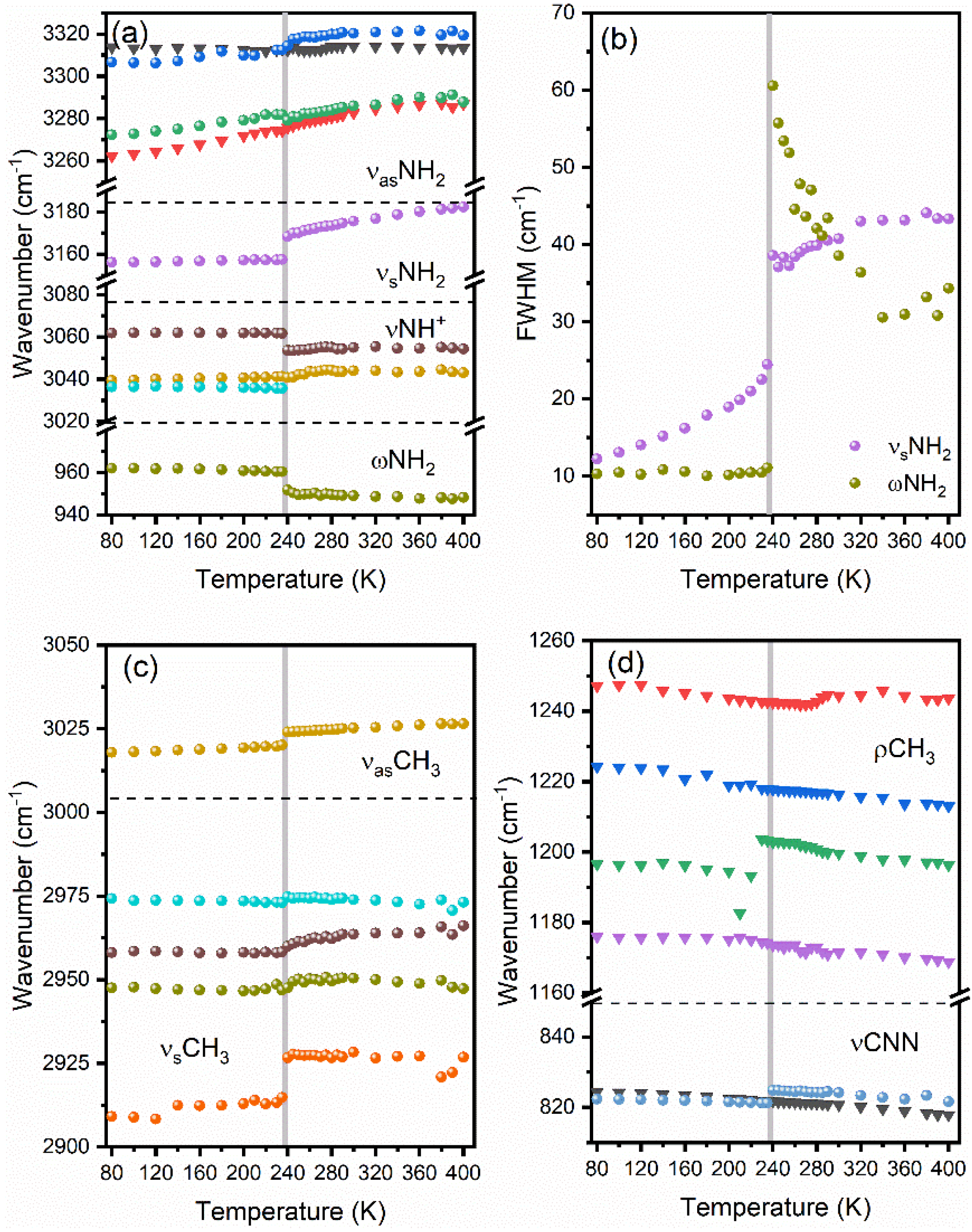
3.5.2. Internal Modes of DMHy+ Cation
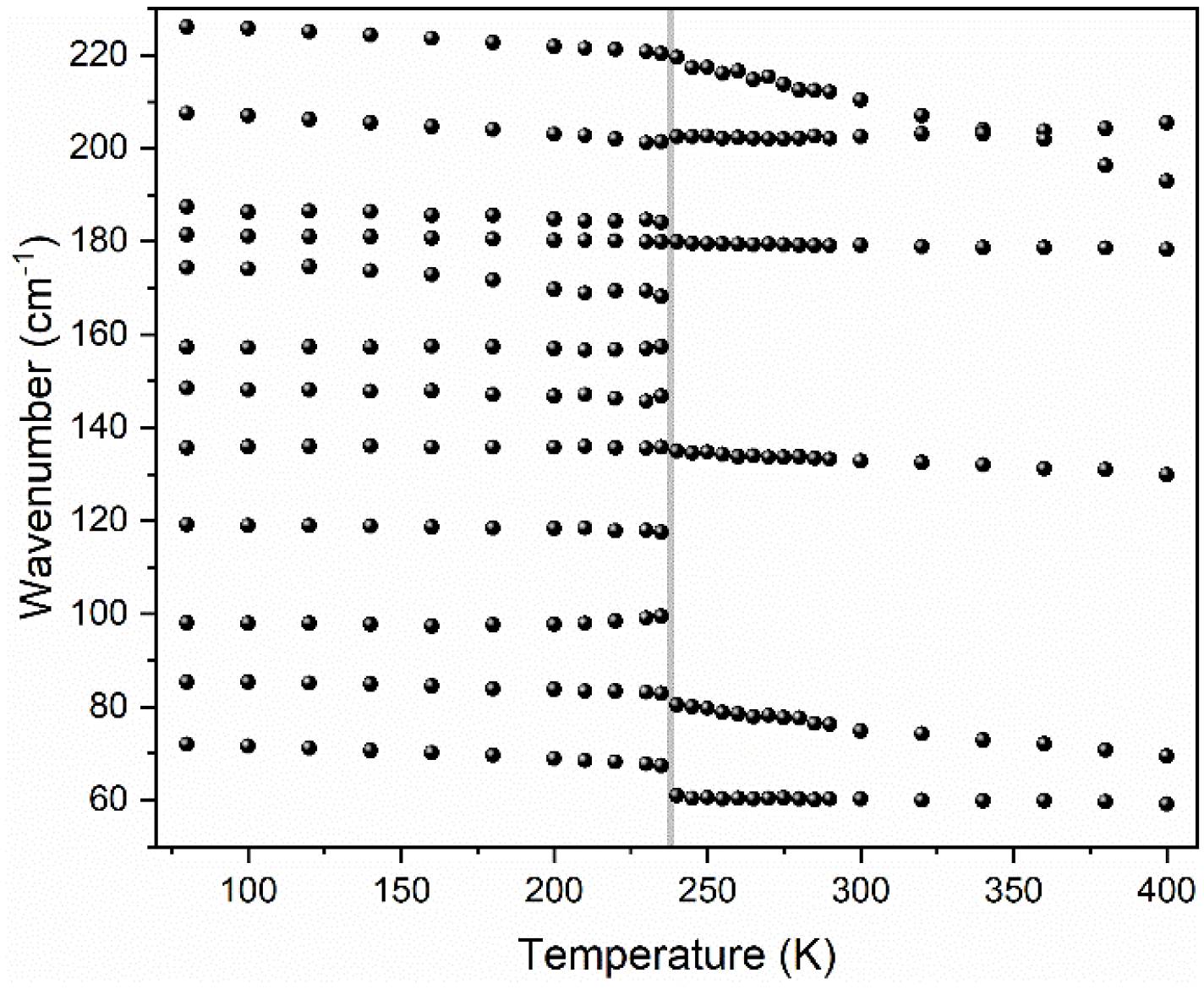
3.5.3. Lattice Modes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mączka, M.; Bondzior, B.; Dereń, P.; Sieradzki, A.; Trzmiel, J.; Pietraszko, A.; Hanuza, J. Synthesis and characterization of [(CH3)2NH2][Na0.5Cr0.5(HCOO)3]: A rare example of luminescent metal-organic frameworks based on Cr(III) ions. Dalt. Trans. 2015, 44, 6871–6879. [Google Scholar] [CrossRef]
- Zienkiewicz, J.A.; Kowalska, D.A.; Fedoruk, K.; Stefański, M.; Pikul, A.; Ptak, M. Unusual isosymmetric order-disorder phase transition in the new perovskite-type dimethylhydrazinium manganese formate exhibiting ferrimagnetic and photoluminescent properties. J. Mater. Chem. C 2021, 9, 6841–6851. [Google Scholar] [CrossRef]
- Li, W.; Zhang, Z.; Bithell, E.G.; Batsanov, A.S.; Barton, P.T.; Saines, P.J.; Jain, P.; Howard, C.J.; Carpenter, M.A.; Cheetham, A.K. Ferroelasticity in a metal-organic framework perovskite; Towards a new class of multiferroics. Acta Mater. 2013, 61, 4928–4938. [Google Scholar] [CrossRef]
- Wang, K.; Xiong, J.B.; Xia, B.; Wang, Q.L.; Tong, Y.Z.; Ma, Y.; Bu, X.H. Ferroelastic phase transition and switchable dielectric constant in heterometallic niccolite formate frameworks. Inorg. Chem. 2018, 57, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Dalal, N.S.; Toby, B.H.; Kroto, H.W.; Cheetham, A.K. Order-disorder antiferroelectric phase transition in a hybrid inorganic-organic framework with the perovskite architecture. J. Am. Chem. Soc. 2008, 130, 10450–10451. [Google Scholar] [CrossRef] [PubMed]
- Abhyankar, N.; Bertaina, S.; Dalal, N.S. On Mn2+ EPR probing of the ferroelectric transition and absence of magnetoelectric coupling in dimethylammonium manganese formate (CH3)2NH2Mn(HCOO)3, a metal-organic complex with the Pb-free perovskite framework. J. Phys. Chem. C 2015, 119, 28143–28147. [Google Scholar] [CrossRef] [Green Version]
- Kieslich, G.; Forse, A.C.; Sun, S.; Butler, K.T.; Kumagai, S.; Wu, Y.; Warren, M.R.; Walsh, A.; Grey, C.P.; Cheetham, A.K. Role of amine-cavity interactions in determining the structure and mechanical properties of the ferroelectric hybrid perovskite [NH3NH2]Zn(HCOO)3. Chem. Mater. 2016, 28, 312–317. [Google Scholar] [CrossRef]
- Yu, Y.; Shang, R.; Chen, S.; Wang, B.-W.; Wang, Z.-M.; Gao, S. A Series of bimetallic ammonium AlNa formates. Chem. A Eur. J. 2017, 23, 9857–9871. [Google Scholar] [CrossRef]
- Wang, Z.; Hu, K.; Gao, S.; Kobayashi, H. Formate-based magnetic metal-organic frameworks templated by protonated amines. Adv. Mater. 2010, 22, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Mączka, M.; Gągor, A.; Hermanowicz, K.; Sieradzki, A.; Macalik, L.; Pikul, A. Structural, magnetic and phonon properties of Cr(III)-doped perovskite metal formate framework [(CH3)2NH2][Mn(HCOO)3]. J. Solid State Chem. 2016, 237, 150–158. [Google Scholar] [CrossRef]
- Nagabhushana, G.P.; Shivaramaiah, R.; Navrotsky, A. Thermochemistry of multiferroic organic-inorganic hybrid perovskites [(CH3)2NH2][M(HCOO)3] (M = Mn, Co, Ni, and Zn). J. Am. Chem. Soc. 2015, 137, 10351–10356. [Google Scholar] [CrossRef]
- Jain, P.; Ramachandran, V.; Clark, R.J.; Hai, D.Z.; Toby, B.H.; Dalal, N.S.; Kroto, H.W.; Cheetham, A.K. Multiferroic behavior associated with an order-disorder hydrogen bonding transition in metal-organic frameworks (MOFs) with the perovskite ABX3 architecture. J. Am. Chem. Soc. 2009, 131, 13625–13627. [Google Scholar] [CrossRef]
- Thomson, R.I.; Jain, P.; Cheetham, A.K.; Carpenter, M.A. Elastic relaxation behavior, magnetoelastic coupling and order-disorder processes in multiferroic metal-organic frameworks. Phys. Rev. B 2012, 214304, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Brenes, R.; Eames, C.; Bulović, V.; Islam, M.S.; Stranks, S.D. The impact of atmosphere on the local luminescence properties of metal halide perovskite grains. Adv. Mater. 2018, 30, 1–8. [Google Scholar] [CrossRef]
- Sun, D.-S.; Zhang, Y.-Z.; Gao, J.-X.; Hua, X.-N.; Chen, X.-G.; Mei, G.-Q.; Liao, W.-Q. Reversible high temperature dielectric switching Reversible high temperature dielectric switching in a 2H-perovskite compound: [Me3NCH2CH3]CdCl3. CrystEngComm 2019, 21, 2669–2674. [Google Scholar] [CrossRef]
- Rok, M.; Bator, G.; Zarychta, B.; Dziuk, B.; Medycki, W.; Zamponi, M.; Banys, J. Isostructural phase transition, quasielastic neutron scattering and magnetic resonance studies of a bistable dielectric ion-pair crystal [(CH3)2NH2]2KCr(CN)6. Dalt. Trans. 2019, 48, 4190–4202. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Shang, R.; Hu, K.-L.L.; Wang, Z.-M.M.; Gao, S. [NH2NH3][M(HCOO)3] (M = Mn2+, Zn2+, Co2+ and Mg2+): Structural phase transitions, prominent dielectric anomalies and negative thermal expansion, and magnetic ordering. Inorg. Chem. Front. 2014, 1, 83–98. [Google Scholar] [CrossRef]
- Mączka, M.; Pasińska, K.; Ptak, M.; Paraguassu, W.; da Silva, T.A.; Sieradzki, A.; Pikul, A. Effect of solvent, temperature and pressure on the stability of chiral and perovskite metal formate frameworks of [NH2NH3][M(HCOO)3] (M = Mn, Fe, Zn). Phys. Chem. Chem. Phys. 2016, 18, 31653–31663. [Google Scholar] [CrossRef] [PubMed]
- Kieslich, G.; Kumagai, S.; Butler, K.T.; Okamura, T.; Hendon, C.H.; Sun, S.; Yamashita, M.; Walsh, A.; Cheetham, A.K. Role of entropic effects in controlling the polymorphism in formate ABX3 metal-organic frameworks. Chem. Commun. 2015, 51, 15538–15541. [Google Scholar] [CrossRef] [Green Version]
- Mączka, M.; Gągor, A.; Ptak, M.; Paraguassu, W.; Da Silva, T.A.; Sieradzki, A.; Pikul, A. Phase transitions and coexistence of magnetic and electric orders in the methylhydrazinium metal formate frameworks. Chem. Mater. 2017, 29, 2264–2275. [Google Scholar] [CrossRef]
- Kieslich, G.; Sun, S.; Cheetham, A.K. Solid-state principles applied to organic-inorganic perovskites: New tricks for an old dog. Chem. Sci. 2014, 5, 4712–4715. [Google Scholar] [CrossRef]
- Christy, A.G. Isosymmetric structural phase transitions: Phenomenology and examples. Acta Crystallogr. Sect. B Struct. Sci. 1995, 51, 753–757. [Google Scholar] [CrossRef]
- Zhang, W.; Ye, H.-Y.; Graf, R.; Spiess, H.W.; Yao, Y.-F.; Zhu, R.-Q.; Xiong, R.-G. Tunable and switchable dielectric constant in an mmphidynamic crystal. J. Am. Chem. Soc. 2013, 135, 5230–5233. [Google Scholar] [CrossRef]
- Rok, M.; Bator, G.; Medycki, W.; Zamponi, M.; Balčiūnas, S.; Šimėnas, M.; Banys, J. Reorientational dynamics of organic cations in perovskite-like coordination polymers. Dalt. Trans. 2018, 47, 17329–17341. [Google Scholar] [CrossRef]
- Xu, W.-J.; Chen, S.-L.; Hu, Z.-T.; Lin, R.-B.; Su, Y.-J.; Zhang, W.-X.; Chen, X.-M. The cation-dependent structural phase transition and dielectric response in a family of cyano-bridged perovskite-like coordination polymers. Dalt. Trans. 2016, 45, 4224–4229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Shao, X.D.; Li, S.C.; Cai, Y.; Yao, Y.F.; Xiong, R.G.; Zhang, W. Dynamics of a caged imidazolium cation-toward understanding the order-disorder phase transition and the switchable dielectric constant. Chem. Commun. 2015, 51, 4568–4571. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Cai, Y.; Xiong, R.-G.; Yoshikawa, H.; Awaga, K. Exceptional dielectric phase transitions in a perovskite-type cage compound. Angew. Chem. Int. Ed. 2010, 49, 6608–6610. [Google Scholar] [CrossRef]
- Zhao, X.H.; Huang, X.C.; Zhang, S.L.; Shao, D.; Wei, H.Y.; Wang, X.Y. Cation-dependent magnetic ordering and room-temperature bistability in azido-bridged perovskite-type compounds. J. Am. Chem. Soc. 2013, 135, 16006–16009. [Google Scholar] [CrossRef] [PubMed]
- Mączka, M.; Marinho Costa, N.L.; Gągor, A.; Paraguassu, W.; Sieradzki, A.; Hanuza, J. Structural, thermal, dielectric and phonon properties of perovskite-like imidazolium magnesium formate. Phys. Chem. Chem. Phys. 2016, 18, 13993–14000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision A.1; Gaussian, Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. IV. A new dynamical correlation functional and implications for exact-exchange mixing. J. Chem. Phys. 1996, 104, 1040–1046. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Springer: Dordrecht, The Netherlands, 1989. [Google Scholar]
- Mclean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Rostkowska, H.; Lapinski, L.; Nowak, M.J. Analysis of the normal modes of molecules with D3h symmetry: Infrared spectra of monomeric s-triazine and cyanuric acid. Vib. Spectrosc. 2009, 49, 43–51. [Google Scholar] [CrossRef]
- Zhurko, G.A.; Zhurko, D.A. Chemcraft Graphical Program of Visualization of Computed Results. Available online: http://chemcraftprog.com (accessed on 9 March 2021).
- Ptak, M.; Mączka, M.M.; Gągor, A.; Sieradzki, A.; Stroppa, A.; Di Sante, D.; Perez-Mato, J.M.; Macalik, L. Experimental and theoretical studies of structural phase transition in a novel polar perovskite-like [C2H5NH3][Na0.5Fe0.5(HCOO)3] formate. Dalt. Trans. 2016, 45, 2574–2583. [Google Scholar] [CrossRef] [PubMed]
- Zeroka, D.; Jensen, J.O.; Samuels, A.C. Infrared spectra of some isotopomers of isopropylamine: A theoretical study. J. Mol. Struct. Theochem 1999, 465, 119–139. [Google Scholar] [CrossRef]
- Mączka, M.; Gągor, A.; Macalik, B.; Pikul, A.; Ptak, M.; Hanuza, J. Order-disorder transition and weak ferromagnetism in the perovskite metal formate frameworks of [(CH3)2NH2][M(HCOO)3] and [(CH3)2ND2][M(HCOO)3] (M = Ni, Mn). Inorg. Chem. 2014, 53, 457–467. [Google Scholar] [CrossRef]
- Durig, J.R.; Zheng, C. On the vibrational spectra and conformational stability of 1,1-dimethylhydrazine from temperature dependent FT-IR spectra of krypton solutions and ab initio calculations. J. Mol. Struct. 2004, 690, 31–44. [Google Scholar] [CrossRef]
- Ciupa-Litwa, A.; Ptak, M.; Kucharska, E.; Hanuza, J.; Mączka, M. Vibrational properties and DFT calculations of perovskite-type methylhydrazinium manganese hypophosphite. Molecules 2020, 25, 5215. [Google Scholar] [CrossRef]
- Mączka, M.; Ptak, M.; Gągor, A.; Stefańska, D.; Zaręba, J.K.; Sieradzki, A. Methylhydrazinium lead bromide: Noncentrosymmetric three-dimensional perovskite with exceptionally large framework distortion and green photoluminescence. Chem. Mater. 2020, 32, 1667–1673. [Google Scholar] [CrossRef]
- Mączka, M.; Ptak, M.; Vasconcelos, D.L.M.; Giriunas, L.; Freire, P.T.C.; Bertmer, M.; Banys, J.; Simenas, M. NMR and Raman scattering studies of temperature-and pressure-driven phase transitions in CH3NH2NH2PbCl3 perovskite. J. Phys. Chem. C 2020, 124, 26999–27008. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Raman | IR (ATR) | IR (Nujol) | IR (Fluorolube) | Assignment |
|---|---|---|---|---|
| 3286vw | 3306sh, 3279w | 3312w, 3283w | 3310w, 3283w | νasNH2 |
| 3173w | 3157w | 3171w | 3170w | νsNH2 |
| 3052sh, 3041w | 3054vw, 3040vw | 3054vw, 3039vw | 3052sh, 3039sh | νNH+ |
| 3025m | 3027vw | 3025vw | 3025w | νasCH3 |
| 2968sh, 2963m, 2926m | 2952sh, 2936vw | * | 2953sh, 2931w | νsCH3 |
| 2851sh, 2844w, 2831sh | 2832w | * | 2858w, 2843sh, 2826sh | ν1 |
| 2732sh, 2712w, 2684sh | 2710w, 2676sh, 2534vw, 2498vw | 2715w, 2674w, 2639sh, 2540vw, 2495vw | 2717w, 2675vw, 2639sh, 2540vw, 2494vw | νNH2 + νNH+ + o + cb |
| 1654vw | 1639sh | 1644sh | 1642sh | δNH2 |
| 1578vw, 1560vw | 1574vw | 1591vs | 1593vs | ν4 + δNH+ |
| 1480w, 1469w, 1445w | 1478m, 1468m, 1447sh | * | 1477w, 1467w, 1446vw | δasCH3 |
| 1439sh, 1412w | 1436vw, 1411vw | * | 1436vw, 1411vw | δsCH3 + γNH+ |
| 1365vs | 1363sh | * | * | ν5 |
| 1345sh | 1347vs | 1352s, 1342sh | * | ν2 + ρNH2 |
| 1243vw, 1217vw, 1202vw, 1145vw | 1244w, 1217w, 1202w, 1149vw | 1246w, 1217w, 1201vw, 1146sh | * | ρCH3 |
| 1098vw | 1089w | 1093w | * | ρCH3 + νNN |
| 1065w | 1056w | 1058w | * | ν6 |
| 1002vw, 991w | 1002w, 991w | 1002w, 990vw | * | νasCNC |
| 957w, 946w, 937vw | 957w, 946vw | 958w, 946w | * | ωNH2 |
| 832m, 823m | 821w | 820vw | 820vw | νsCNN |
| 789m | 788vs | 792s | 792m | ν3 |
| 507vw | 506w | 504w | * | δCNC |
| 442vw, 421w | 442sh, 421w | 441w, 419w | * | δCNN |
| 219sh | 282s, 233m | τNH2 + τCH3 + lm | ||
| 198sh, 177s, 144s | 191s, 161sh | lm |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zienkiewicz, J.A.; Kucharska, E.; Ptak, M. Mechanism of Unusual Isosymmetric Order-Disorder Phase Transition in [Dimethylhydrazinium]Mn(HCOO)3 Hybrid Perovskite Probed by Vibrational Spectroscopy. Materials 2021, 14, 3984. https://doi.org/10.3390/ma14143984
Zienkiewicz JA, Kucharska E, Ptak M. Mechanism of Unusual Isosymmetric Order-Disorder Phase Transition in [Dimethylhydrazinium]Mn(HCOO)3 Hybrid Perovskite Probed by Vibrational Spectroscopy. Materials. 2021; 14(14):3984. https://doi.org/10.3390/ma14143984
Chicago/Turabian StyleZienkiewicz, Jan Albert, Edyta Kucharska, and Maciej Ptak. 2021. "Mechanism of Unusual Isosymmetric Order-Disorder Phase Transition in [Dimethylhydrazinium]Mn(HCOO)3 Hybrid Perovskite Probed by Vibrational Spectroscopy" Materials 14, no. 14: 3984. https://doi.org/10.3390/ma14143984
APA StyleZienkiewicz, J. A., Kucharska, E., & Ptak, M. (2021). Mechanism of Unusual Isosymmetric Order-Disorder Phase Transition in [Dimethylhydrazinium]Mn(HCOO)3 Hybrid Perovskite Probed by Vibrational Spectroscopy. Materials, 14(14), 3984. https://doi.org/10.3390/ma14143984