
Organosulfur Materials with High Photo- and Photo-Oxidation Stability: 10-Anthryl Sulfoxides and Sulfones and Their Photophysical Properties Dependent on the Sulfur Oxidation State

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthetic Procedures and Spectroscopic Techniques
2.2. Photostability and Photooxidation Stability in Ethanol Solutions
2.3. HRMS-(+)-APCI Spectra
3. Results and Discussion
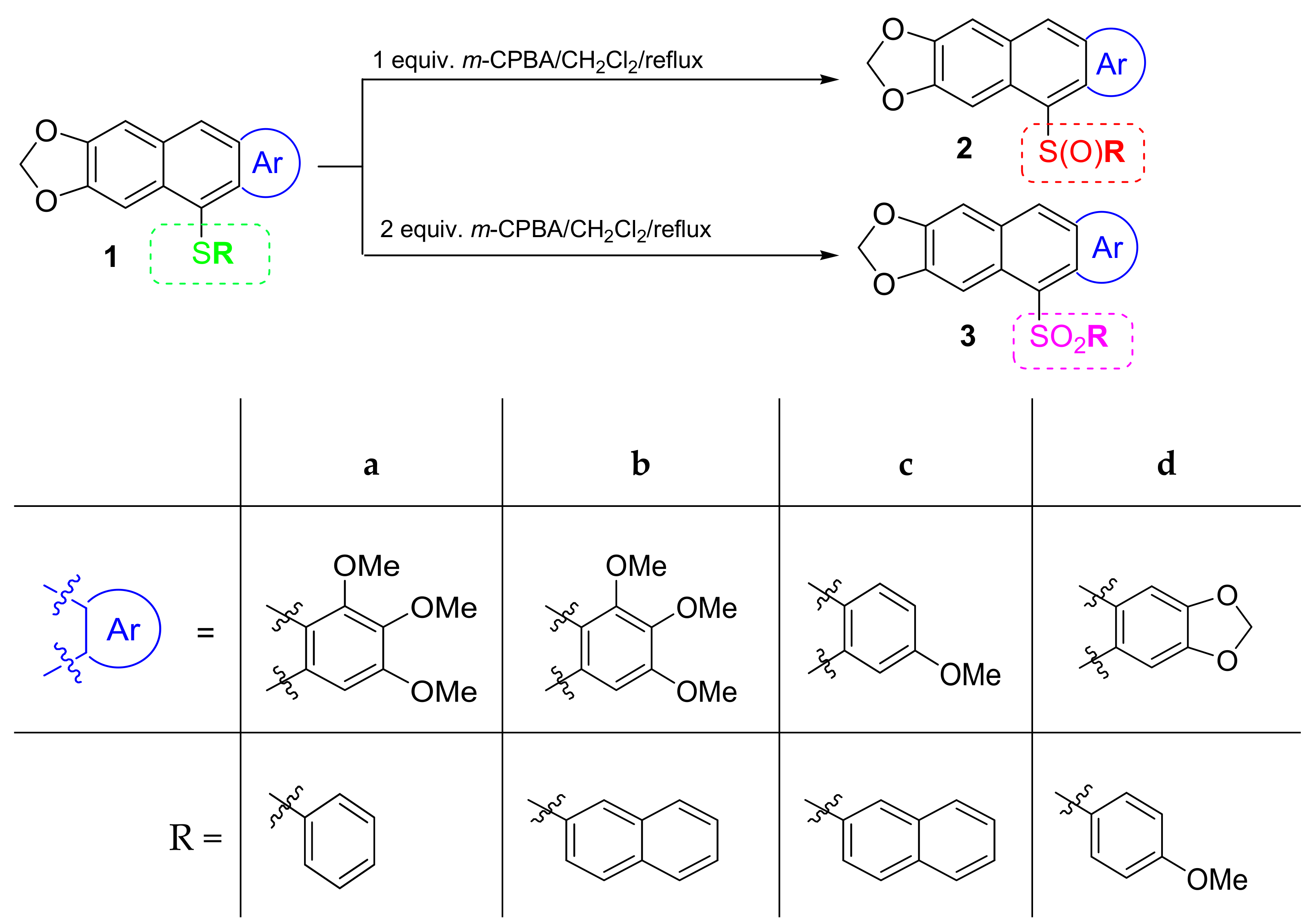
3.1. Chemical Synthesis
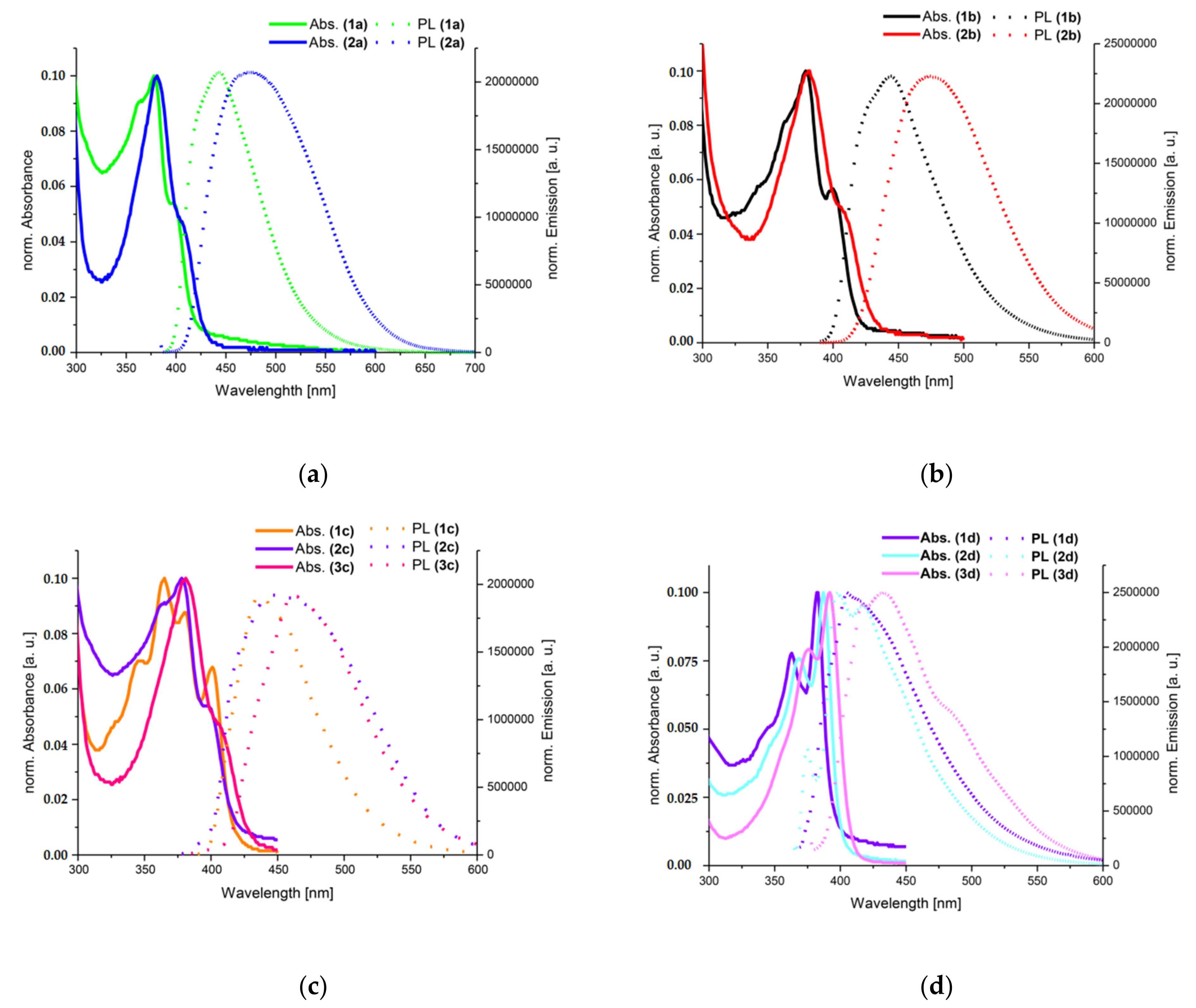
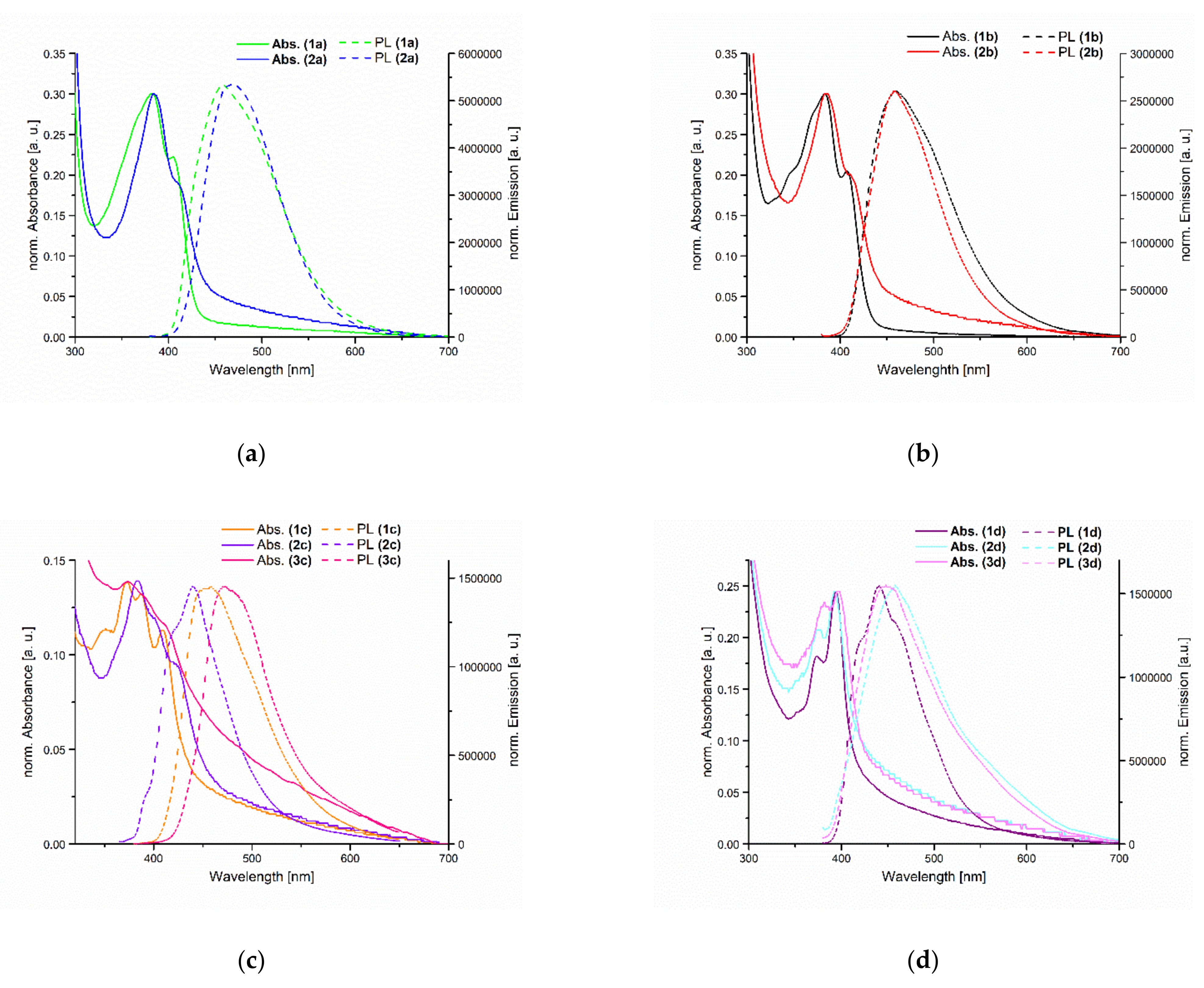
3.2. Photophysical Properties
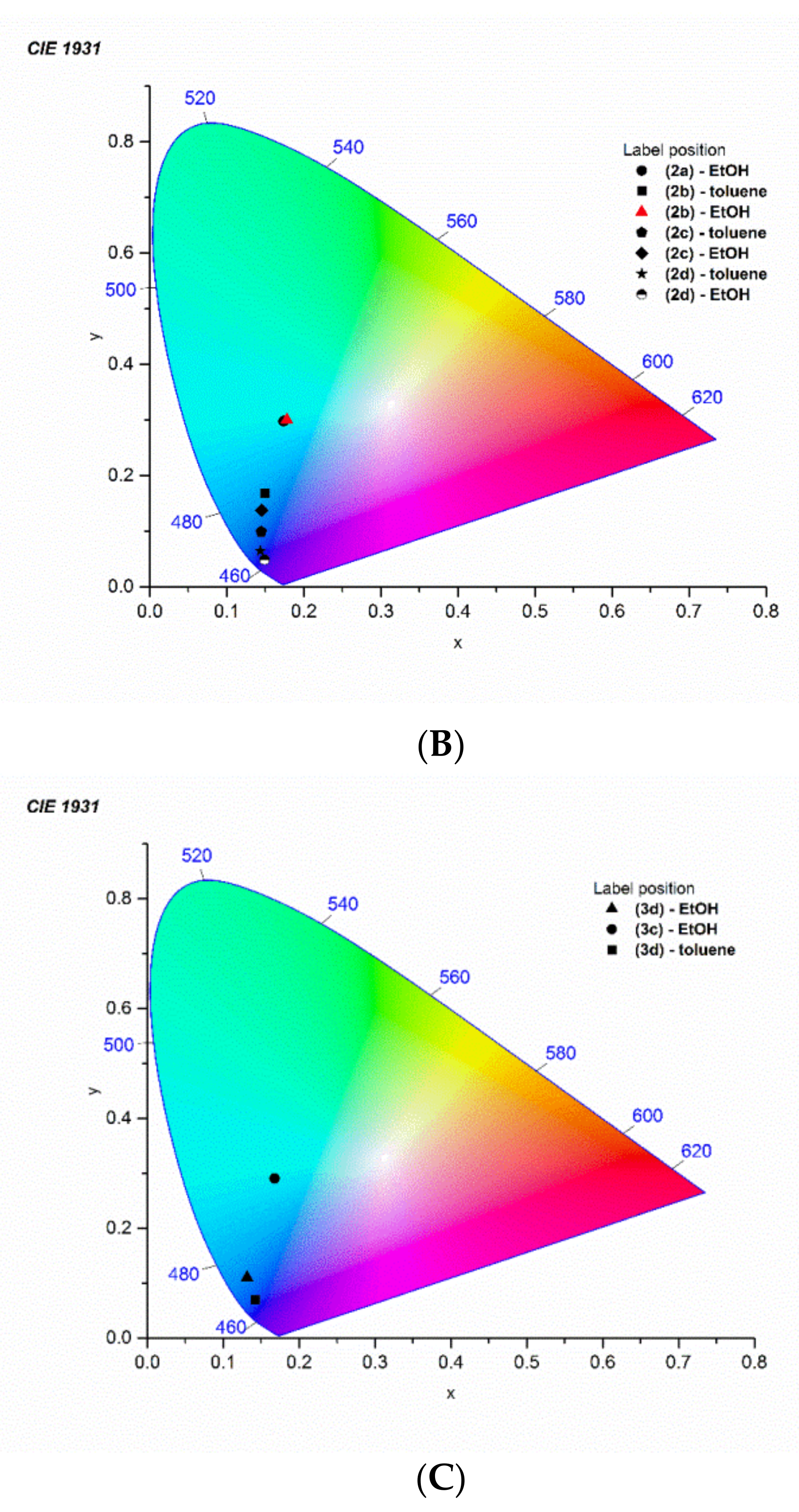
3.3. Structural Determinants of the CIE 1931 Chromaticity Coordinates
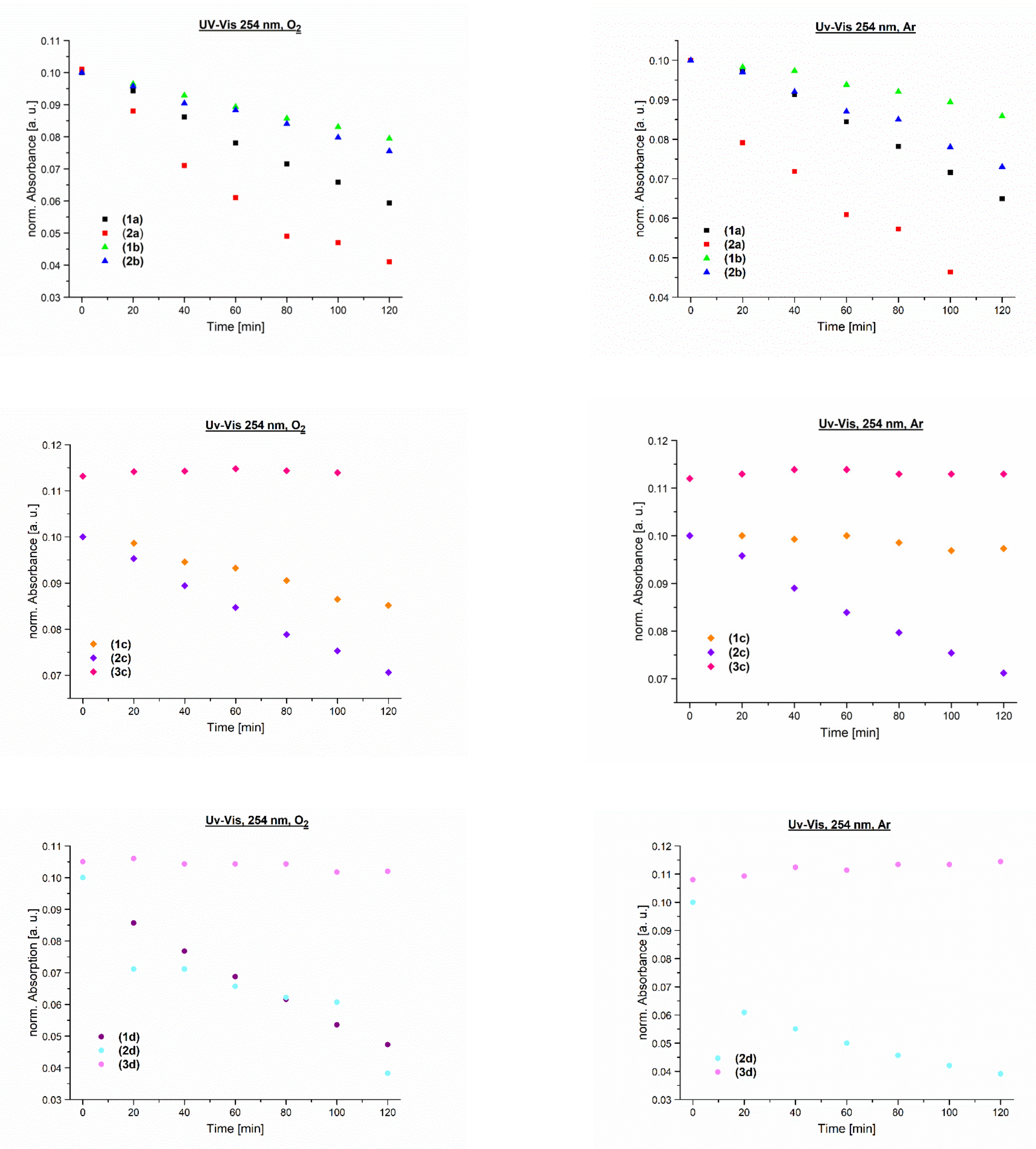
3.4. Photochemical Stability
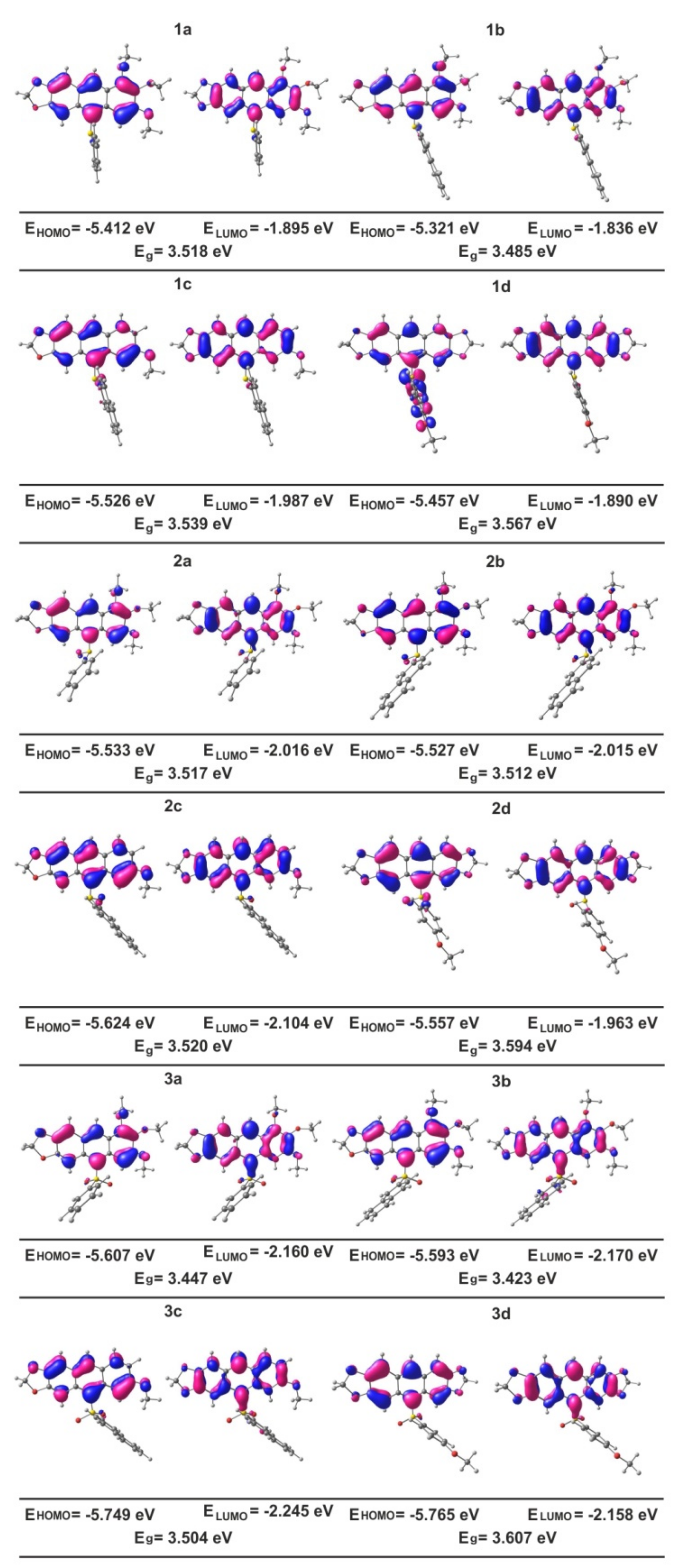
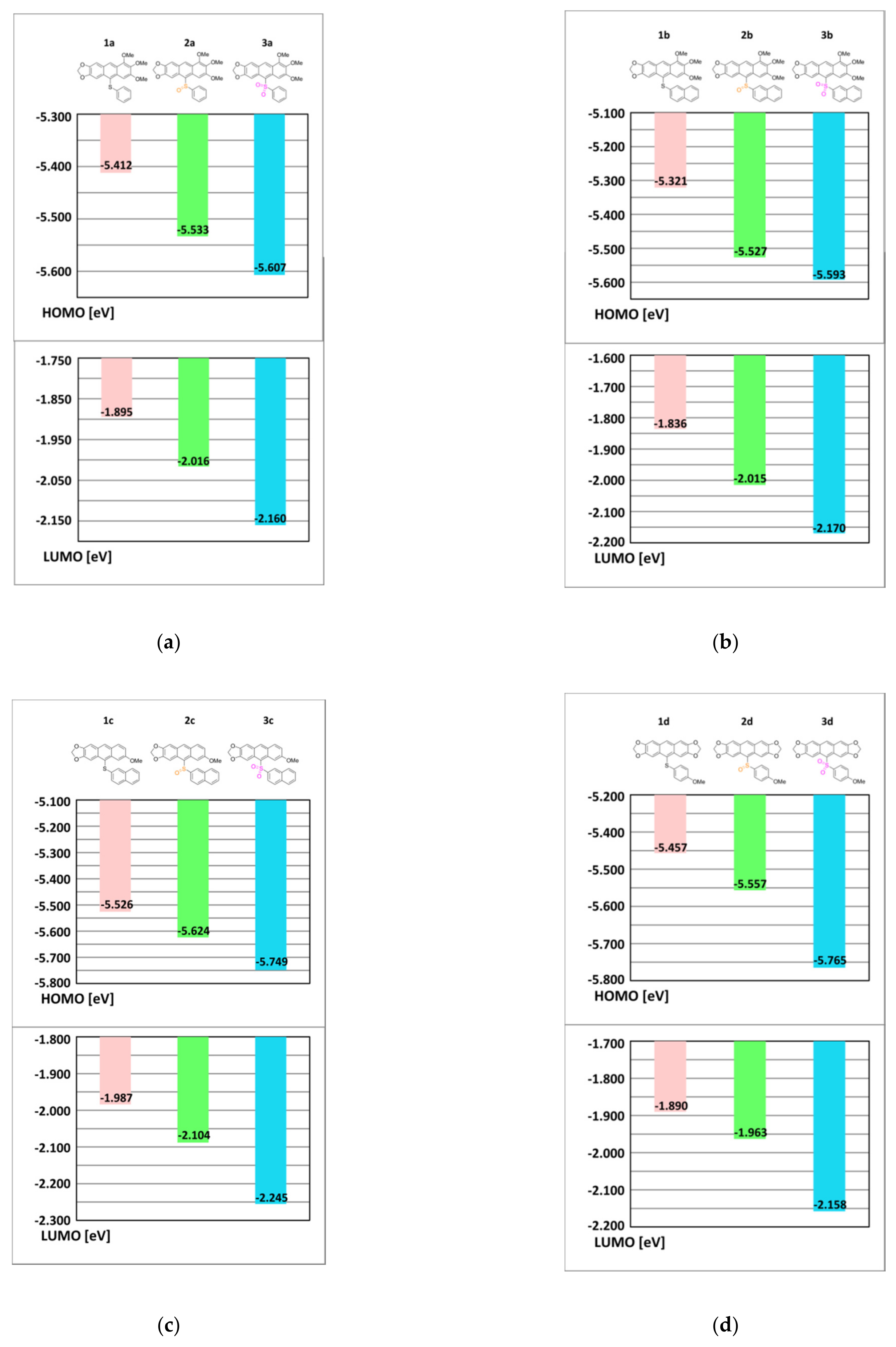
3.5. DFT Calculations of Molecular and Electronic Structures of 1, 2 and 3. Influence of Sulfur Oxidation State on Stabilization of Frontier Orbitals
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, Y.; Xu, P.; Hu, D.; Ma, Y. Recent progress in hot exciton materials for organic light-emitting diodes. Chem. Soc. Rev. 2021, 50, 1030–1069. [Google Scholar] [CrossRef]
- Sasabe, H.; Seino, Y.; Kimura, M.; Kido, J. A m-Terphenyl-Modifed Sulfone Derivative as a Host Material for High-Efficiency Blue and Green Phosphorescent OLEDs. Chem. Mater. 2012, 24, 1404–1406. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, J.; Shizu, K.; Huang, S.; Hirata, S.; Miyazaki, H.; Adachi, C. Design of Efficient Thermally Activated Delayed Fluorescence Materials for Pure Blue Organic Light Emitting Diodes. J. Am. Chem. Soc. 2012, 134, 14706–14709. [Google Scholar] [CrossRef]
- Yang, S.-Y.; Tian, Q.-S.; Yu, Y.-J.; Zou, S.-N.; Li, H.-C.; Khan, A.; Wu, Q.-H.; Jiang, Z.-Q.; Liao, L.-S. Sky-Blue Thermally Activated Delayed Fluorescence with Intramolecular Spatial Charge Transfer Based on a Dibenzothiophene Sulfone Emitter. J. Org. Chem. 2020, 85, 10628–10637. [Google Scholar] [CrossRef]
- Jeon, S.O.; Earmme, T.; Jenekhe, S.A. New sulfone-based electron-transport materials with high triplet energy for highly efficient blue phosphorescent organic light-emitting diodes. J. Mater. Chem. C 2014, 2, 10129–10137. [Google Scholar] [CrossRef]
- Di, Z.; Tingting, Y.; Zhengqin, W.; Huixia, X.; Hua, W.; Junsheng, Y.; Bingshe, X. Photoelectric properties of host materials based on diphenyl sulfone as acceptor and the performances in green phosphorescent OLEDs. Opt. Mater. 2020, 109, 110313. [Google Scholar] [CrossRef]
- Jürgensen, N.; Kretzschmar, A.; Höfle, S.; Freudenberg, J.; Bunz, U.H.F.; Hernandez-Sosa, G. Sulfone-Based Deep Blue Thermally Activated Delayed Fluorescence Emitters: Solution-Processed Organic Light-Emitting Diodes with High Efficiency and Brightness. Chem. Mater. 2017, 29, 9154–9161. [Google Scholar] [CrossRef]
- Tonge, C.M.; Zeng, J.; Zhao, Z.; Tang, B.Z.; Hudson, Z.M. Bis(hexamethylazatriangulene)sulfone: A high-stability deep blue-violet fluorophore with 100% quantum yield and CIE y <0.07. J. Mater. Chem. C 2020, 8, 5150–5155. [Google Scholar] [CrossRef]
- Liu, X.; Xu, R.; Duan, C.; Huang, F.; Cao, Y. Non-conjugated water/alcohol soluble polymers with different oxidation states of sulfide as cathode interlayers for high-performance polymer solar cells. J. Mater. Chem. C 2016, 4, 4288–4295. [Google Scholar] [CrossRef]
- Li, S.; Ye, L.; Wang, Q.; Zhang, S.; Zhao, W.; Hou, J. Improving the open-circuit voltage of alkylthio-substituted photovoltaic polymers via post-oxidation. Org. Electron. 2016, 28, 39–46. [Google Scholar] [CrossRef]
- Hirose, A.; Tanaka, K.; Yoshii, R.; Chujo, Y. Film-type chemosensors based on boron diiminate polymers having oxidation-induced emission properties. Polym. Chem. 2015, 6, 5590–5595. [Google Scholar] [CrossRef]
- Lamberth, C. Sulfur chemistry in crop protection. J. Sulfur Chemistry 2004, 25, 39–62. [Google Scholar] [CrossRef]
- Kathayat, R.S.; Yang, L.; Sattasathuchana, T.; Zoppi, L.; Baldridge, K.K.; Linden, A.; Finney, N.S. On the Origins of Nonradiative Excited State Relaxation in Aryl Sulfoxides Relevant to Fluorescent Chemosensing. J. Am. Chem. Soc. 2016, 138, 15889–15895. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Ma, X.; Yu, J.; Jia, X.; Han, D.; Zhou, T.; Yang, J.; Nie, J.; Wang, T. Aromatic amine–sulfone/sulfoxide conjugated D–π-A–π-D-type dyes in photopolymerization under 405 nm and 455 nm laser beams. Polym. Chem. 2015, 6, 4424–4435. [Google Scholar] [CrossRef]
- Christensen, P.R.; Wolf, M.O. Photopatterned Multidimensional Fluorescent Images. Adv. Funct. Mater. 2016, 26, 8471–8477. [Google Scholar] [CrossRef]
- Mísek, J.; Jentzsch, A.V.; Sakurai, S.; Emery, D.; Mareda, J.; Matile, S. A Chiral and Colorful Redox Switch: Enhanced π-Acidity in Action. Angew. Chem. Int. Ed. 2010, 49, 7680–7683. [Google Scholar] [CrossRef]
- Monçalves, M.; da Silveira Rampon, D.; Schneider, P.H.; Rodembusch, F.S.; da Cruz Silveira, C. Divinyl sulfides/sulfones-based D–π–A–π–D dyes as efficient non-aromatic bridges for π-conjugated compounds. Dyes. Pigm. 2014, 102, 71–78. [Google Scholar] [CrossRef]
- Benz, S.; Mareda, J.; Besnard, C.; Sakai, N.; Matile, S. Catalysis with chalcogen bonds: Neutral benzodiselenazole scaffolds with high-precision selenium donors of variable strength. Chem. Sci. 2017, 8, 8164–8169. [Google Scholar] [CrossRef] [Green Version]
- Caron, E.; Wolf, M.O. Soluble Oligo- and Polythienyl Sulfides and Sulfones: Synthesis and Photophysics. Macromolecules 2017, 50, 7543–7549. [Google Scholar] [CrossRef]
- Monçalves, M.; Zanotto, G.M.; Toldo, J.M.; Rampon, D.S.; Schneider, P.H.; Gonçalves, P.F.B.; Rodembusch, F.S.; Silveira, C.C. Dipolar vinyl sulfur fluorescent dyes. Synthesis and photophysics of sulfide, sulfoxide and sulfone based D–π–A compounds. RSC Adv. 2017, 7, 8832–8842. [Google Scholar] [CrossRef] [Green Version]
- Verolet, Q.; Rosspeintner, A.; Soleimanpour, S.; Sakai, N.; Vauthey, E.; Matile, S. Turn-On Sulfide π Donors: An Ultrafast Push for Twisted Mechanophores. J. Am. Chem. Soc. 2015, 137, 15644–15647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piazza, G.A.; Rahm, A.L.K.; Krutzsch, M.; Sperl, G.; Paranka, N.S.; Gross, P.H.; Brendel, K.; Burt, R.W.; Alberts, D.S.; Pamukcu, R.; et al. Antineoplastic Drugs Sulindac Sulfide and Sulfone Inhibit Cell Growth by Inducing Apoptosis. Cancer Res. 1995, 55, 3110–3116. [Google Scholar] [PubMed]
- Liu, L.; Stelmach, J.E.; Natarajan, S.R.; Chen, M.H.; Singh, S.B.; Schwatrz, C.D.; Fitzgerald, C.E.; O’Keefe, S.J.; Zaller, D.M.; Schmatz, D.B.; et al. SAR of 3,4-Dihydropyrido [3,2-d] pyrimidone p38 inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 3979–3982. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Tang, B.; Liang, S.H.; Jiang, X. Sulfur Containing Scaffolds in Drugs: Synthesis and Application in Medicinal Chemistry. Curr. Top. Med. Chem. 2016, 16, 1200–1216. [Google Scholar] [CrossRef]
- Godumala, M.; Choi, S.; Cho, M.J.; Choi, D.H. Thermally activated delayed fluorescence blue dopants and hosts: From the design strategy to organic light-emitting diode applications. J. Mater. Chem. C 2016, 4, 11355–11381. [Google Scholar] [CrossRef]
- Christensen, P.R.; Nagle, J.K.; Bhatti, A.; Wolf, M.O. Enhanced Photoluminescence of Sulfur-Bridged Organic Chromophores. J. Am. Chem. Soc. 2013, 135, 8109–8112. [Google Scholar] [CrossRef]
- Cruz, C.D.; Christensen, P.R.; Chronister, E.L.; Casanova, D.; Wolf, M.O.; Bardeen, C.J. Sulfur-Bridged Terthiophene Dimers: How Sulfur Oxidation State Controls Interchromophore Electronic Coupling. J. Am. Chem. Soc. 2015, 137, 12552–12564. [Google Scholar] [CrossRef]
- Climent, C.; Barbatti, M.; Wolf, M.O.; Bardeend, C.J.; Casanova, D. The photophysics of naphthalene dimers controlled by sulfur bridge oxidation. Chem. Sci. 2017, 8, 4941–4950. [Google Scholar] [CrossRef] [Green Version]
- Malashikhin, S.; Finney, N.S. Fluorescent Signaling Based on Sulfoxide Profluorophores: Application to the Visual Detection of the Explosive TATP. J. Am. Chem. Soc. 2008, 130, 12846–12847. [Google Scholar] [CrossRef]
- Malashikhin, S. Efficient Discovery of Fluorescent Chemosensors Based on the Biarylpyridine Scaffold: Visual Detection of the Explosive TATP the Origin of Sulfoxides “Non Emission”. Ph.D. Thesis, Faculty of Science, University of Zurich (CH), Zürich, Switzerland, 2010. [Google Scholar]
- Christensen, P.R.; Patrick, B.O.; Caron, E.; Wolf, M.O. Oxidation-State-Dependent Photochemistry of Sulfur-Bridged Anthracenes. Angew. Chem. Int. Ed. 2013, 52, 12946–12950. [Google Scholar] [CrossRef]
- Zhao, C.; Wang, X.; Cao, J.; Feng, P.; Zhang, J.; Zhang, Y.; Yang, Y.; Yang, Z. BODIPY-based sulfoxide: Synthesis, photophysical characterization and response to benzenethiols. Dye. Pigm. 2013, 96, 328–332. [Google Scholar] [CrossRef]
- Bałczewski, P.; Kowalska, E.; Różycka-Sokołowska, E.; Skalik, J.; Owsianik, K.; Koprowski, M.; Marciniak, B.; Guziejewski, D.; Ciesielski, W. Mono-Aryl/Alkylthio-Substituted (Hetero)acenes of Exceptional Thermal and Photochemical Stability by the Thio-Friedel–Crafts/Bradsher Cyclization Reaction. Chem. Eur. J. 2019, 25, 14148–14161. [Google Scholar] [CrossRef]
- Bałczewski, P.; Kowalska, E.; Skalik, J. 10-Thiosubstituted Pentahydroxyanthracene Derivatives, a Method of Their Preparation and Intermediate Compounds. EPO appl., No. EP-14460003.8, 06.02.2014. EPO patent No. 28896539, 1 February 2017. [Google Scholar]
- Gurria, G.M.; Posner, G.H. Photochemical deoxygenation of aryl sulfoxides. J. Org. Chem. 1973, 88, 2419–2420. [Google Scholar] [CrossRef]
- ACD/Percepta, Version 14.0.0; Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2015.
- Zhang, J.; Sarrafpour, S.; Haas, T.E.; Müller, P.; Thomas, S.W. Structure, photophysics and photooxidation of crowded diethynyltetracenes. J. Mater. Chem. 2012, 22, 6182–6189. [Google Scholar] [CrossRef]
- Maliakal, A.; Raghavachari, K.; Katz, H.; Chandross, E.; Siegrist, T. Photochemical Stability of Pentacene and a Substituted Pentacene in Solution and in Thin Films. Chem. Mater. 2004, 16, 4980–4986. [Google Scholar] [CrossRef]
- Bałczewski, P.; Koprowski, M.; Bodzioch, A.; Marciniak, B.; Różycka-Sokołowska, E. Unusual Transformation of the Diarylmethanol Derivative into an Unknown 1,2,3,6,7,10-Hexahydroxylated Anthracene System. J. Org. Chem. 2006, 71, 2899–2902. [Google Scholar] [CrossRef]
- Bałczewski, P.; Bodzioch, A.; Różycka-Sokołowska, E.; Marciniak, B.; Uznański, P. First Approach to Nitrogen-Containing Fused Aromatic Hydrocarbons as Targets for Organoelectronics Utilizing a New Transformation of O-Protected Diaryl Methanols. Chem. Eur. J. 2010, 16, 2392–2400. [Google Scholar] [CrossRef]
- Bodzioch, A.; Marciniak, B.; Różycka-Sokołowska, E.; Jeszka, J.K.; Uznański, P.; Kania, S.; Kuliński, J.; Bałczewski, P. Synthesis and Optoelectronic Properties of Hexahydroxylated 10-RO-Substituted Anthracenes via a New Modification of the Friedel–Crafts Reaction Using O-Protected ortho-Acetal Diarylmethanols. Chem. Eur. J. 2012, 18, 4866–4876, Synfacts—Highlights in Curr. Synth. Org. Chem. 2012, 8, 619. [Google Scholar] [CrossRef]
- 10-Thiosubstituted Pentahydroxyanthracene Derivatives, a Method of Their Preparation and Intermediate Compounds; EP 288539; European Patent Office: München, Germany, 2014.
- Tio-Sfunkcjonalizowane aceny i ich Zastosowanie; Pat. Appl. 429239; UPRP: Warszawa, Poland, 2019.
- Bałczewski, P.; Skalik, J.; Uznański, P.; Guziejewski, D.; Ciesielski, W. Use of isomeric, aromatic dialdehydes in the synthesis of photoactive, positional isomers of higher analogs of o-bromo (hetero) acenaldehydes. RSC Adv. 2015, 5, 24700–24704. [Google Scholar] [CrossRef]
- Kowalska, E.; Bałczewski, P. Ultrasound assisted Bradsher reaction in aqueous and non-aqueous media: First use of ultrasounds in electrophilic aromatic cyclisation leading to polyacenes. Ultrason. Sonochem. 2017, 34, 743–753. [Google Scholar] [CrossRef]
- Bałczewski, P.; Bodzioch, A.; Koprowski, M. New Condensed Polyaromatic and Polyheteroaromatic Hydrocarbons, Method of Their Manufacturing and the Intermediate Compounds. UPRP PL 219334B1, 25 August 2014. [Google Scholar]
- thioBodzioch, A.; Kowalska, E.; Skalik, J.; Bałczewski, P. Synthesis of Polycyclic (Hetero)Aromatic Hydrocarbons via the Friedel–Crafts/Bradsher Cyclization. Chem. Heterocycl. Compd. 2017, 53, 11–20. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound. | Chemical Structure | Yield (%) |
|---|---|---|
| 1a |  | 62 |
| 1b | 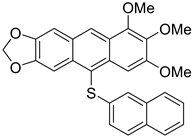 | 51 |
| 1c |  | 50 |
| 1d | 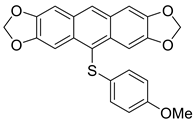 | 52 |
| 2a |  | 55 |
| 2b | 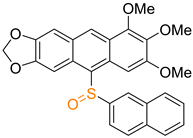 | 42 |
| 2c | 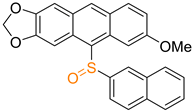 | 36 |
| 2d | 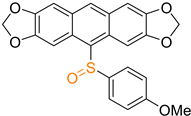 | 32 |
| 3c | 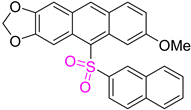 | 26 |
| 3d |  | 17 |
| Compound | Absorption λmax (nm) | Photoluminescence λmax (nm) | Stokes Shift (cm−1) | τ (a) (ns) | χ2 (b) | Φ (c) (%) |
|---|---|---|---|---|---|---|
| In toluene | ||||||
| 1a | 380, 401 | 445 | 3840 | 5.43 | 1.03 | 31.5 |
| 2a | 380, 405 | 467 | 4900 | 8.29 | 1.20 | 40.6 |
| 1b | 380, 403 | 447 | 3940 | 2.23 | 1.17 | 18.3 |
| 2b | 380, 405 | 458 | 4480 | 8.70 | 1.17 | 55.6 |
| 1c | 330, 348, 367, 383, 403 | 440 | 4520 | 1.94 | 1.10 | 15.0 |
| 2c | 368, 386, 407 | 442 | 3280 | 4.06 | 1.01 | 21.6 |
| 3c | 416, 395, 378, 328 | 460 | 4720 | 0.98 | 1.13 | 10.0 |
| 1d | 364, 384 | 424 | 2460 | 0.23; 2.17 (0.95) | 1.12 | 0.63 |
| 2d | 367, 387 | 397 | 650 | 0.34; 4.75 (3.90) | 1.09 | 1.91 |
| 3d | 375, 392 | 433 | 3840 | 0.47; 3.98; 9.46 (7.50) | 1.15 | 2.02 |
| In ethanol | ||||||
| 1a | 379, 399 | 442 | 3760 | 6.08 | 1.14 | 33.3 |
| 2a | 379 | 473 | 5240 | 15.28 | 1.23 | 58.2 |
| 1b | 379, 400 | 444 | 3860 | 2.24 | 1.01 | 16.6 |
| 2b | 380 | 477 | 5350 | 14.23 | 1.20 | 54.2 |
| 1c | 345, 365, 380, 401 | 437 | 4510 | 2.10 | 1.06 | 12.9 |
| 2c | 368, 370, 386, 406 | 449 | 4900 | 1.91; 8.36 (5.15) | 1.12 | 32.4 |
| 3c | 376, 391, 413 | 466 | 5140 | 0.41; 3.80 (1.55) | 1.04 | 1.9 |
| 1d | 362, 382 | 405 | 1490 | 0.47; 2.81 (1.15) | 1.24 | 0.1 |
| 2d | 368, 387 | 398 | 710 | 0.28; 5.47 (4.20) | 1.09 | 2.5 |
| 3d | 376, 392 | 433 | 2420 | 0.16; 0.77; 5.64 (4.90) | 1.45 | 0.53 |
| Compound | Absorption λmax (nm) | Photoluminescence λmax (nm) | Stokes Shift (cm−1) |
|---|---|---|---|
| 1a | 383 | 459 | 4320 |
| 2a | 384 | 469 | 4720 |
| 1b | 383 | 460 | 4370 |
| 2b | 385 | 458 | 4140 |
| 1c | 373 | 458 | 4980 |
| 2c | 373 | 439 | 4030 |
| 3c | 383 | 473 | 4970 |
| 1d | 394 | 441 | 2700 |
| 2d | 393 | 458 | 3610 |
| 3d | 397 | 448 | 2870 |
| The CIE 1931 Color Spaces-Luminosity Function | ||
|---|---|---|
| Compound | Toluene (x, y) | Ethanol (x, y) |
| 1a | x = 0.1534, y = 0.0891 | x = 0.15148, y = 0.09148 |
| 2a | (a) | x = 0.17369, y = 0.29827 |
| 1b | x = 0.15012, y = 0.0855 | x = 0.16057, y = 0.10734 |
| 2b | x = 0.14996, y = 0.16765 | x = 0.17792, y = 0.3006 |
| 1c | x = 0.1627, y = 0.10657 | x = 0.16889, y = 0.1198 |
| 2c | x = 0.1447, y = 0.09946 | x = 0.14522, y = 0.13796 |
| 3c | (a) | x = 0.16769, y = 0.29084 |
| 1d | x = 0.1526, y = 0.03397 | x = 0.14725, y = 0.05423 |
| 2d | x = 0.14357, y = 0.06482 | x = 0.14886, y = 0.04898 |
| 3d | x = 0.14217, y = 0.06996 | x = 0.13137, y = 0.11087 |
| Compound | The Time of 50% Loss of Absorbance Measured for the Lowest Energy Absorption Band (min) | ||
|---|---|---|---|
| UV-Vis 254 nm/O2 | UV-Vis 254 nm/Ar | UV-Vis 365 nm/O2 | |
| 1a | 166 | 175 | n/o (a) |
| 2a | 80 | 88 | n/o (a) |
| 1b | 249 | 519 | n/o (a) |
| 2b | 249 | 251 | n/o (a) |
| 1c | 210 | 276 | n/o (a) |
| 2c | 247 | 245 | 254 |
| 1d | 115 | n/o | 812 |
| 2d | 111 | 93 | 124 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bałczewski, P.; Kowalska, E.; Różycka-Sokołowska, E.; Uznański, P.; Wilk, J.; Koprowski, M.; Owsianik, K.; Marciniak, B. Organosulfur Materials with High Photo- and Photo-Oxidation Stability: 10-Anthryl Sulfoxides and Sulfones and Their Photophysical Properties Dependent on the Sulfur Oxidation State. Materials 2021, 14, 3506. https://doi.org/10.3390/ma14133506
Bałczewski P, Kowalska E, Różycka-Sokołowska E, Uznański P, Wilk J, Koprowski M, Owsianik K, Marciniak B. Organosulfur Materials with High Photo- and Photo-Oxidation Stability: 10-Anthryl Sulfoxides and Sulfones and Their Photophysical Properties Dependent on the Sulfur Oxidation State. Materials. 2021; 14(13):3506. https://doi.org/10.3390/ma14133506
Chicago/Turabian StyleBałczewski, Piotr, Emilia Kowalska, Ewa Różycka-Sokołowska, Paweł Uznański, Joanna Wilk, Marek Koprowski, Krzysztof Owsianik, and Bernard Marciniak. 2021. "Organosulfur Materials with High Photo- and Photo-Oxidation Stability: 10-Anthryl Sulfoxides and Sulfones and Their Photophysical Properties Dependent on the Sulfur Oxidation State" Materials 14, no. 13: 3506. https://doi.org/10.3390/ma14133506
APA StyleBałczewski, P., Kowalska, E., Różycka-Sokołowska, E., Uznański, P., Wilk, J., Koprowski, M., Owsianik, K., & Marciniak, B. (2021). Organosulfur Materials with High Photo- and Photo-Oxidation Stability: 10-Anthryl Sulfoxides and Sulfones and Their Photophysical Properties Dependent on the Sulfur Oxidation State. Materials, 14(13), 3506. https://doi.org/10.3390/ma14133506