
Peptides and Peptidomimetics as Inhibitors of Enzymes Involved in Fibrillar Collagen Degradation



Abstract
1. Introduction
2. Collagen Turnover in Cells
2.1. Methods of Collagen Turnover Measurements
2.1.1. Indirect Methods
2.1.2. Direct Methods
2.1.3. Others
3. Inhibitors of Collagen-Degrading Enzymes
3.1. Collagenases
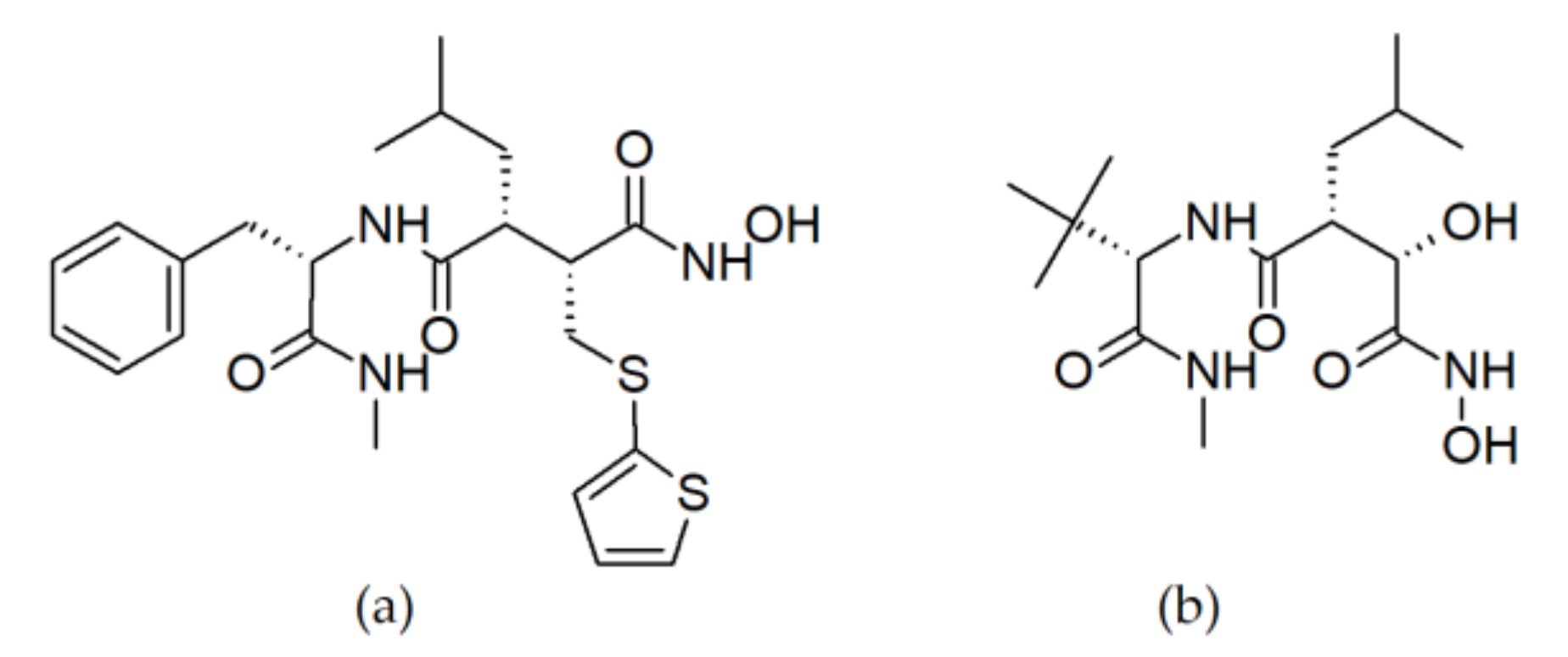
3.1.1. Peptide Hydroxamates
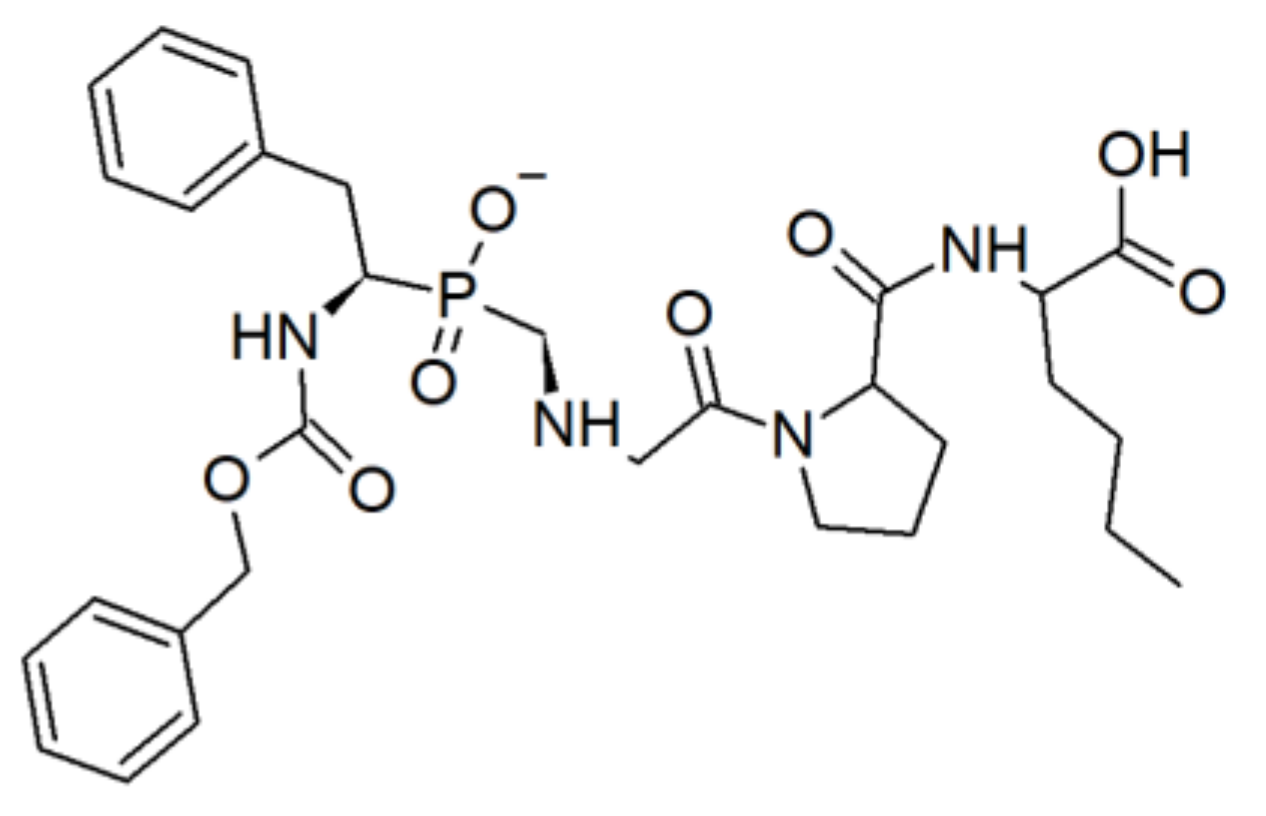
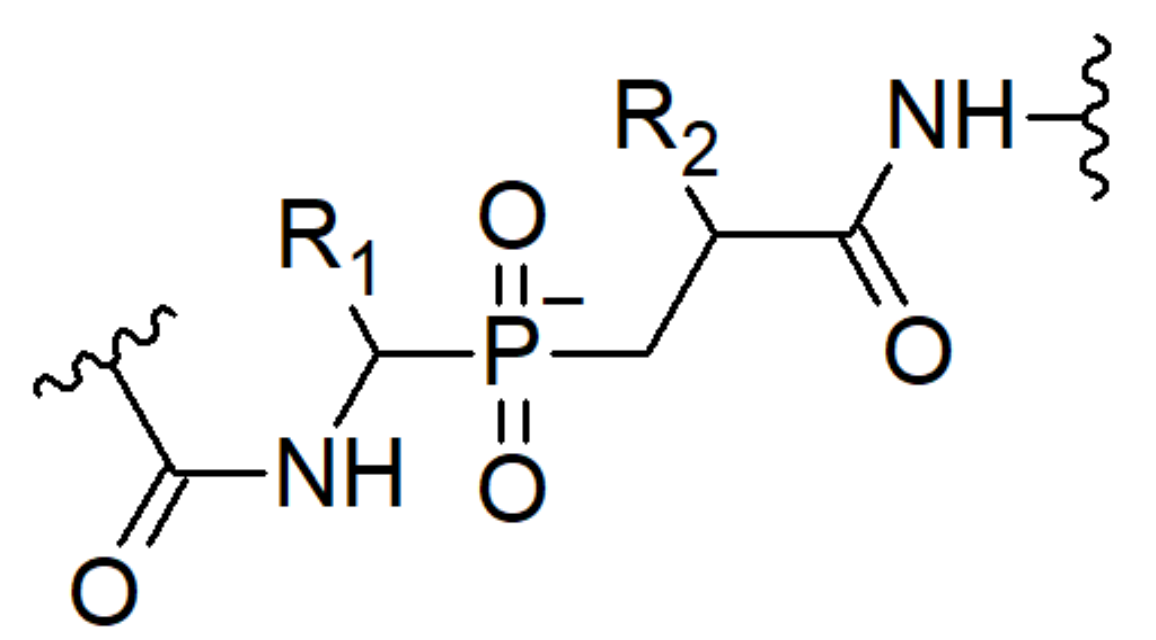
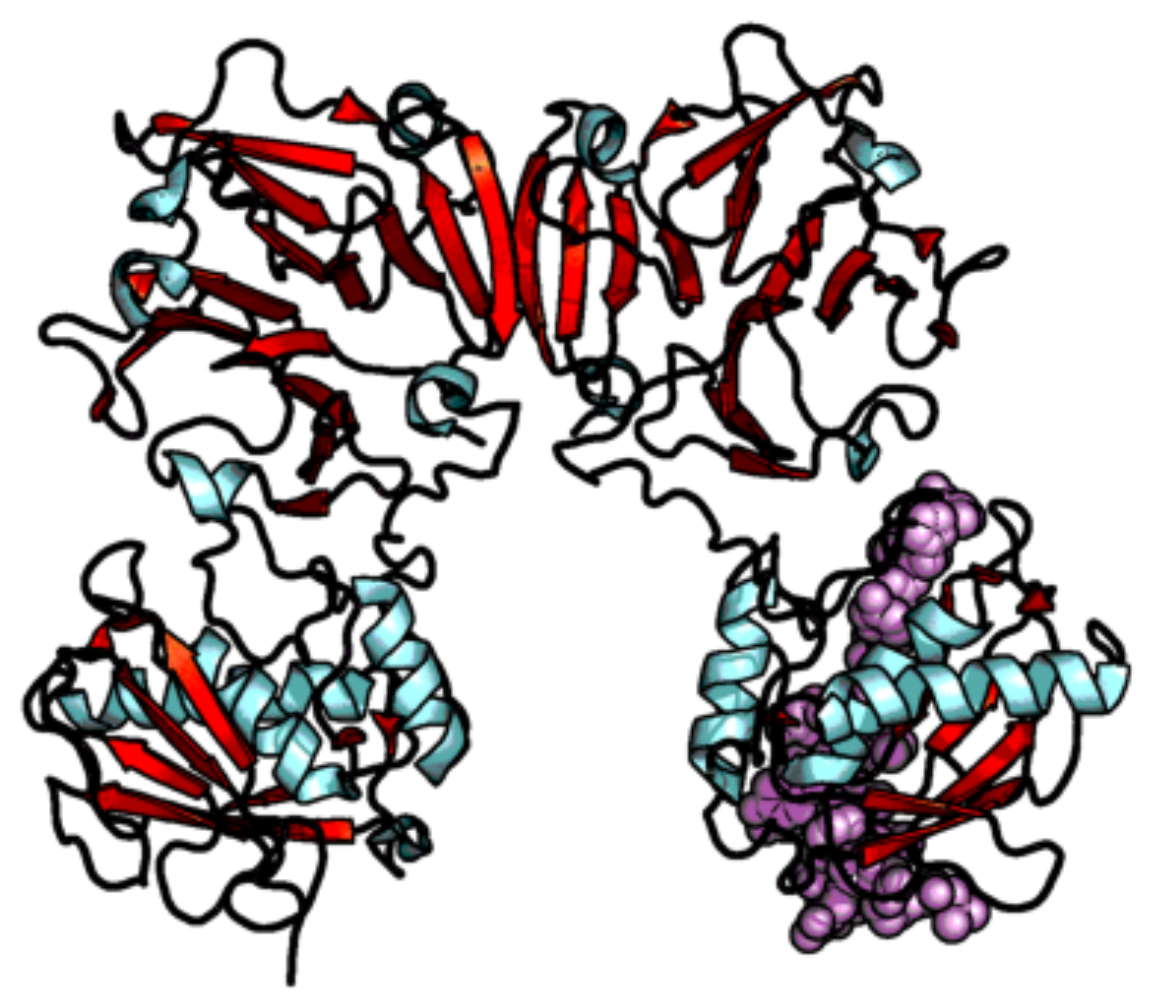
3.1.2. Phosphinic acid Derivatives
3.1.3. Unmodified Peptides
Synthetic Peptides
Peptides from Natural Sources
3.1.4. Amino Acids and Peptide Derivatives
3.2. Elastase
3.2.1. Chloromethyl Ketones
3.2.2. Cyclic Depsipeptides
3.2.3. Unmodified Peptides
3.2.4. Peptide Derivatives and Cyclic Compounds
3.3. Cathepsins
3.3.1. Peptide Aldehydes
3.3.2. Peptidic Nitriles
3.3.3. Peptide Ketoheterocycles
4. Conclusions and Future Prospect
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yue, B. Biology of the Extracellular Matrix: An Overview. J. Glaucoma 2014, 23, S20–S23. [Google Scholar] [CrossRef] [PubMed]
- Żelaszczyk, D.; Waszkielewicz, A.; Marona, H. Kolagen—Struktura Oraz Zastosowanie w Kosmetologii i Medycynie Estetycznej. Estetol. Med. Kosmetol. 2012, 14–20. [Google Scholar] [CrossRef]
- Veit, G.; Kobbe, B.; Keene, D.R.; Paulsson, M.; Koch, M.; Wagener, R. Collagen XXVIII, a Novel von Willebrand Factor A Domain-Containing Protein with Many Imperfections in the Collagenous Domain. J. Biol. Chem. 2006, 281, 3494–3504. [Google Scholar] [CrossRef]
- Smith, K.; Rennie, M.J. New Approaches and Recent Results Concerning Human-Tissue Collagen Synthesis. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Burgeson, R.E.; Nimni, M.E. Collagen Types. Molecular Structure and Tissue Distribution. Clin. Orthop Relat Res. 1992, 250–272. [Google Scholar]
- Bhattacharjee, A.; Bansal, M. Collagen Structure: The Madras Triple Helix and the Current Scenario. IUBMB Life Int. Union Biochem. Mol. Biol. Life 2005, 57, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, B.; Thiagarajan, G.; Madhan, B.; Kar, K. Triple-Helical Peptides: An Approach to Collagen Conformation, Stability, and Self-Association. Biopolymers 2008, 89, 345–353. [Google Scholar] [CrossRef]
- Fields, G.B. Interstitial Collagen Catabolism. J. Biol. Chem. 2013, 288, 8785–8793. [Google Scholar] [CrossRef]
- Bishop, J.E.; Laurent, G.J. Collagen Turnover and Its Regulation in the Normal and Hypertrophying Heart. Eur. Heart J. 1995, 16, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Manon-Jensen, T.; Kjeld, N.G.; Karsdal, M.A. Collagen-Mediated Hemostasis. J. Thromb Haemost. 2016, 14, 438–448. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Genovese, F.; Madsen, E.A.; Manon-Jensen, T.; Schuppan, D. Collagen and Tissue Turnover as a Function of Age: Implications for Fibrosis. J. Hepatol. 2016, 64, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Sprangers, S.; Everts, V. Molecular Pathways of Cell-Mediated Degradation of Fibrillar Collagen. Matrix Biol. 2019, 75–76, 190–200. [Google Scholar] [CrossRef]
- Kim, M.; Park, H.J. Molecular Mechanisms of Skin Aging and Rejuvenation; InTech: Seoul, Korea, 2016; pp. 57–76. [Google Scholar]
- Rodriguez-Feo, J.; Sluijter, J.; Kleijn, D.; Pasterkamp, G. Modulation of Collagen Turnover in Cardiovascular Disease. Curr. Pharm. Des. 2005, 11, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Gelse, K. Collagens—Structure, Function, and Biosynthesis. Adv. Drug Deliv. Rev. 2003, 55, 1531–1546. [Google Scholar] [CrossRef]
- Vannella, K.M.; Wynn, T.A. Mechanisms of Organ Injury and Repair by Macrophages. Annu. Rev. Physiol. 2017, 79, 593–617. [Google Scholar] [CrossRef]
- Rennard, S.I.; Stier, L.E.; Crystal, R.G. Intracellular Degradation of Newly Synthesized Collagen. J. Investig. Dermatol. 1982, 79, 77–82. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular Matrix Degradation and Remodeling in Development and Disease. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Zhong, S.; Khalil, R.A. A Disintegrin and Metalloproteinase (ADAM) and ADAM with Thrombospondin Motifs (ADAMTS) Family in Vascular Biology and Disease. Biochem. Pharmacol. 2019, 164, 188–204. [Google Scholar] [CrossRef]
- McAnulty, R.J.; Laurent, G.J. Collagen Synthesis and Degradation In Vivo. Evidence for Rapid Rates of Collagen Turnover with Extensive Degradation of Newly Synthesized Collagen in Tissues of the Adult Rat. Collagen Relat. Res. 1987, 7, 93–104. [Google Scholar] [CrossRef]
- Laurent, G.J. Dynamic State of Collagen: Pathways of Collagen Degradation in Vivo and Their Possible Role in Regulation of Collagen Mass. Am. J. Physiol. Cell Physiol. 1987, 252, C1–C9. [Google Scholar] [CrossRef]
- McClung, J.M.; Davis, J.M.; Wilson, M.A.; Goldsmith, E.C.; Carson, J.A. Estrogen Status and Skeletal Muscle Recovery from Disuse Atrophy. J. Appl. Physiol. 2006, 100, 2012–2023. [Google Scholar] [CrossRef]
- Miller, B.F.; Hansen, M.; Olesen, J.L.; Flyvbjerg, A.; Schwarz, P.; Babraj, J.A.; Smith, K.; Rennie, M.J.; Kjaer, M. No Effect of Menstrual Cycle on Myofibrillar and Connective Tissue Protein Synthesis in Contracting Skeletal Muscle. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E163–E168. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.F.; Hansen, M.; Olesen, J.L.; Schwarz, P.; Babraj, J.A.; Smith, K.; Rennie, M.J.; Kjaer, M. Tendon Collagen Synthesis at Rest and after Exercise in Women. J. Appl. Physiol. 2007, 102, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Kehlet, S.N.; Willumsen, N.; Armbrecht, G.; Dietzel, R.; Brix, S.; Henriksen, K.; Karsdal, M.A. Age-Related Collagen Turnover of the Interstitial Matrix and Basement Membrane: Implications of Age- and Sex-Dependent Remodeling of the Extracellular Matrix. PLoS ONE 2018, 13, e0194458. [Google Scholar] [CrossRef]
- Bohn, G.; Liden, B.; Schultz, G.; Yang, Q.; Gibson, D.J. Ovine-Based Collagen Matrix Dressing: Next-Generation Collagen Dressing for Wound Care. Adv. Wound Care 2016, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, C.; Pascarella, S.; Errante, F.; Menicacci, B.; Magnelli, L.; Mocali, A.; Rovero, P.; Giovannelli, L. Serpin A1 and the Modulation of Type I Collagen Turnover: Effect of the C-Terminal Peptide 409-418 (SA1-III) in Human Dermal Fibroblasts: Serpin-A1 C-Terminal Modulates Collagen Levels. Cell Biol. Int. 2018, 42, 1340–1348. [Google Scholar] [CrossRef]
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix Metalloproteinases (MMPs), the Main Extracellular Matrix (ECM) Enzymes in Collagen Degradation, as a Target for Anticancer Drugs. J. Enzym. Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef]
- Williams, K.E.; Olsen, D.R. Matrix Metalloproteinase-1 Cleavage Site Recognition and Binding in Full-Length Human Type III Collagen. Matrix Biol. 2009, 28, 373–379. [Google Scholar] [CrossRef]
- Ruettger, A.; Schueler, S.; Mollenhauer, J.A.; Wiederanders, B. Cathepsins B, K, and L Are Regulated by a Defined Collagen Type II Peptide via Activation of Classical Protein Kinase C and P38 MAP Kinase in Articular Chondrocytes. J. Biol. Chem. 2008, 283, 1043–1051. [Google Scholar] [CrossRef]
- Klaus, V.; Schmies, F.; Reeps, C.; Trenner, M.; Geisbüsch, S.; Lohoefer, F.; Eckstein, H.-H.; Pelisek, J. Cathepsin S Is Associated with Degradation of Collagen I in Abdominal Aortic Aneurysm. Vasa 2018, 47, 285–293. [Google Scholar] [CrossRef]
- Parks, A.N.; Nahata, J.; Edouard, N.-E.; Temenoff, J.S.; Platt, M.O. Sequential, but Not Concurrent, Incubation of Cathepsin K and L with Type I Collagen Results in Extended Proteolysis. Sci. Rep. 2019, 9, 5399. [Google Scholar] [CrossRef]
- Kafienah, W.; Buttle, J.D.; Burnett, D.; Hollander, P.A. Cleavage of Native Type I Collagen by Human Neutrophil Elastase. Biochem. J. 1998, 330, 897–902. [Google Scholar] [CrossRef]
- Bertini, I.; Fragai, M.; Luchinat, C.; Melikian, M.; Toccafondi, M.; Lauer, J.L.; Fields, G.B. Structural Basis for Matrix Metalloproteinase 1-Catalyzed Collagenolysis. J. Am. Chem. Soc. 2012, 134, 2100–2110. [Google Scholar] [CrossRef]
- Song, F. Matrix Metalloproteinase Dependent and Independent Collagen Degradation. Front. Biosci. 2006, 11, 3100. [Google Scholar] [CrossRef] [PubMed]
- Moilanen, M.; Sorsa, T.; Stenman, M.; Nyberg, P.; Lindy, O.; Vesterinen, J.; Paju, A.; Konttinen, Y.T.; Stenman, U.-H.; Salo, T. Tumor-Associated Trypsinogen-2 (Trypsinogen-2) Activates Procollagenases (MMP-1, -8, -13) and Stromelysin-1 (MMP-3) and Degrades Type I Collagen. Biochemistry 2003, 42, 5414–5420. [Google Scholar] [CrossRef] [PubMed]
- Kafienah, W.; Brömme, D.; Buttle, D.J.; Croucher, L.J.; Hollander, A.P. Human Cathepsin K Cleaves Native Type I and II Collagens at the N-Terminal End of the Triple Helix. Biochem. J. 1998, 331, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, X.; Sköld, C.M.; Wang, H.; Kohyama, T.; Wen, F.-Q.; Ertl, R.F.; Rennard, S.I. Collaborative Interactions between Neutrophil Elastase and Metalloproteinases in Extracellular Matrix Degradation in Three-Dimensional Collagen Gels. Respir. Res. 2001, 2, 300. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.K.; Liu, X.D.; Sköld, C.M.; Umino, T.; Wang, H.J.; Spurzem, J.R.; Kohyama, T.; Ertl, R.F.; Rennard, S.I. Synergistic Neutrophil Elastase-Cytokine Interaction Degrades Collagen in Three-Dimensional Culture. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L868–L878. [Google Scholar] [CrossRef] [PubMed]
- Stenman, M.; Ainola, M.; Valmu, L.; Bjartell, A.; Ma, G.; Stenman, U.-H.; Sorsa, T.; Luukkainen, R.; Konttinen, Y.T. Trypsin-2 Degrades Human Type II Collagen and Is Expressed and Activated in Mesenchymally Transformed Rheumatoid Arthritis Synovitis Tissue. Am. J. Pathol. 2005, 167, 1119–1124. [Google Scholar] [CrossRef][Green Version]
- van Deemter, M.; Kuijer, R.; Harm Pas, H.; Jacoba van der Worp, R.; Hooymans, J.M.M.; Los, L.I. Trypsin-Mediated Enzymatic Degradation of Type II Collagen in the Human Vitreous. Mol. Vis. 2013, 19, 1591–1599. [Google Scholar]
- Pascarella, S.; Tiberi, C.; Sabatino, G.; Nuti, F.; Papini, A.M.; Giovannelli, L.; Rovero, P. Serpin A1 C-Terminal Peptides as Collagen Turnover Modulators. ChemMedChem 2016, 11, 1850–1855. [Google Scholar] [CrossRef]
- Nielsen, R.H.; Stoop, R.; Leeming, D.J.; Stolina, M.; Qvist, P.; Christiansen, C.; Karsdal, M.A. Evaluation of Cartilage Damage by Measuring Collagen Degradation Products in Joint Extracts in a Traumatic Model of Osteoarthritis. Biomarkers 2008, 13, 79–87. [Google Scholar] [CrossRef]
- Veidal, S.S.; Karsdal, M.A.; Nawrocki, A.; Larsen, M.R.; Dai, Y.; Zheng, Q.; Hägglund, P.; Vainer, B.; Skjøt-Arkil, H.; Leeming, D.J. Assessment of Proteolytic Degradation of the Basement Membrane: A Fragment of Type IV Collagen as a Biochemical Marker for Liver Fibrosis. Fibrogenesis Tissue Repair 2011, 4, 22. [Google Scholar] [CrossRef]
- McAnulty, R.J. Methods for Measuring Hydroxyproline and Estimating in Vivo Rates of Collagen Synthesis and Degradation. Methods Mol. Med. 2005, 117, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Gorres, K.L.; Raines, R.T. Prolyl 4-Hydroxylase. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 106–124. [Google Scholar] [CrossRef]
- Gorres, K.L.; Raines, R.T. Direct and Continuous Assay for Prolyl 4-Hydroxylase. Anal. Biochem. 2009, 386, 181–185. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Poobalarahi, F.; Baicu, C.F.; Bradshaw, A.D. Cardiac Myofibroblasts Differentiated in 3D Culture Exhibit Distinct Changes in Collagen I Production, Processing, and Matrix Deposition. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2924–H2932. [Google Scholar] [CrossRef]
- Errante, F.; Ledwoń, P.; Latajka, R.; Rovero, P.; Papini, A.M. Cosmeceutical Peptides in the Framework of Sustainable Wellness Economy. Front. Chem. 2020, 8, 572923. [Google Scholar] [CrossRef]
- Draelos, Z.D. The Cosmeceutical Realm. Clin. Dermatol. 2008, 26, 627–632. [Google Scholar] [CrossRef]
- Ledwoń, P.; Errante, F.; Papini, A.M.; Rovero, P.; Latajka, R. Peptides as Active Ingredients: A Challenge for Cosmeceutical Industry. Chem. Biodivers. 2021, 18, e2000833. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, 2nd ed.; Wiley: New York, NY, USA, 2000; ISBN 978-0-471-35929-6. [Google Scholar]
- Aljoundi, A.; Bjij, I.; El Rashedy, A.; Soliman, M.E.S. Covalent Versus Non-Covalent Enzyme Inhibition: Which Route Should We Take? A Justification of the Good and Bad from Molecular Modelling Perspective. Protein J. 2020, 39, 97–105. [Google Scholar] [CrossRef]
- Adeniyi, A.A.; Muthusamy, R.; Soliman, M.E. New Drug Design with Covalent Modifiers. Expert Opin. Drug Discov. 2016, 11, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.D. Ongoing Trials with Matrix Metalloproteinase Inhibitors. Expert Opin. Investig. Drugs 2000, 9, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Cuniasse, P.; Devel, L.; Makaritis, A.; Beau, F.; Georgiadis, D.; Matziari, M.; Yiotakis, A.; Dive, V. Future Challenges Facing the Development of Specific Active-Site-Directed Synthetic Inhibitors of MMPs. Biochimie 2005, 87, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, H.S.; McCann, P.P. Matrix Metalloproteinase Inhibition as a Novel Anticancer Strategy: A Review with Special Focus on Batimastat and Marimastat. Pharmacol. Ther. 1997, 75, 69–75. [Google Scholar] [CrossRef]
- Ottl, J.; Gabriel, D.; Murphy, G.; Knäuper, V.; Tominaga, Y.; Nagase, H.; Kröger, M.; Tschesche, H.; Bode, W.; Moroder, L. Recognition and Catabolism of Synthetic Heterotrimeric Collagen Peptides by Matrix Metalloproteinases. Chem. Biol. 2000, 7, 119–132. [Google Scholar] [CrossRef]
- Bode, W.; Maskos, K. Structural Basis of the Matrix Metalloproteinases and Their Physiological Inhibitors, the Tissue Inhibitors of Metalloproteinases. Biol. Chem. 2003, 384. [Google Scholar] [CrossRef]
- Maskos, K. Crystal Structures of MMPs in Complex with Physiological and Pharmacological Inhibitors. Biochimie 2005, 87, 249–263. [Google Scholar] [CrossRef]
- Choi, J.Y.; Fuerst, R.; Knapinska, A.M.; Taylor, A.B.; Smith, L.; Cao, X.; Hart, P.J.; Fields, G.B.; Roush, W.R. Structure-Based Design and Synthesis of Potent and Selective Matrix Metalloproteinase 13 Inhibitors. J. Med. Chem. 2017, 60, 5816–5825. [Google Scholar] [CrossRef]
- Fuerst, R.; Yong Choi, J.; Knapinska, A.M.; Smith, L.; Cameron, M.D.; Ruiz, C.; Fields, G.B.; Roush, W.R. Development of Matrix Metalloproteinase-13 Inhibitors—A Structure-Activity/Structure-Property Relationship Study. Bioorg. Med. Chem. 2018, 26, 4984–4995. [Google Scholar] [CrossRef]
- Moore, W.M.; Spilburg, C.A. Peptide Hydroxamic Acids Inhibit Skin Collagenase. Biochem. Biophys. Res. Commun. 1986, 136, 390–395. [Google Scholar] [CrossRef]
- Grobelny, D.; Poncz, L.; Galardy, R.E. Inhibition of Human Skin Fibroblast Collagenase, Thermolysin, and Pseudomonas Aeruginosa Elastase by Peptide Hydroxamic Acids. Biochemistry 1992, 31, 7152–7154. [Google Scholar] [CrossRef] [PubMed]
- Grams, F.; Reinemer, P.; Powers, J.C.; Kleine, T.; Pieper, M.; Tschesche, H.; Huber, R.; Bode, W. X-Ray Structures of Human Neutrophil Collagenase Complexed with Peptide Hydroxamate and Peptide Thiol Inhibitors. Implications for Substrate Binding and Rational Drug Design. Eur. J. Biochem. 1995, 228, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Bode, W.; Reinemer, P.; Huber, R.; Kleine, T.; Schnierer, S.; Tschesche, H. The X-Ray Crystal Structure of the Catalytic Domain of Human Neutrophil Collagenase Inhibited by a Substrate Analogue Reveals the Essentials for Catalysis and Specificity. EMBO J. 1994, 13, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, H.S. Batimastat and Marimastat in Cancer. In Antiangiogenic Agents in Cancer Therapy; Teicher, B.A., Ed.; Humana Press: Totowa, NJ, USA, 1999; pp. 399–405. ISBN 978-1-4757-4518-4. [Google Scholar]
- Gooljarsingh, L.T.; Lakdawala, A.; Coppo, F.; Luo, L.; Fields, G.B.; Tummino, P.J.; Gontarek, R.R. Characterization of an Exosite Binding Inhibitor of Matrix Metalloproteinase 13. Protein Sci. 2007, 17, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Yiotakis, A.; Lecoq, A.; Nicolaou, A.; Labadie, J.; Dive, V. Phosphinic Peptide Analogues as Potent Inhibitors of Corynebacterium Rathayii Bacterial Collagenase. Biochem. J. 1994, 303, 323–327. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yiotakis, A.; Lecoq, A.; Vassiliou, S.; Raynal, I.; Cuniasse, P.; Dive, V. Cyclic Peptides with a Phosphinic Bond as Potent Inhibitors of a Zinc Bacterial Collagenase. J. Med. Chem. 1994, 37, 2713–2720. [Google Scholar] [CrossRef] [PubMed]
- Cantel, S.; Le Chevalier Isaad, A.; Scrima, M.; Levy, J.J.; DiMarchi, R.D.; Rovero, P.; Halperin, J.A.; D’Ursi, A.M.; Papini, A.M.; Chorev, M. Synthesis and Conformational Analysis of a Cyclic Peptide Obtained via i to i +4 Intramolecular Side-Chain to Side-Chain Azide−Alkyne 1,3-Dipolar Cycloaddition. J. Org. Chem. 2008, 73, 5663–5674. [Google Scholar] [CrossRef]
- Kracker, O.; Góra, J.; Krzciuk-Gula, J.; Marion, A.; Neumann, B.; Stammler, H.-G.; Nieß, A.; Antes, I.; Latajka, R.; Sewald, N. 1,5-Disubstituted 1,2,3-Triazole-Containing Peptidotriazolamers: Design Principles for a Class of Versatile Peptidomimetics. Chem. Eur. J. 2018, 24, 953–961. [Google Scholar] [CrossRef]
- Schröder, D.C.; Kracker, O.; Fröhr, T.; Góra, J.; Jewginski, M.; Nieß, A.; Antes, I.; Latajka, R.; Marion, A.; Sewald, N. 1,4-Disubstituted 1H-1,2,3-Triazole Containing Peptidotriazolamers: A New Class of Peptidomimetics With Interesting Foldamer Properties. Front. Chem. 2019, 7, 155. [Google Scholar] [CrossRef]
- D’Ercole, A.; Sabatino, G.; Pacini, L.; Impresari, E.; Capecchi, I.; Papini, A.M.; Rovero, P. On-resin Microwave-assisted Copper-catalyzed Azide-alkyne Cycloaddition of H1-relaxin B Single Chain ‘Stapled’ Analogues. Pept. Sci. 2020, 112. [Google Scholar] [CrossRef]
- Staśkiewicz, A.; Ledwoń, P.; Rovero, P.; Papini, A.M.; Latajka, R. Triazole-Modified Peptidomimetics: An Opportunity for Drug Discovery and Development. Front. Chem. 2021, 9, 674705. [Google Scholar] [CrossRef]
- Bhowmick, M.; Sappidi, R.R.; Fields, G.B.; Lepore, S.D. Efficient Synthesis of Fmoc-Protected Phosphinic Pseudodipeptides: Building Blocks for the Synthesis of Matrix Metalloproteinase Inhibitors. Biopolymers 2011, 96, 1–3. [Google Scholar] [CrossRef]
- Bhowmick, M.; Fields, G.B. Synthesis of Fmoc-Gly-Ile Phosphinic Pseudodipeptide: Residue Specific Conditions for Construction of Matrix Metalloproteinase Inhibitor Building Blocks. Int. J. Pept. Res. 2012, 18, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Yiotakis, A.; Georgiadis, D.; Matziari, M.; Makaritis, A.; Dive, V. Phosphinic Peptides: Synthetic Approaches and Biochemical Evaluation as Zn-Metalloprotease Inhibitors. Curr. Org. Chem. 2004, 8, 1135–1158. [Google Scholar] [CrossRef]
- Gall, A.-L.; Ruff, M.; Kannan, R.; Cuniasse, P.; Yiotakis, A.; Dive, V.; Rio, M.-C.; Basset, P.; Moras, D. Crystal Structure of the Stromelysin-3 (MMP-11) Catalytic Domain Complexed with a Phosphinic Inhibitor Mimicking the Transition-State11Edited by R. Huber. J. Mol. Biol. 2001, 307, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Lauer-Fields, J.; Brew, K.; Whitehead, J.K.; Li, S.; Hammer, R.P.; Fields, G.B. Triple-Helical Transition State Analogues: A New Class of Selective Matrix Metalloproteinase Inhibitors. J. Am. Chem. Soc. 2007, 129, 10408–10417. [Google Scholar] [CrossRef]
- Oono, T.; Shirafuji, Y.; Huh, W.-K.; Akiyama, H.; Iwatsuki, K. Effects of Human Neutrophil Peptide-1 on the Expression of Interstitial Collagenase and Type I Collagen in Human Dermal Fibroblasts. Arch. Derm. Res. 2002, 294, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Defensin HNP-1 Human D2043 (Merck/Sigma-Aldrich Catalogue Website). Available online: https://www.sigmaaldrich.com/catalog/product/sigma/d2043 (accessed on 21 April 2021).
- Ndinguri, M.W.; Bhowmick, M.; Tokmina-Roszyk, D.; Robichaud, T.K.; Fields, G.B. Peptide-Based Selective Inhibitors of Matrix Metalloproteinase-Mediated Activities. Molecules 2012, 17, 14230–14248. [Google Scholar] [CrossRef]
- Lauer-Fields, J.L.; Cudic, M.; Wei, S.; Mari, F.; Fields, G.B.; Brew, K. Engineered Sarafotoxins as Tissue Inhibitor of Metalloproteinases-like Matrix Metalloproteinase Inhibitors. J. Biol. Chem. 2007, 282, 26948–26955. [Google Scholar] [CrossRef]
- Hu, J.; Van den Steen, P.E.; Dillen, C.; Opdenakker, G. Targeting Neutrophil Collagenase/Matrix Metalloproteinase-8 and Gelatinase B/Matrix Metalloproteinase-9 with a Peptidomimetic Inhibitor Protects against Endotoxin Shock. Biochem. Pharm. 2005, 70, 535–544. [Google Scholar] [CrossRef]
- Bahudhanapati, H.; Zhang, Y.; Sidhu, S.S.; Brew, K. Phage Display of Tissue Inhibitor of Metalloproteinases-2 (TIMP-2). J. Biol. Chem. 2011, 286, 31761–31770. [Google Scholar] [CrossRef]
- Stura, E.A.; Visse, R.; Cuniasse, P.; Dive, V.; Nagase, H. Crystal Structure of Full-length Human Collagenase 3 (MMP-13) with Peptides in the Active Site Defines Exosites in the Catalytic Domain. FASEB J. 2013, 27, 4395–4405. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Version 2.2.3; Schrödinger, LLC.: New York, NY, USA, 2018.
- Howes, J.-M.; Bihan, D.; Slatter, D.A.; Hamaia, S.W.; Packman, L.C.; Knauper, V.; Visse, R.; Farndale, R.W. The Recognition of Collagen and Triple-Helical Toolkit Peptides by MMP-13: Sequence Specificity for Binding and Cleavage. J. Biol. Chem. 2014, 289, 24091–24101. [Google Scholar] [CrossRef]
- Bhowmick, M.; Stawikowska, R.; Tokmina-Roszyk, D.; Fields, G.B. Matrix Metalloproteinase Inhibition by Heterotrimeric Triple-Helical Peptide Transition State Analogues. Chembiochem 2015, 16, 1084–1092. [Google Scholar] [CrossRef]
- Park, S.-H.; Lee, J.-K.; Jeon, J.-K.; Byun, H.-G. Characterization of a Collagenase-1 Inhibitory Peptide Purified from Skate Dipturus Chilensis Skin. Kor. J. Fish. Aquat. Sci. 2011, 44, 456–463. [Google Scholar] [CrossRef]
- Quillard, T.; Tesmenitsky, Y.; Croce, K.; Travers, R.; Shvartz, E.; Koskinas, K.C.; Sukhova, G.K.; Aikawa, E.; Aikawa, M.; Libby, P. Selective Inhibition of Matrix Metalloproteinase-13 Increases Collagen Content of Established Mouse Atherosclerosis. Arter. Thromb. Vasc. Biol. 2011, 31, 2464–2472. [Google Scholar] [CrossRef]
- Hartmann, A.; Gostner, J.; Fuchs, J.; Chaita, E.; Aligiannis, N.; Skaltsounis, L.; Ganzera, M. Inhibition of Collagenase by Mycosporine-like Amino Acids from Marine Sources. Planta Med. 2015, 81, 813–820. [Google Scholar] [CrossRef]
- Raeeszadeh-Sarmazdeh, M.; Do, L.; Hritz, B. Metalloproteinases and Their Inhibitors: Potential for the Development of New Therapeutics. Cells 2020, 9, 1313. [Google Scholar] [CrossRef] [PubMed]
- Baló, J.; Banga, I. Elastase and Elastase-Inhibitor. Nature 1949, 164, 491. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, N.; Moriwaki, S.; Suzuki, Y.; Takema, Y.; Imokawa, G. The Role of Elastases Secreted by Fibroblasts in Wrinkle Formation: Implication through Selective Inhibition of Elastase Activity. Photochem. Photobiol. 2001, 74, 283–290. [Google Scholar] [CrossRef]
- Godeau, G.; Hornebeck, W. Morphometric Analysis of the Degradation of Human Skin Elastic Fibres by Human Leukocyte Elastase (EC 3-4-21-37) and Human Skin Fibroblast Elastase (EC 3-4-24). Pathol. Biol. 1988, 36, 1133–1138. [Google Scholar]
- Azmi, N.; Hashim, P.; Hashim, D.M.; Halimoon, N.; Majid, N.M.N. Anti–Elastase, Anti–Tyrosinase and Matrix Metalloproteinase–1 Inhibitory Activity of Earthworm Extracts as Potential New Anti–Aging Agent. Asian Pac. J. Trop. Biomed. 2014, 4, S348–S352. [Google Scholar] [CrossRef]
- Ohbayashi, H. Current Synthetic Inhibitors of Human Neutrophil Elastase in 2005. Expert Opin. Ther. Pat. 2005, 15, 759–771. [Google Scholar] [CrossRef]
- Kelly, E.; Greene, C.M.; McElvaney, N.G. Targeting Neutrophil Elastase in Cystic Fibrosis. Expert Opin. Ther. Targets 2008, 12, 145–157. [Google Scholar] [CrossRef]
- Groutas, W.C.; Dou, D.; Alliston, K.R. Neutrophil Elastase Inhibitors. Expert Opin. Ther. Pat. 2011, 21, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Saleem, M.; Riaz, N.; Lee, Y.S.; Diri, R.; Noor, A.; Almasri, D.; Bagalagel, A.; Elsebai, M.F. The Natural Polypeptides as Significant Elastase Inhibitors. Front. Pharmacol. 2020, 11, 688. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.C.; Gupton, B.F.; Harley, A.D.; Nishino, N.; Whitley, R.J. Specificity of Porcine Pancreatic Elastase, Human Leukocyte Elastase and Cathepsin G Inhibition with Peptide Chloromethyl Ketones. Biochim. Biophys. Acta BBA Enzymol. 1977, 485, 156–166. [Google Scholar] [CrossRef]
- Navia, M.A.; McKeever, B.M.; Springer, J.P.; Lin, T.Y.; Williams, H.R.; Fluder, E.M.; Dorn, C.P.; Hoogsteen, K. Structure of Human Neutrophil Elastase in Complex with a Peptide Chloromethyl Ketone Inhibitor at 1.84-A Resolution. Proc. Natl. Acad. Sci. USA 1989, 86, 7–11. [Google Scholar] [CrossRef]
- Cowan, K.N.; Heilbut, A.; Humpl, T.; Lam, C.; Ito, S.; Rabinovitch, M. Complete Reversal of Fatal Pulmonary Hypertension in Rats by a Serine Elastase Inhibitor. Nat. Med. 2000, 6, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Matern, U.; Oberer, L.; Falchetto, R.A.; Erhard, M.; König, W.A.; Herdman, M.; Weckesser, J. Scyptolin A and B, Cyclic Depsipeptides from Axenic Cultures of Scytonema Hofmanni PCC 7110. Phytochemistry 2001, 58, 1087–1095. [Google Scholar] [CrossRef]
- Matern, U.; Schleberger, C.; Jelakovic, S.; Weckesser, J.; Schulz, G.E. Binding Structure of Elastase Inhibitor Scyptolin A. Chem. Biol. 2003, 10, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Keller, L.; Canuto, K.M.; Liu, C.; Suzuki, B.M.; Almaliti, J.; Sikandar, A.; Naman, C.B.; Glukhov, E.; Luo, D.; Duggan, B.M.; et al. Tutuilamides A-C: Vinyl-Chloride-Containing Cyclodepsipeptides from Marine Cyanobacteria with Potent Elastase Inhibitory Properties. ACS Chem. Biol. 2020, 15, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Toth, I.; Christodoulou, M.; Bankowsky, K.; Flinn, N.; Gibbons, W.A.; Godeau, G.; Moczar, E.; Hornebeck, W. Design of Potent Lipophilic-Peptide Inhibitors of Human Neutrophil Elastase: In Vitro and in Vivo Studies. Int. J. Pharm. 1995, 125, 117–122. [Google Scholar] [CrossRef]
- Hilpert, K.; Hansen, G.; Wessner, H.; Schneider-Mergener, J.; Hohne, W. Characterizing and Optimizing Protease/Peptide Inhibitor Interactions, a New Application for Spot Synthesis. J. Biochem. 2000, 128, 1051–1057. [Google Scholar] [CrossRef]
- Hilpert, K.; Wessner, H.; Schneider-Mergener, J.; Welfle, K.; Misselwitz, R.; Welfle, H.; Hocke, A.C.; Hippenstiel, S.; Höhne, W. Design and Characterization of a Hybrid Miniprotein That Specifically Inhibits Porcine Pancreatic Elastase. J. Biol. Chem. 2003, 278, 24986–24993. [Google Scholar] [CrossRef]
- Vasconcelos, A.; Azoia, N.G.; Carvalho, A.C.; Gomes, A.C.; Güebitz, G.; Cavaco-Paulo, A. Tailoring Elastase Inhibition with Synthetic Peptides. Eur. J. Pharm. 2011, 666, 53–60. [Google Scholar] [CrossRef][Green Version]
- McBride, J.D.; Watson, E.M.; Brauer, A.B.E.; Jaulent, A.M.; Leatherbarrow, R.J. Peptide Mimics of the Bowman-Birk Inhibitor Reactive Site Loop. Biopolymers 2002, 66, 79–92. [Google Scholar] [CrossRef]
- Wan, H.; Lee, K.S.; Kim, B.Y.; Zou, F.M.; Yoon, H.J.; Je, Y.H.; Li, J.; Jin, B.R. A Spider-Derived Kunitz-Type Serine Protease Inhibitor That Acts as a Plasmin Inhibitor and an Elastase Inhibitor. PLoS ONE 2013, 8, e53343. [Google Scholar] [CrossRef]
- Nakanishi, I.; Kinoshita, T.; Sato, A.; Tada, T. Structure of Porcine Pancreatic Elastase Complexed with FR901277, a Novel Macrocyclic Inhibitor of Elastases, at 1.6 A Resolution. Biopolymers 2000, 53, 434–445. [Google Scholar] [CrossRef]
- Fujita, T.; Hatanaka, H.; Hayashi, K.; Shigematsu, N.; Takase, S.; Okamoto, M.; Okuhara, M.; Shimatani, K.; Satoh, A. FR901451, a Novel Inhibitor of Human Leukocyte Elastase from Flexibacter Sp. I. Producing Organism, Fermentation, Isolation, Physico-Chemical and Biological Properties. J. Antibiot. 1994, 47, 1359–1364. [Google Scholar] [CrossRef][Green Version]
- Tsukahara, K.; Takema, Y.; Moriwaki, S.; Tsuji, N.; Suzuki, Y.; Fujimura, T.; Imokawa, G. Selective Inhibition of Skin Fibroblast Elastase Elicits a Concentration-Dependent Prevention of Ultraviolet B-Induced Wrinkle Formation. J. Investig. Dermatol. 2001, 117, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Schepetkin, I.A.; Khlebnikov, A.I.; Quinn, M.T. N-Benzoylpyrazoles Are Novel Small-Molecule Inhibitors of Human Neutrophil Elastase. J. Med. Chem. 2007, 50, 4928–4938. [Google Scholar] [CrossRef] [PubMed]
- Crocetti, L.; Giovannoni, M.P.; Schepetkin, I.A.; Quinn, M.T.; Khlebnikov, A.I.; Cilibrizzi, A.; Piaz, V.D.; Graziano, A.; Vergelli, C. Design, Synthesis and Evaluation of N-Benzoylindazole Derivatives and Analogues as Inhibitors of Human Neutrophil Elastase. Bioorganic Med. Chem. 2011, 19, 4460–4472. [Google Scholar] [CrossRef] [PubMed]
- Crocetti, L.; Schepetkin, I.A.; Cilibrizzi, A.; Graziano, A.; Vergelli, C.; Giomi, D.; Khlebnikov, A.I.; Quinn, M.T.; Giovannoni, M.P. Optimization of N-Benzoylindazole Derivatives as Inhibitors of Human Neutrophil Elastase. J. Med. Chem. 2013, 56, 6259–6272. [Google Scholar] [CrossRef] [PubMed]
- Vidak, E.; Javoršek, U.; Vizovišek, M.; Turk, B. Cysteine Cathepsins and Their Extracellular Roles: Shaping the Microenvironment. Cells 2019, 8, 264. [Google Scholar] [CrossRef]
- Brömme, D.; Lecaille, F. Cathepsin K Inhibitors for Osteoporosis and Potential Off-Target Effects. Expert Opin. Investig. Drugs 2009, 18, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Aguda, A.H.; Panwar, P.; Du, X.; Nguyen, N.T.; Brayer, G.D.; Brömme, D. Structural Basis of Collagen Fiber Degradation by Cathepsin K. Proc. Natl. Acad. Sci. USA 2014, 111, 17474–17479. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, T.; Takeuchi, T.; Matsuzaki, A.; Kawamura, K.; Kondo, S. Leupeptins, New Protease Inhibitors from Actinomycetes. J. Antibiot. 1969, 22, 283–286. [Google Scholar] [CrossRef]
- Suda, H.; Aoyagi, T.; Hamada, M.; Takeuchi, T.; Umezawa, H. Antipain, a New Protease Inhibitor Isolated from Actinomycetes. J. Antibiot. 1972, 25, 263–266. [Google Scholar] [CrossRef]
- Sasaki, T.; Kishi, M.; Saito, M.; Tanaka, T.; Higuchi, N.; Kominami, E.; Katunuma, N.; Murachi, T. Inhibitory Effect of Di- and Tripeptidyl Aldehydes on Calpains and Cathepsins. J. Enzym. Inhib. 1990, 3, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Votta, B.J.; Levy, M.A.; Badger, A.; Bradbeer, J.; Dodds, R.A.; James, I.E.; Thompson, S.; Bossard, M.J.; Carr, T.; Connor, J.R.; et al. Peptide Aldehyde Inhibitors of Cathepsin K Inhibit Bone Resorption Both in Vitro and in Vivo. J. Bone Miner. Res. 1997, 12, 1396–1406. [Google Scholar] [CrossRef]
- Walker, B.; Lynas, J.F.; Meighan, M.A.; Brömme, D. Evaluation of Dipeptide α-Keto-β-Aldehydes as New Inhibitors of Cathepsin S. Biochem. Biophys. Res. Commun. 2000, 275, 401–405. [Google Scholar] [CrossRef]
- Palmer, J.T.; Bryant, C.; Wang, D.-X.; Davis, D.E.; Setti, E.L.; Rydzewski, R.M.; Venkatraman, S.; Tian, Z.-Q.; Burrill, L.C.; Mendonca, R.V.; et al. Design and Synthesis of Tri-Ring P Benzamide-Containing Aminonitriles as Potent, Selective, Orally Effective Inhibitors of Cathepsin K. J. Med. Chem. 2005, 48, 7520–7534. [Google Scholar] [CrossRef]
- Jerome, C.; Missbach, M.; Gamse, R. Balicatib, a Cathepsin K Inhibitor, Stimulates Periosteal Bone Formation in Monkeys. Osteoporos Int. 2011, 22, 3001–3011. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.Y.; Chauret, N.; Cromlish, W.; Desmarais, S.; Duong, L.T.; Falgueyret, J.-P.; Kimmel, D.B.; Lamontagne, S.; Léger, S.; LeRiche, T.; et al. The Discovery of Odanacatib (MK-0822), a Selective Inhibitor of Cathepsin K. Bioorg. Med. Chem. Lett. 2008, 18, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Robichaud, J.; Black, W.C.; Thérien, M.; Paquet, J.; Oballa, R.M.; Bayly, C.I.; McKay, D.J.; Wang, Q.; Isabel, E.; Léger, S.; et al. Identification of a Nonbasic, Nitrile-Containing Cathepsin K Inhibitor (MK-1256) That Is Efficacious in a Monkey Model of Osteoporosis. J. Med. Chem. 2008, 51, 6410–6420. [Google Scholar] [CrossRef]
- Burtoloso, A.C.B.; de Albuquerque, S.; Furber, M.; Gomes, J.C.; Gonçalez, C.; Kenny, P.W.; Leitão, A.; Montanari, C.A.; Quilles, J.C.; Ribeiro, J.F.R.; et al. Anti-Trypanosomal Activity of Non-Peptidic Nitrile-Based Cysteine Protease Inhibitors. PLoS Negl. Trop. Dis. 2017, 11, e0005343. [Google Scholar] [CrossRef]
- Frizler, M.; Stirnberg, M.; Sisay, M.; Gutschow, M. Development of Nitrile-Based Peptidic Inhibitors of Cysteine Cathepsins. Curr. Top. Med. Chem. 2010, 10, 294–322. [Google Scholar] [CrossRef]
- McGrath, M.E.; Sprengeler, P.A.; Hill, C.M.; Martichonok, V.; Cheung, H.; Somoza, J.R.; Palmer, J.T.; Janc, J.W. Peptide Ketobenzoxazole Inhibitors Bound to Cathepsin K. Biochemistry 2003, 42, 15018–15028. [Google Scholar] [CrossRef] [PubMed]
- Quibell, M.; Benn, A.; Flinn, N.; Monk, T.; Ramjee, M.; Wang, Y.; Watts, J. Bicyclic Peptidomimetic Tetrahydrofuro[3,2-b]Pyrrol-3-One and Hexahydrofuro[3,2-b]Pyridine-3-One Based Scaffolds: Synthesis and Cysteinyl Proteinase Inhibition. Bioorg. Med. Chem. 2004, 12, 5689–5710. [Google Scholar] [CrossRef] [PubMed]
- Quibell, M.; Benn, A.; Flinn, N.; Monk, T.; Ramjee, M.; Ray, P.; Wang, Y.; Watts, J. Synthesis and Evaluation of Cis-Hexahydropyrrolo[3,2-b]Pyrrol-3-One Peptidomimetic Inhibitors of CAC1 Cysteinyl Proteinases. Bioorg. Med. Chem. 2005, 13, 609–625. [Google Scholar] [CrossRef] [PubMed]
- Wijkmans, J.; Gossen, J. Inhibitors of Cathepsin K: A Patent Review (2004–2010). Expert Opin. Ther. Pat. 2011, 21, 1611–1629. [Google Scholar] [CrossRef] [PubMed]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
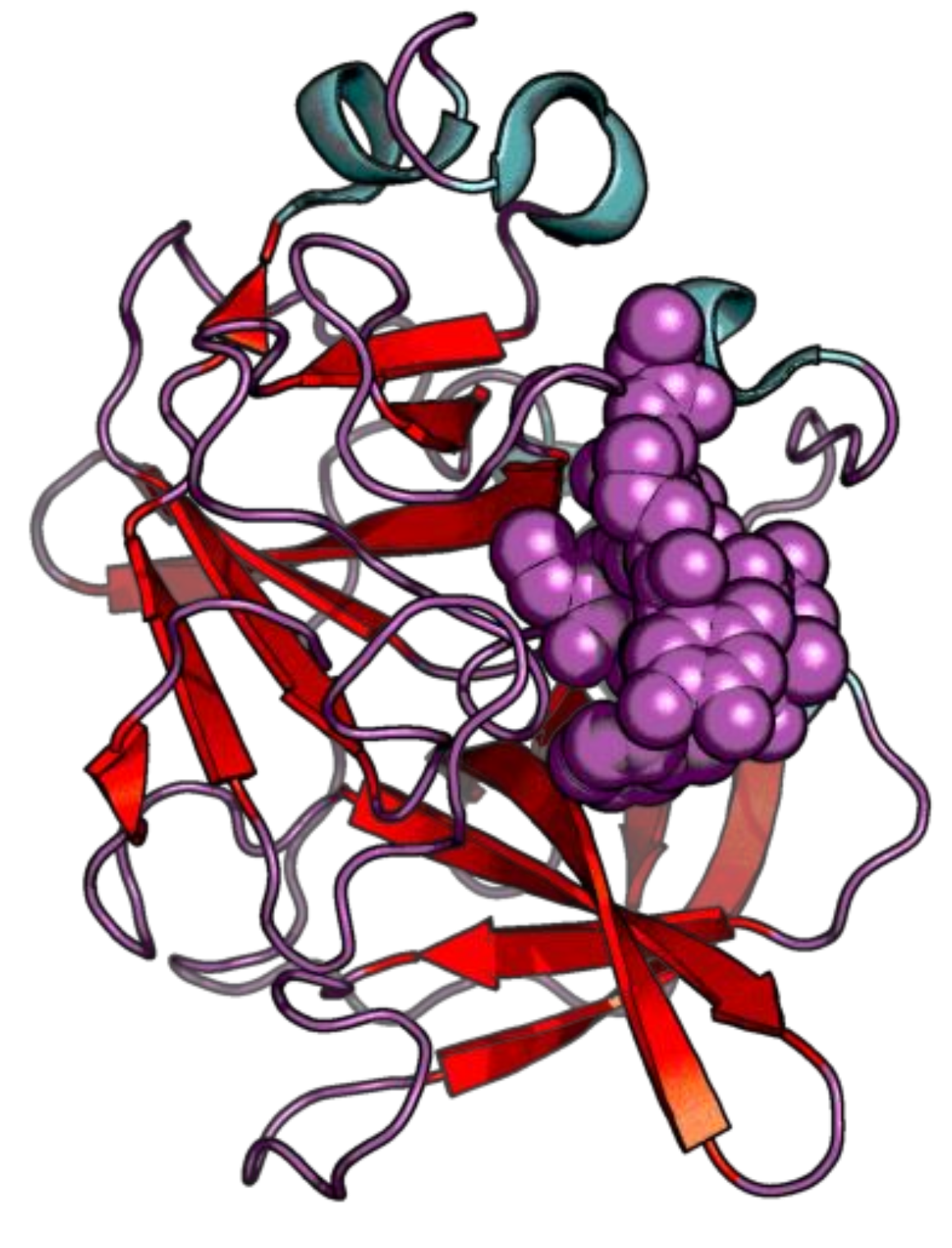{kind=link}
{kind=link}
{kind=link}
{kind=link}
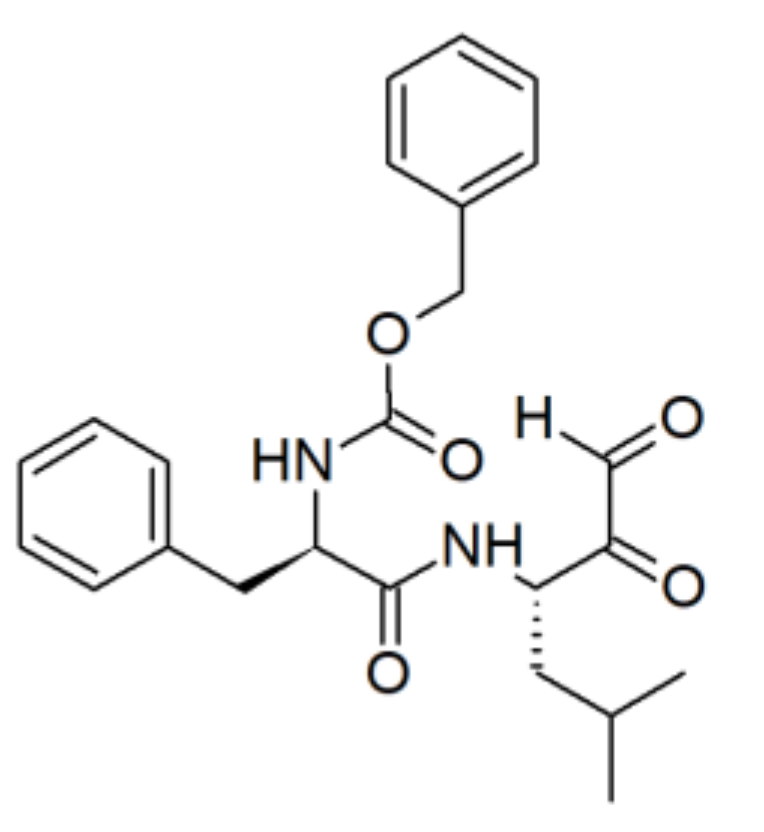{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme Family | Enzyme Common Name | Enzyme Classification or Abbreviation | Degraded Collagen Type | Type of Preferentially Degraded Fibrillar Collagen Type 1 | Preferential Cleavage Site3 |
|---|---|---|---|---|---|
| Matrix metalloproteinases | Collagenase-1 | MMP-1 | I, II, III, VII, VIII, X | I and III | Pro-neutral amino acid- -Gly-Pro -Gly-Pro |
| Collagenase-2 | MMP-8 | I–III, V, VII, VIII, X | I | ||
| Collagenase-3 | MMP-13 | II, III | II, III | ||
| Gelatinase A | MMP-2 | IV–VI, X | n.a. | Pro-Gln-Gly- -Ile/Leu-Ala-Gly-Gln -Ile/Leu-Ala-Gly-Gln | |
| Gelatinase B | MMP-9 | IV, V, VII, X, XIV | n.a. | ||
| Stromelysin-1 | MMP-3 | III | III (indirectly; by the MMP-1 activation) | Ser- -Met -Met | |
| Stromelysin-2 | MMP-10 | III–V | III (indirectly; by the MMP-1 activation) | ||
| Matrilysin | MMP-7 | IV–X | n.a. | Ser- -Leu -LeuAla-  -Leu -LeuTyr-  -Leu -Leu | |
| Metalloelastase | MMP-12 | I, IV | n.a. | Pro-X-X- -Leu -Leu | |
| Matrilysin-2 | MMP-26 | IV | n.a. | Ser- -Leu -Leu | |
| MT-MMP-12 | MMP-14 | I, II, III | n.a. | Ser- -Leu -Leu | |
| MT-MMP-3 | MMP-16 | III | n.a. | n.a. | |
| Serine proteases | Elastase | - | I, III | I (independently and enhancing MMPs activity) | X- -Gly/Ala/Ser -Gly/Ala/Ser |
| Trypsin-2 | - | I, II, III | I (independently and enhancing MMPs activity) | Lys/Arg- -X -X | |
| Cysteine proteases | Cathepsin K | CatK | I, II | I, II | n.a. |
| Cathepsin B | CatB | II | n.a. | n.a. (carboxypeptidase) | |
| Cathepsin L | CatL | I, II | I | Arg- -X -X | |
| Cathepsin S | CatS | I | I | n.a. |
 : cleaved bond.
: cleaved bond.| Trade Name | IUPAC or PubChem Name | Structure | % of Inhibition |
|---|---|---|---|
| Amastatin | (2S)-2-[(2S)-2-[(2S)-2-[(2S,3R)-3-Amino-2-hydroxy-5-methylhexanoyl]amino]-3-methylbutanoyl]amino]-3-methylbutanoyl]amino]butanedioic acid |  | 0 |
| Captopril | (2S)-1-[(2S)-2-methyl-3-sulfanylpropanoyl]pyrrolidine-2-carboxylic acid |  | 0 |
| Phosphoramidon | (2S)-2-[(2S)-2-[hydroxy-[(2S,3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxyphosphoryl]amino]-4-methylpentanoyl]amino]-3-(1H-indol-3-yl)propanoic acid N-[N-[(6-deoxy-α-L-mannoopyranosyl)oxy]hydroxyphosphinyl]-L-leucyl]-L-tryptophan |  | 0 |
| Zincov | 2-(N-hydroxycarboxamido)-4-methylpentanoyl-alanyl-glycinamide |  | 27 |
| Name | IC50 Value |
|---|---|
| Shinorine | 104.0 ± 3.7 |
| Porphyra | 105.9 ± 2.3 |
| Palythine | 158.9 ± 3.2 |
| Phosphoramidone 1 | 18.8 ± 1.6 |
| 1,10-phenanthroline 1 | 238.1 ± 3.4 |
| Name | Structure |
|---|---|
| 1a | CH3(CH2)7CH=CH(CH2)7CO-AAPV-OH |
| 1b | (CH3)3COCO-DL-NH[CH(CH2)11CH3]-AAPV-OH |
| 1c | (CH3)3COCO{DL-NH[CH(CH2)11CH3]CO}2-AAPV-OH |
| 1d | (CH3)3COCO{DL-NH[CH(CH2)11CH3]CO}3-AAPV-OH |
| 1e | (CH3)3COCO{DL-NH[CH(CH2)11CH3]CO}3-AAPV-OCH3 |
| Compound | Structure | IC50 Value for CatK | Reference |
|---|---|---|---|
| Balicatib (AAE581) |  | 0.0014 μM | [129] |
| Odanacatib (MK 0822) |  | 0.2 nM | [131] |
| MK-1256 |  | 0.62 nM | [132] |
| Compound | Structure | Cathepsin K Ki [nM] | Cathepsin L Ki [nM] | Cathepsin S Ki [nM] | Reference |
|---|---|---|---|---|---|
| 4 |  | 0.0029 | 0.02 | 0.0066 | [135] |
| 10 |  | 87.4 +/− 0.8 | >25,000 | >41,000 | [136] |
| 22 |  | 10.1 +/− 6.7 | >3500 | >4500 | [137] |
| Peptide Symbol | Enzyme Class | Sequence of an Inhibitor | Reference |
|---|---|---|---|
| A | Collagenases | ACYCRIPACIAGERRYGTCIYQGRLWAFCC | [81] |
| B | NLDVLEVF | [91] | |
| C | CSCSDMTDKECLYFCMSEMS | [84] | |
| D | PRCBCGE | [85] | |
| E | CDCAPV | [86] | |
| F | GGDEDDLSEEDLQFAERYLRSYYHPT DLSEEDLQFAERYLRSYYHPT | [87] | |
| G | |||
| H | GLAGQRGIVGLOGQRGERGFOGLOGRS GLOGERGRTGPAGAAGARGNDGQOGPA | [89] | |
| I | |||
| J |  | [90] | |
| K | |||
| L | |||
| M | Elastase | (CH3)3COCO{DL-NH[CH(CH2)11CH3]CO}3-AAPV-OH | [109] |
| N | PMTLEYR | [110] | |
| O | MGWCTASVPPQCYG MGWCTASVPPQCYG(GA)7 | [112] | |
| P | |||
| R | KDRCLLPKVTGPCKASLTRYYYDKDTKACVEFIYGGCRGNRNNFKQKDECEKACTDH | [114] | |
| S | OT-dehydroxyT-AA1-F-AA2-V 2 | [115,116] | |
| T | Cathepsins | Ac-LLR-CHO | [124] |
| U | N-[Nα-carbonyl-RVR(aldehyde)]-F | [125] | |
| V | Ac-LLnorL-CHO Ac-LLM-CHO | [126] | |
| X | |||
| Y | Cbz-LLL-CHO | [127] | |
| Z | Cbz-FL-COCHO | [128] |
 – indicates the sites of click ligation; O – Hyp. 2 AA1, AA2—non-abbreviated, unnatural amino acids. For cyclic and detailed structures, please see the references.
– indicates the sites of click ligation; O – Hyp. 2 AA1, AA2—non-abbreviated, unnatural amino acids. For cyclic and detailed structures, please see the references.Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ledwoń, P.; Papini, A.M.; Rovero, P.; Latajka, R. Peptides and Peptidomimetics as Inhibitors of Enzymes Involved in Fibrillar Collagen Degradation. Materials 2021, 14, 3217. https://doi.org/10.3390/ma14123217
Ledwoń P, Papini AM, Rovero P, Latajka R. Peptides and Peptidomimetics as Inhibitors of Enzymes Involved in Fibrillar Collagen Degradation. Materials. 2021; 14(12):3217. https://doi.org/10.3390/ma14123217
Chicago/Turabian StyleLedwoń, Patrycja, Anna Maria Papini, Paolo Rovero, and Rafal Latajka. 2021. "Peptides and Peptidomimetics as Inhibitors of Enzymes Involved in Fibrillar Collagen Degradation" Materials 14, no. 12: 3217. https://doi.org/10.3390/ma14123217
APA StyleLedwoń, P., Papini, A. M., Rovero, P., & Latajka, R. (2021). Peptides and Peptidomimetics as Inhibitors of Enzymes Involved in Fibrillar Collagen Degradation. Materials, 14(12), 3217. https://doi.org/10.3390/ma14123217