NiO Pseudocapacitance and Optical Properties: Does The Shape Win?

 ,
,  ,
,  ,
,  and
and
Abstract

1. Introduction
2. Experimental
2.1. Synthetic Procedure of NiO Nano- and Microstructures
2.2. Materials and Equipment
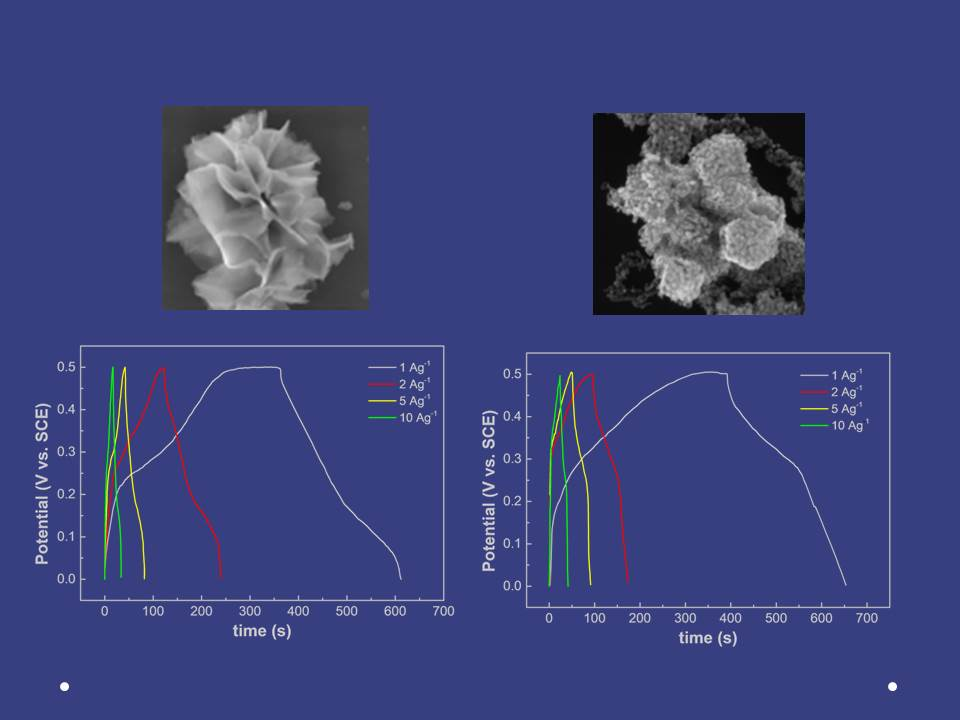
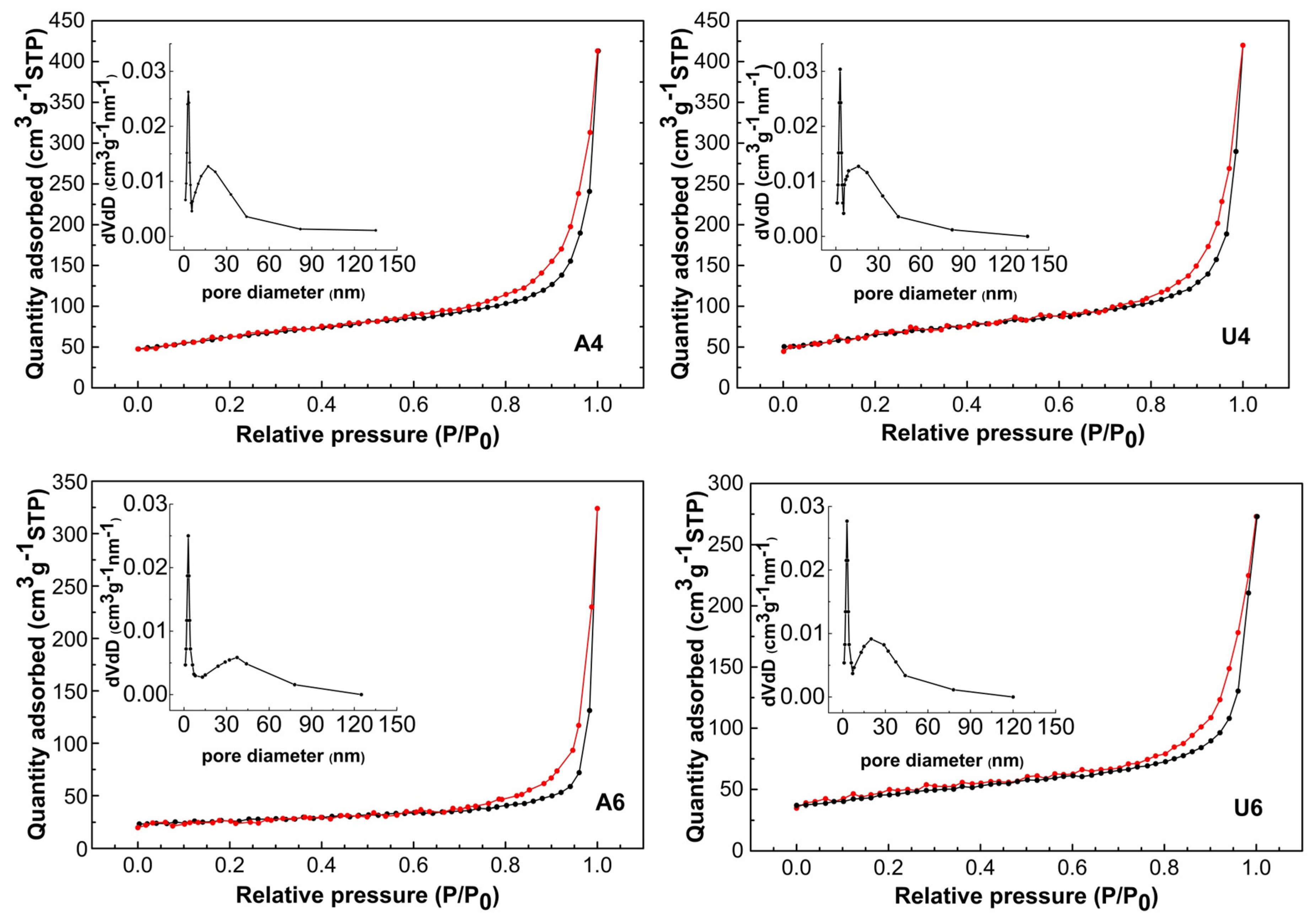
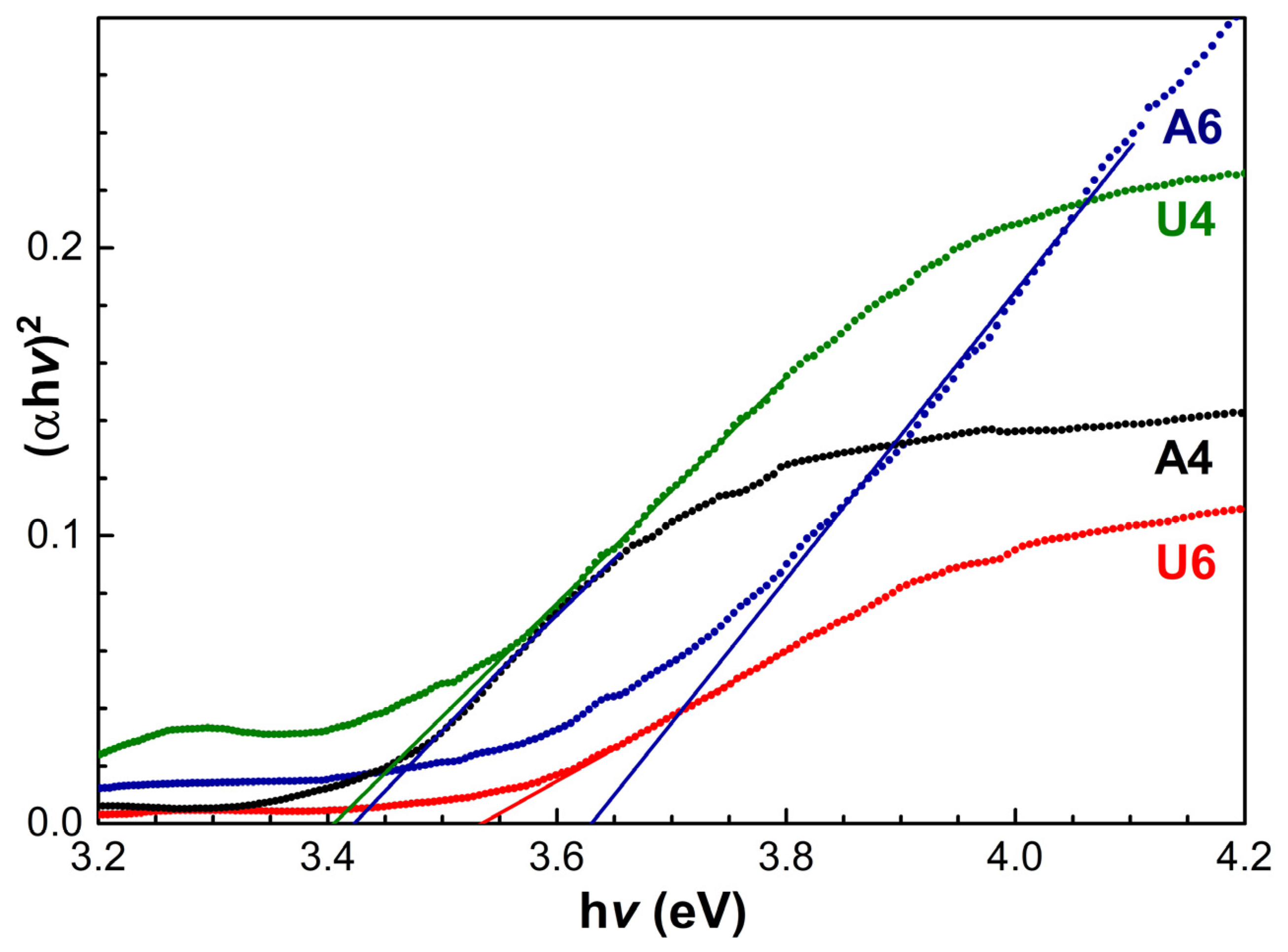
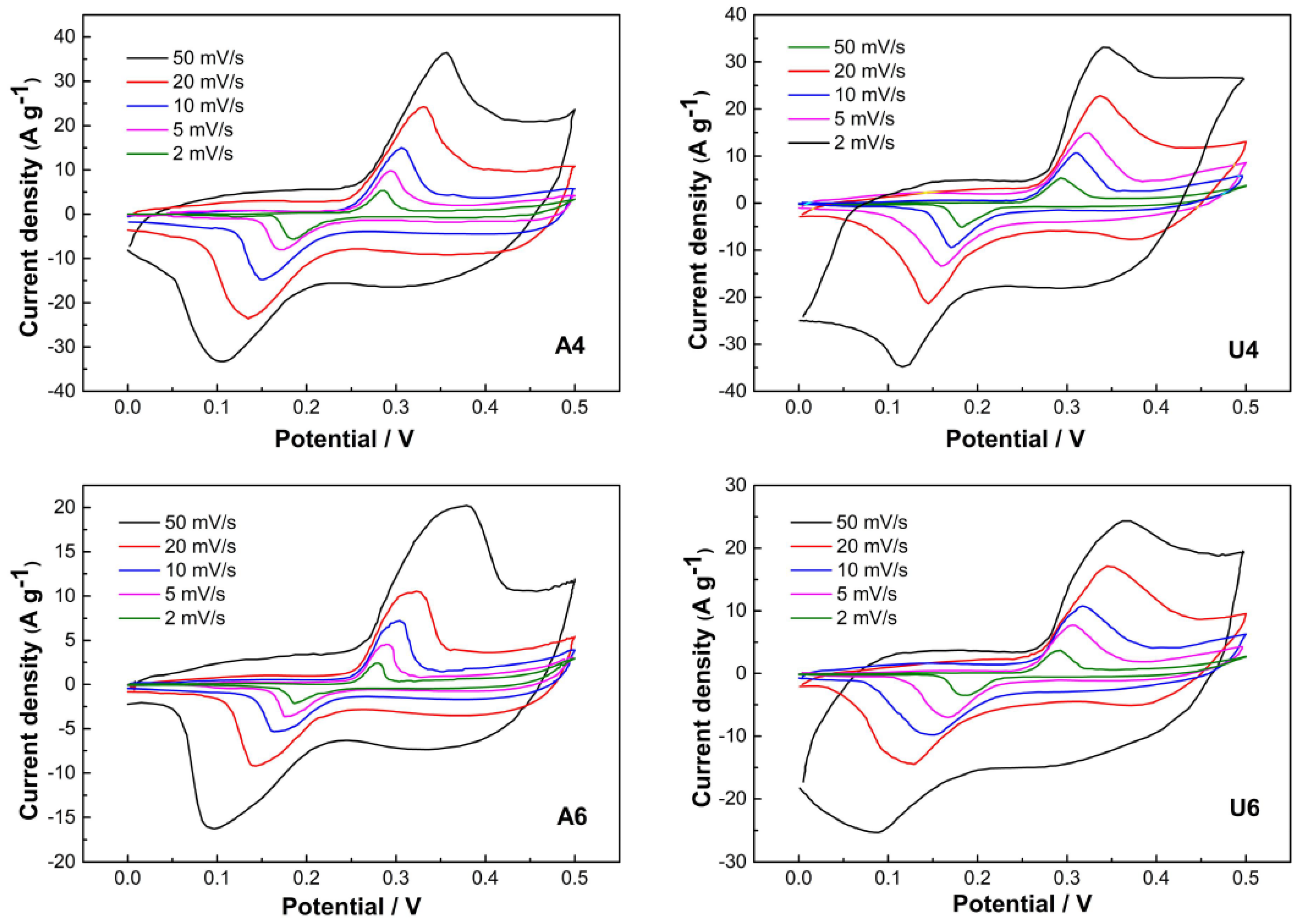
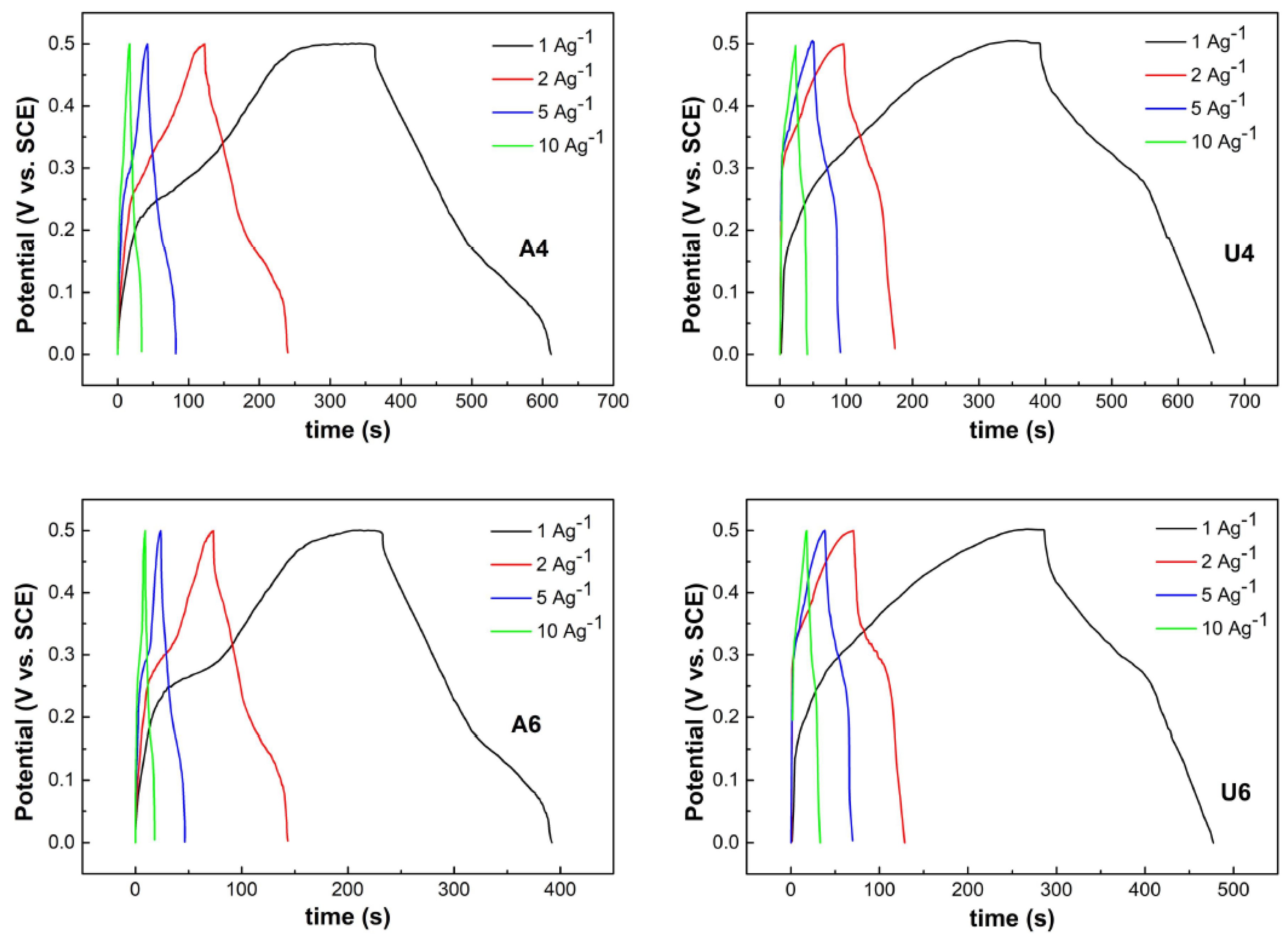
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, G.; Zhang, L.; Zhang, J. A review of electrode materials for electrochemical supercapacitors. Chem. Soc. Rev. 2012, 41, 797–828. [Google Scholar] [CrossRef]
- El-Kemary, M.; Nagy, N.; El-Mehasse, I. Nickel oxide nanoparticles: Synthesis and spectral studies of interactions with glucose. Mat. Sci. Semic. Proc. 2013, 16, 1747–1752. [Google Scholar] [CrossRef]
- Liu, M.; Chang, J.; Sun, J.; Gao, L. Synthesis of porous NiO using NaBH4 dissolved in ethylene glycol as precipitant for high-performance supercapacitor. Electrochim. Acta 2013, 107, 9–15. [Google Scholar] [CrossRef]
- Carbone, M.; Micheli, L.; Nesticò, A.; Bellucci, N.; Palleschi, G. Enhanced performances of sensors based on screen printed electrodes modified with nanosized NiO particles. Electrochim. Acta 2017, 246, 580–587. [Google Scholar] [CrossRef]
- Al-Osta, A.; Jadhav, V.V.; Zate, M.K.; Mane, R.S.; Hui, K.N.; Han, S.H. Electrochemical supercapacitors of anodized-brass-templated NiO nanostrutured electrodes. Scr. Mater. 2015, 99, 29–32. [Google Scholar] [CrossRef]
- Kolathodi, M.S.; Palei, M.; Natarajan, T.S. Electrospun NiO nanofibers as cathode materials for high performance asymmetric supercapacitors. J. Mater. Chem. A 2015, 3, 7513–7522. [Google Scholar] [CrossRef]
- Han, D.; Xu, P.; Jing, X.; Wang, J.; Yang, P.; Shen, Q.; Liu, J.; Song, D.; Gao, Z.; Zhang, M. Trisodium citrate assisted synthesis of hierarchical NiO nanospheres with improved supercapacitor performance. J. Power Sources 2013, 235, 45–53. [Google Scholar] [CrossRef]
- Yuan, C.; Hou, L.; Feng, Y.; Xiong, S.; Zhang, X. Sacrificial template synthesis of short mesoporous NiO nanotubes and their application in electrochemical capacitors. Electrochim. Acta 2013, 88, 507–512. [Google Scholar] [CrossRef]
- Yang, Y.; Liang, Y.; Zhang, Z.; Zhang, Y.; Wu, H.; Hu, Z. Morphology well-controlled synthesis of NiO by solvothermal reaction time and their morphology-dependent pseudocapacitive performances. J. Alloy. Compd. 2016, 658, 621–628. [Google Scholar] [CrossRef]
- Navale, S.T.; Mali, V.V.; Pawar, S.A.; Mane, R.S.; Naushad, M.; Stadler, F.J.; Patil, V.B. Electrochemical supercapacitor development based on electrodeposited nickel oxide film. RSC Adv. 2015, 5, 51961–51965. [Google Scholar] [CrossRef]
- Guo, X.; Yan, K.; Fan, F.; Zhang, Y.; Duan, Y.; Liu, J. Controllable synthesis of a NiO hierarchical microspheres/nanofibers composites assembled on nickel foam for supercapacitor. Mater. Lett. 2019, 240, 60–65. [Google Scholar] [CrossRef]
- Justin, P.; Meher, S.K.; Rao, R. Tuning of Capacitance Behavior of NiO Using Anionic, Cationic, and Nonionic Surfactants by Hydrothermal Synthesis. J. Chem. Phys. C 2010, 114, 5203–5210. [Google Scholar] [CrossRef]
- Carbone, M. Cu-Zn-Co nanosized mixed oxides prepared from hydroxycarbonate precursors. J. Alloy. Compd. 2016, 688, 202–209. [Google Scholar] [CrossRef]
- Carbone, M.; Briancesco, R.; Bonadonna, L. Antimicrobial power of Cu/Zn mixed oxide nanoparticles to Escherichia coli. Environ. Nanotechnol. Monit. Manag. 2017, 7, 97–102. [Google Scholar] [CrossRef]
- Carbone, M. Zn defective ZnCo2O4 nanorods as high capacity anode for lithium ion batteries. J. Electroanal. Chem. 2018, 815, 151–157. [Google Scholar] [CrossRef]
- Caminiti, R.; Carbone, M.; Panero, S.; Sadun, C. Conductivity and structure of poly(ethylene glycol) complexes using energy dispersive X-ray diffraction. J. Chem. Phys. B 1999, 103, 10348–10355. [Google Scholar] [CrossRef]
- Meher, S.K.; Justin, P.; Rao, G.R. Nanoscale morphology dependent pseudocapacitance of NiO: Influence of intercalating anions during synthesis. Nanoscale 2011, 3, 683–692. [Google Scholar] [CrossRef]
- Meng, T.; Ma, P.-P.; Chang, J.-L.; Wang, Z.-H.; Ren, T.-Z. The Electrochemical Capacitive Behaviors of NiO Nanoparticle. Electrochim. Acta 2013, 125, 586–592. [Google Scholar] [CrossRef]
- Kim, S.-I.; Lee, J.-S.; Ahn, H.-J.; Song, H.-K.; Jang, J.-H. Facile Route to an Efficient NiO Supercapacitor with a Three-Dimensional Nanonetwork Morphology. ACS Appl. Mater. Inter. 2013, 5, 1596–1603. [Google Scholar] [CrossRef]
- Carbone, M.; Bauer, E.M.; Micheli, L.; Missori, M. NiO morphology dependent optical and electrochemical properties. Colloid Surf. A 2017, 532, 178–182. [Google Scholar] [CrossRef]
- Duan, W.J.; Lu, S.H.; Wu, Z.L.; Wang, Y.S. Size Effects on Properties of NiO Nanoparticles Grown in Alkalisalts. J. Chem. Phys. C 2012, 116, 26043–26051. [Google Scholar] [CrossRef]
- Varunkumar, K.; Hussain, R.; Hegde, G.; Ethiraj, A.S. Effect of calcination temperature on Cu doped NiO nanoparticles prepared via wet-chemical method: Structural, optical and morphological studies. Mat. Sci. Semicon. Proc. 2017, 66, 149–156. [Google Scholar] [CrossRef]
- Mallick, P.; Sahoo, C.S. Effect of CuO Addition on the Structural and Optical Properties of NiO Nanoparticles. Nanosci. Nanotechol. 2013, 3, 52–55. [Google Scholar]
- Sharma, R.; Yadav, K. Effect of lattice defects on the structural and optical properties of Ni1−XAgXO (where X = 0.0, 0.01, 0.03, 0.05, 0.10 and 0.15) Nanoparticles. Appl. Phys. A 2018, 124, 88. [Google Scholar] [CrossRef]
- Allaedini, G.; Aminayi, P.; Tasirin, S.M. Structural properties and optical characterization of flower-like Mg doped NiO. AIP Adavances 2015, 5, 077161. [Google Scholar] [CrossRef]
- Anandan, K.; Rajendran, V. Morphological and size effects of NiO nanoparticles via solvothermal, process and their optical properties. Mat. Sci. Semicon. Proc. 2011, 14, 43–47. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, H.; Zhang, Q. Preparation of NiO nanoflakes under different calcination temperatures and their supercapacitive and optical properties. Appl. Surf. Sci. 2017, 392, 1097–1106. [Google Scholar] [CrossRef]
- Valentini, F.; Roscioli, D.; Carbone, M.; Conte, V.; Floris, B.; Bauer, E.M.; Ditaranto, N.; Sabbatini, L.; Caponetti, E.; Chillura-Martino, D. Graphene and ionic liquids new gel paste electrodes for caffeic acid quantification. Sens. Actuators B-Chem 2015, 212, 248–255. [Google Scholar] [CrossRef]
- Valentini, F.; Roscioli, D.; Carbone, M.; Conte, V.; Floris, B.; Palleschi, G.; Flammini, R.; Bauer, E.M.; Nasillo, G.; Caponetti, E. Oxidized graphene in ionic liquids for assembling chemically modified electrodes: A structural and electrochemical characterization study. Anal. Chem. 2012, 84, 5823–5831. [Google Scholar] [CrossRef]
- Valentini, F.; Ciambella, E.; Boaretto, A.; Rizzitelli, G.; Carbone, M.; Conte, V.; Cataldo, F.; Russo, V.; Casari, C.S.; Chillura-Martino, D.; et al. Sensor properties of pristine and functionalized carbon nanohorns. Electroanalysis 2016, 28, 2489–2499. [Google Scholar] [CrossRef]
- Valentini, F.; Carbone, M.; Palleschi, G. Graphene oxide nanoribbons (GNO), reduced graphene nanoribbons (GNR), and multi-layers of oxidized graphene functionalized with ionic liquids (GO-IL) for assembly of miniaturized electrochemical devices. Anal. Bioanal. Chem. 2013, 405, 3449–3474. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Micheli, L.; Limosani, F.; Possanza, F.; Abdallah, Y.; Tagliatesta, P. Ruthenium and manganese metalloporphyrins modified screen-printed electrodes for bio-relevant electroactive targets. J. Porphyr. Phthalocya. 2018, 22, 491–500. [Google Scholar] [CrossRef]
- Valentini, F.; Carbone, M.; Palleschi, G. Carbon nanostructured materials for applications in nano-medicine, cultural heritage, and electrochemical biosensors. Anal. Bioanal. Chem. 2013, 405, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Brousse, T.; Bélanger, D.; Long, J.W. To be or not to be pseudocapacitive? J. Electrochem. Soc. 2015, 162, A5185–A5189. [Google Scholar] [CrossRef]
- Simon, P.; Gogotsi, Y.; Dunn, B. Where Do Batteries End and Supercapacitors Begin? Science 2014, 343, 1210–1211. [Google Scholar] [CrossRef]
- Augustyn, V.; Simon, P.; Dunn, B. Pseudocapacitive oxide materials for high-rate electrochemical energy storage. Energ. Environ. Sci. 2014, 7, 1597–1614. [Google Scholar] [CrossRef]
- JCPDS 22-1189; The Joint Committee on Powder Diffraction Standards (JCPDS): Newtown Square, PA, USA.
- Condon, J.B. Surface Area and Porosity Determinations by Physisorption: Measurements and Theory; Elsevier: Amsterdam, The Netherland, 2006; pp. 178–180. [Google Scholar]
- Kubelka, P.; Munk, F. Ein Beitrag Zur Optik Der Farbanstriche. Z. Tech. Phys. 1931, 12, 593–601. [Google Scholar]
- Missori, M.; Pulci, O.; Teodonio, L.; Violante, C.; Kupchak, I.; Bagniuk, J.; Łojewska, J.; Mosca Conte, A. Optical response of strongly absorbing inhomogeneous materials: Application to paper degradation. Phys. Rev. B 2014, 89, 054201. [Google Scholar] [CrossRef]
- Missori, M. Optical spectroscopy of ancient paper and textiles. Il nuovo Cimento C 2016, 39, 293. [Google Scholar]
- Smith, R.A. Semiconductors, 2nd ed.; Cambridge University Press: Cambridge, UK, 1978. [Google Scholar]
- D’Amario, L.; Föhlinger, J.; Boschloo, G.; Hammarström, L. Unveiling hole trapping and surface dynamics of NiO nanoparticles. Chem. Sci. 2018, 9, 223–230. [Google Scholar] [CrossRef]
- Rödl, C.; Schleife, A. Photoemission spectra and effective masses of n- and p-type oxide semiconductors from first principles: ZnO, CdO, SnO2, MnO, and NiO. Phys. Status Solidi A 2014, 211, 74–81. [Google Scholar] [CrossRef]
- Brus, L.E. Electronic wave functions in semiconductor clusters: Experiment and theory. J. Phys. Chem. 1986, 90, 2555–2560. [Google Scholar] [CrossRef]
- Conway, B.E. Transition from “Supercapacitor” to “Battery” behavior in Electrochemical Energy Storage. J. Electrochem. Soc. 1991, 138, 1539–1548. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Label | Alkali | Calcination Temperature |
|---|---|---|
| A4 | (CH3CH2)3N | 400 °C |
| A6 | (CH3CH2)3N | 600 °C |
| U4 | (NH2)2CO | 400 °C |
| U6 | (NH2)2CO | 600 °C |
| Sample | Surface Area (m2 g−1) | Pore Size (nm) | Total Pore Volume (cm3 g−1) |
|---|---|---|---|
| A4 | 203.1 | 17.0 | 0.65 |
| U4 | 210.3 | 16.7 | 0.66 |
| A6 | 90.5 | 23.0 | 0.51 |
| U6 | 160.1 | 19.6 | 0.43 |
| Band Gap (eV) | Diameter (nm) | |
|---|---|---|
| A4 | 3.41 ± 0.02 | 11.4 ± 0.2 |
| U4 | 3.40 ± 0.02 | 11.2 ± 0.2 |
| A6 | 3.63 ± 0.02 | 18.0 ± 1.4 |
| U6 | 3.53 ± 0.02 | 14.0 ± 0.6 |
| Sample | Epa (V) | Epc (V) | ΔEp (V) |
|---|---|---|---|
| A4 | 0.350 | 0.119 | 0.231 |
| U4 | 0.342 | 0.117 | 0.225 |
| A6 | 0.388 | 0.083 | 0.305 |
| U6 | 0.361 | 0.081 | 0.280 |
| Sample | Epa (V) | Epc (V) | ΔEp (V) | Ipa (μA) | Ipc (μA) |
|---|---|---|---|---|---|
| A4 | 0.185 | 0.062 | 0.123 | 26.97 | −19.65 |
| U4 | 0.185 | 0.082 | 0.103 | 29.80 | −23.23 |
| A6 | 0.457 | −0.361 | 0.818 | 15.32 | −5.19 |
| U6 | 0.246 | 0.056 | 0.190 | 15.49 | −8.00 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carbone, M.; Missori, M.; Micheli, L.; Tagliatesta, P.; Bauer, E.M. NiO Pseudocapacitance and Optical Properties: Does The Shape Win? Materials 2020, 13, 1417. https://doi.org/10.3390/ma13061417
Carbone M, Missori M, Micheli L, Tagliatesta P, Bauer EM. NiO Pseudocapacitance and Optical Properties: Does The Shape Win? Materials. 2020; 13(6):1417. https://doi.org/10.3390/ma13061417
Chicago/Turabian StyleCarbone, Marilena, Mauro Missori, Laura Micheli, Pietro Tagliatesta, and Elvira Maria Bauer. 2020. "NiO Pseudocapacitance and Optical Properties: Does The Shape Win?" Materials 13, no. 6: 1417. https://doi.org/10.3390/ma13061417
APA StyleCarbone, M., Missori, M., Micheli, L., Tagliatesta, P., & Bauer, E. M. (2020). NiO Pseudocapacitance and Optical Properties: Does The Shape Win? Materials, 13(6), 1417. https://doi.org/10.3390/ma13061417