Effect of Metal Content on Ethanol Decomposition over Ni-Co Catalysts Supported on La-Ce Oxides

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Catalyst Synthesis and Characterization
2.2. Catalytic Ethanol Decomposition Experiments
2.3. In Situ Diffuse Reflectance Infrared Spectroscopy (DRIFTS) Measurements
2.4. Spent Catalyst Characterization
3. Results
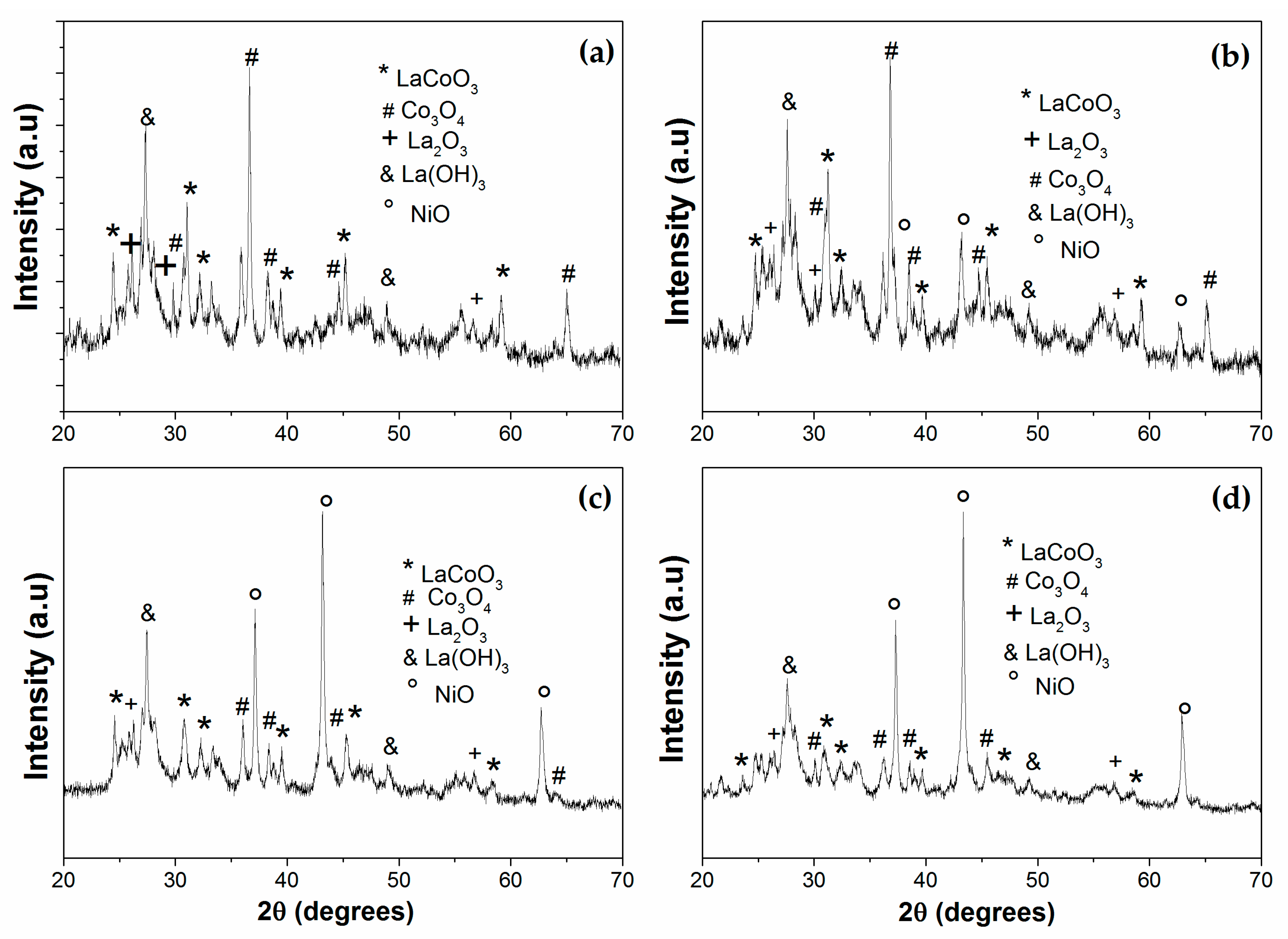
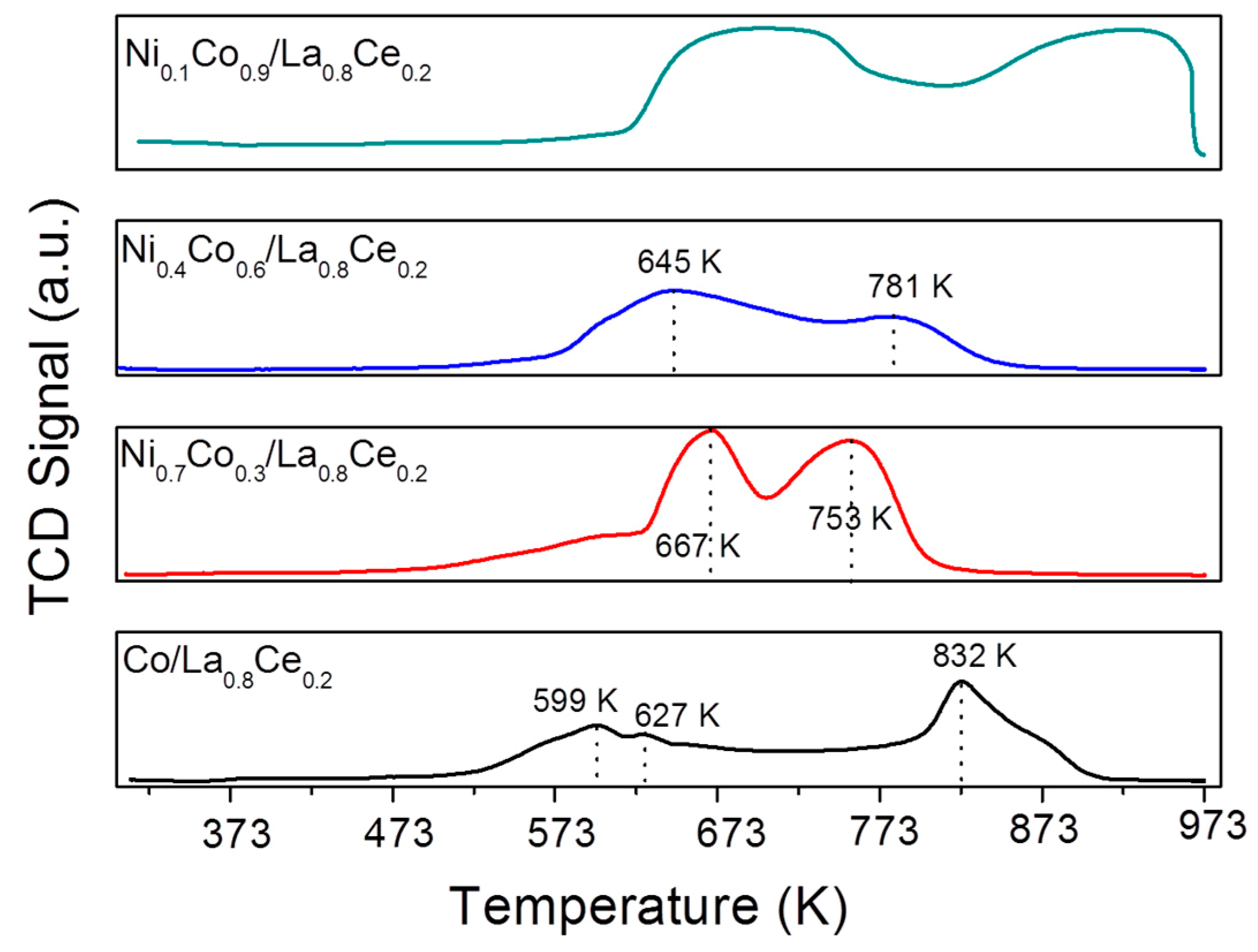
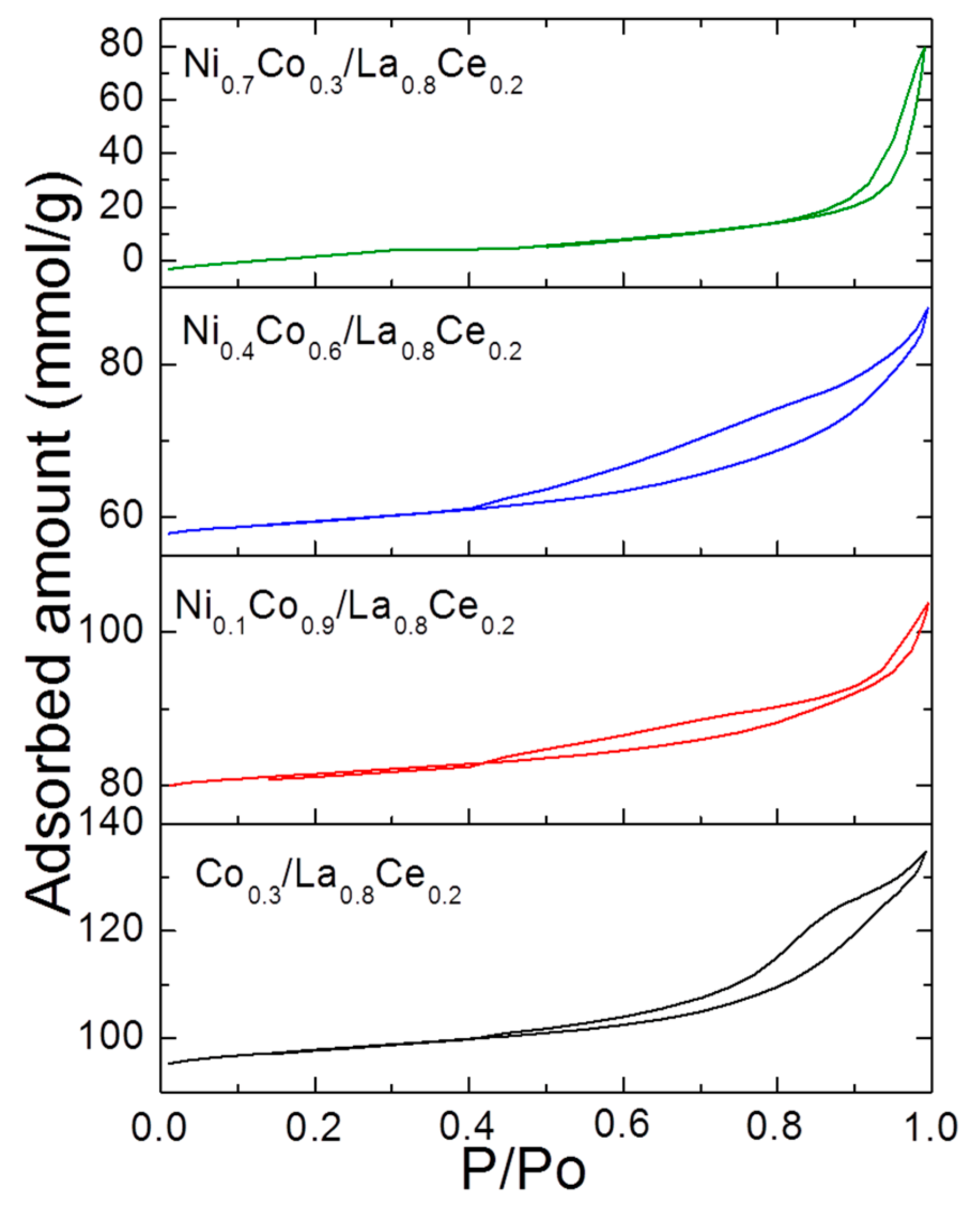
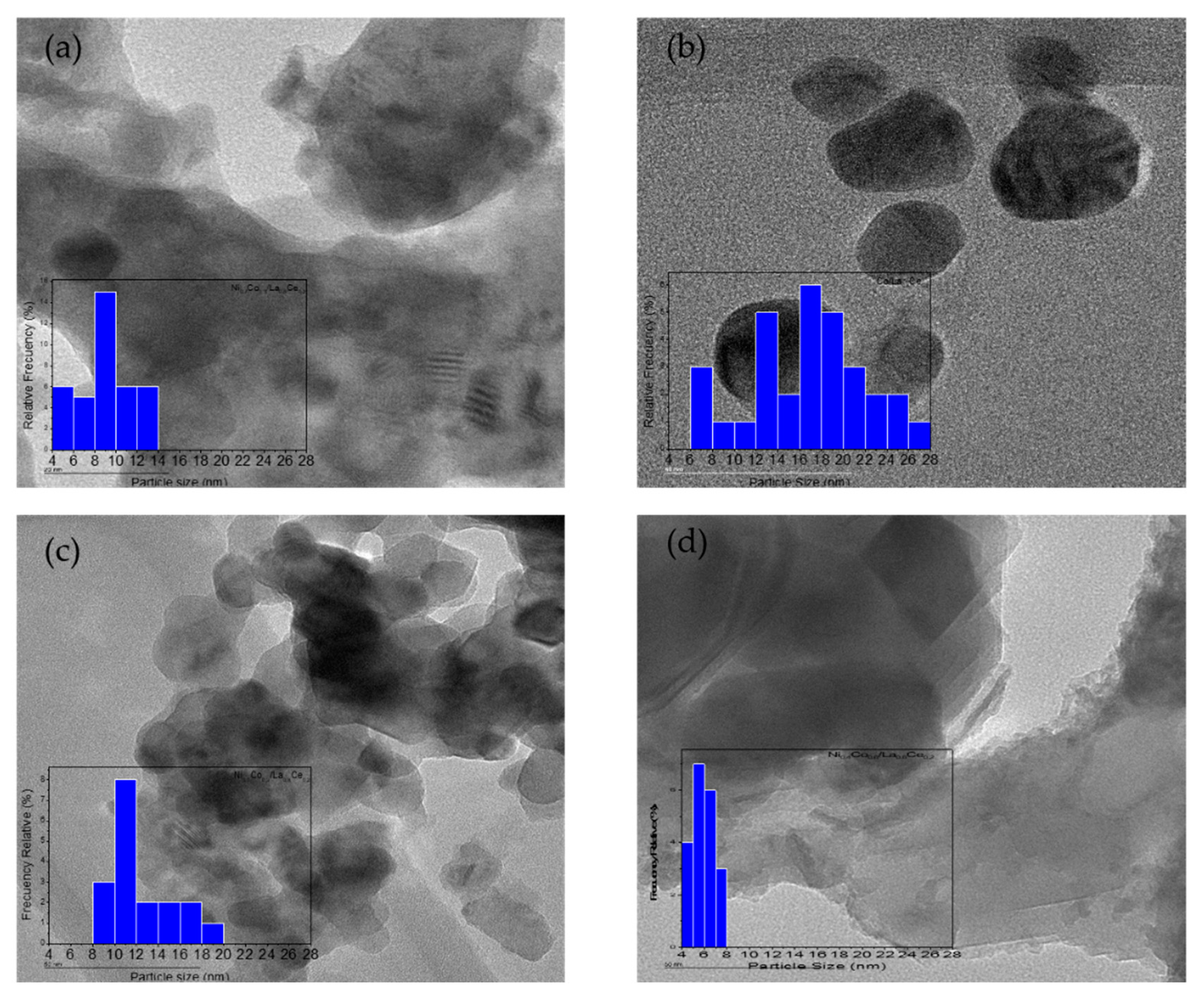
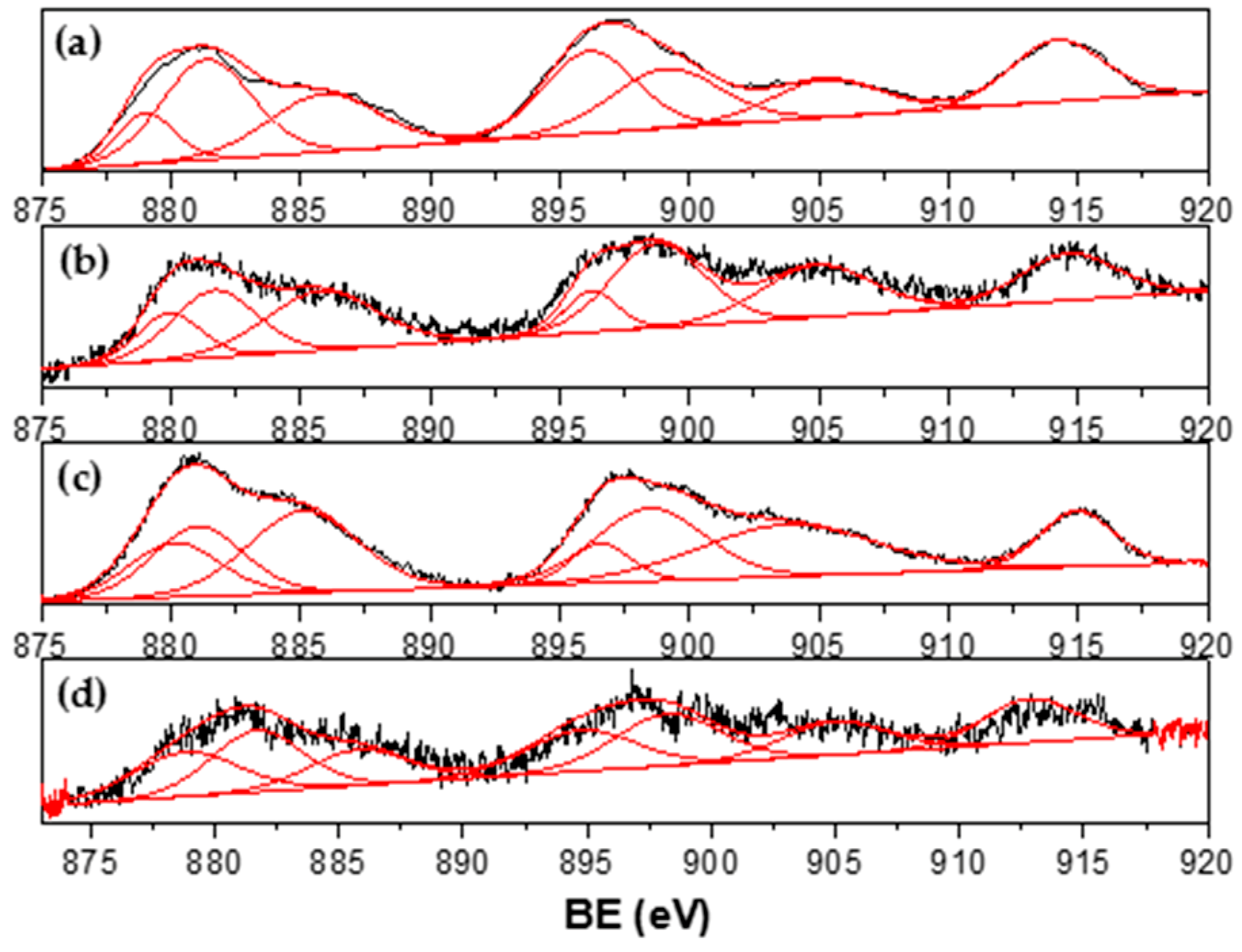
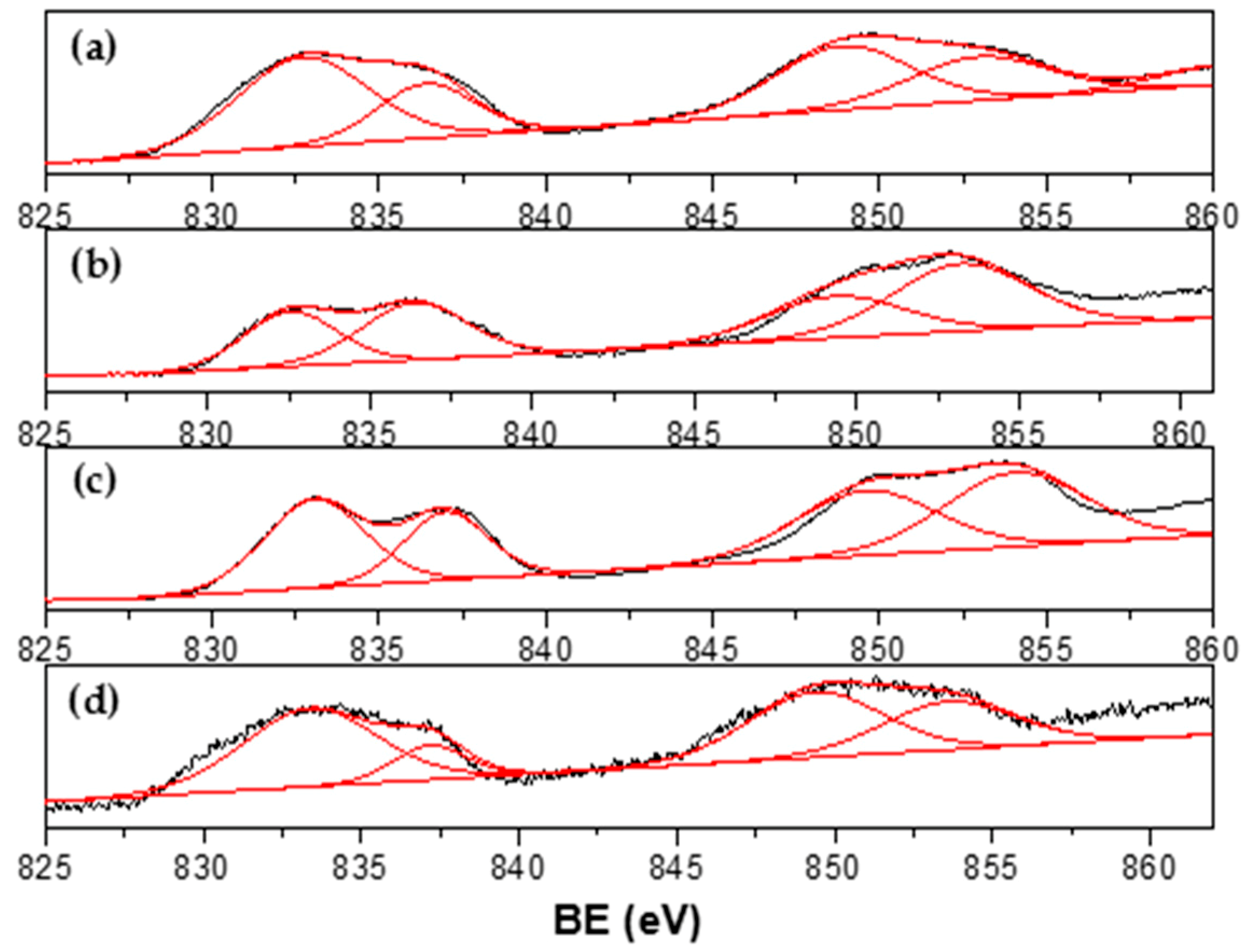
3.1. Catalyst Characterization
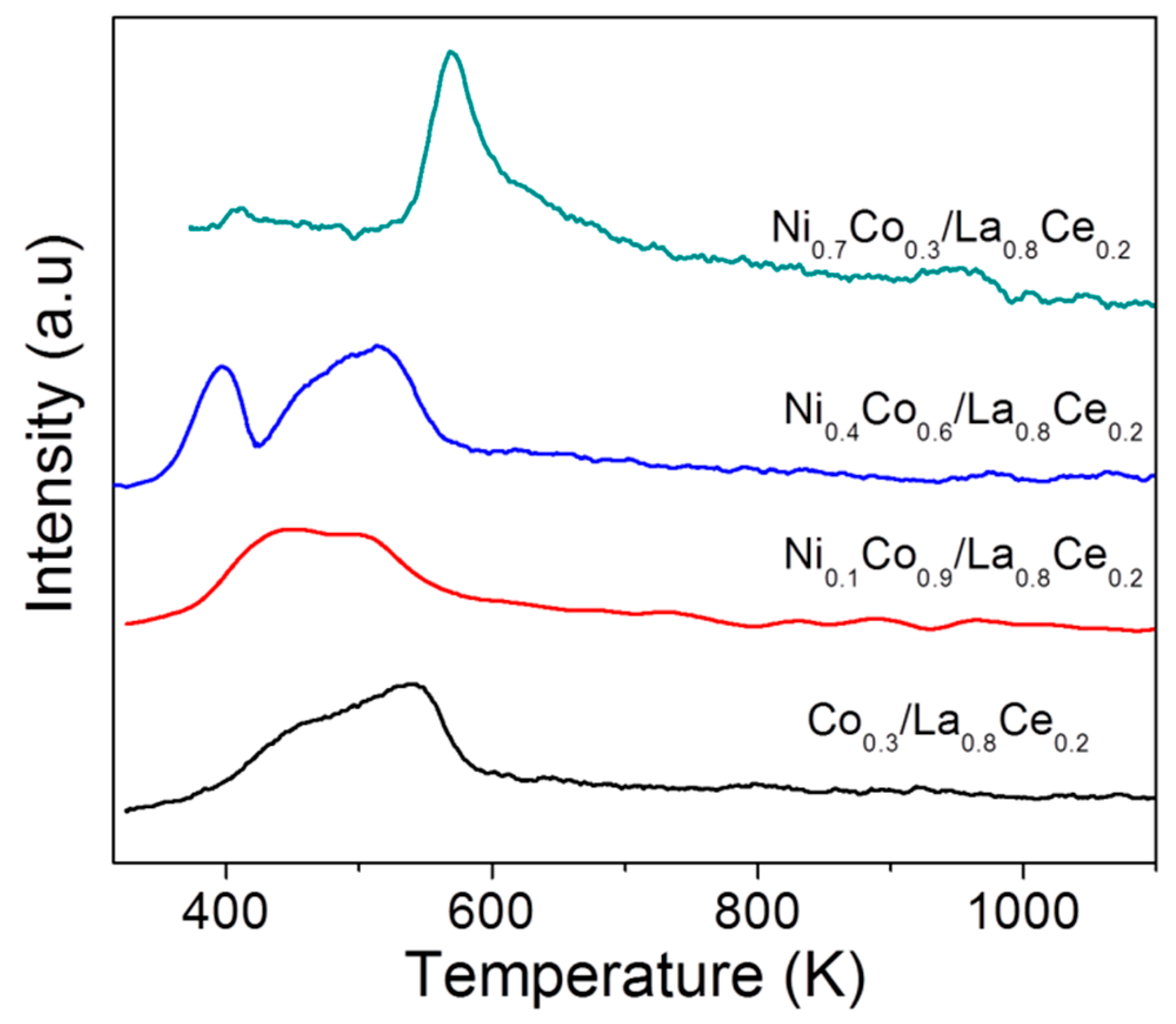
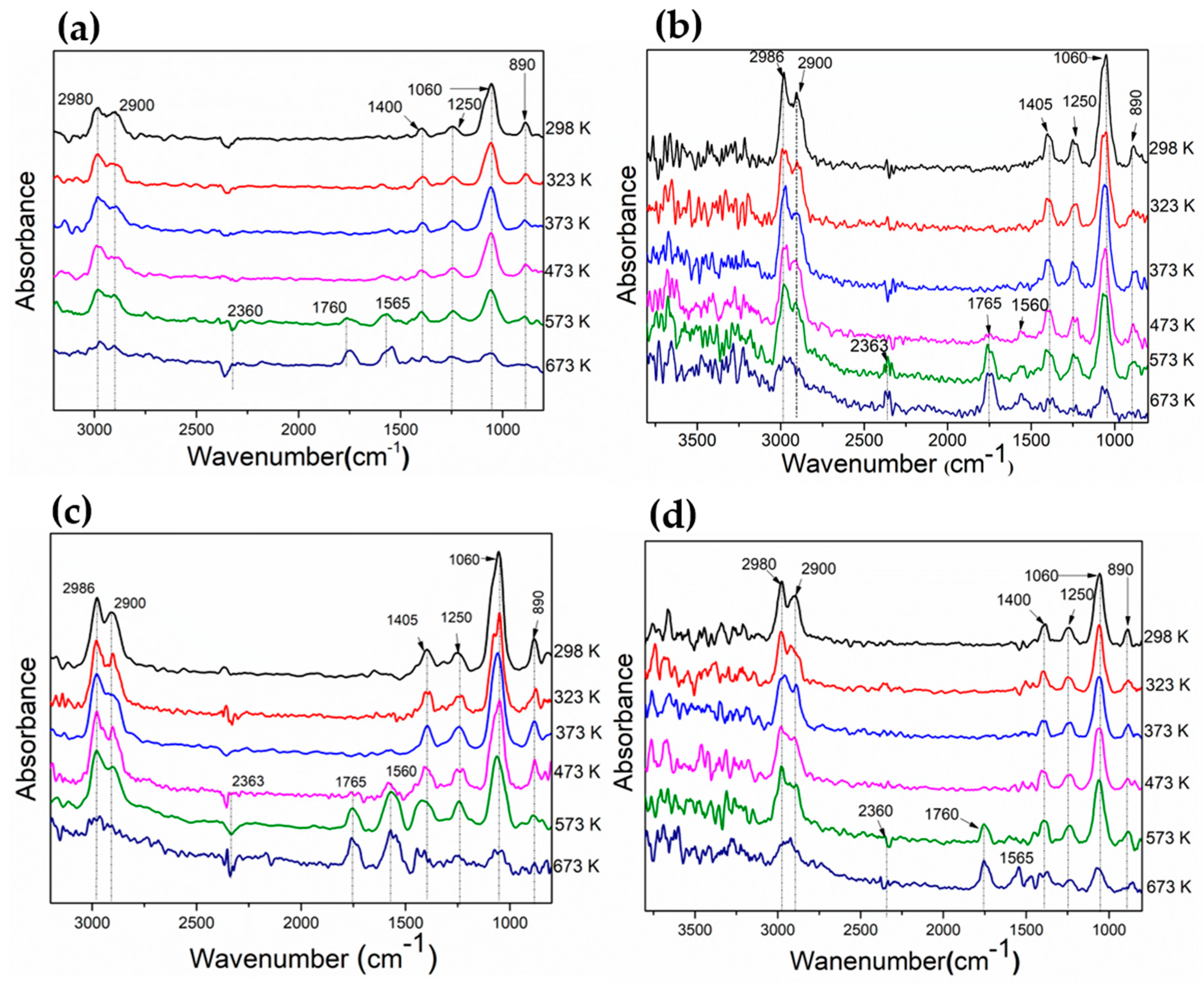
3.2. Evaluation of Adsorbed Species on the Catalyst Surface During Ethanol Decomposition by DRIFT
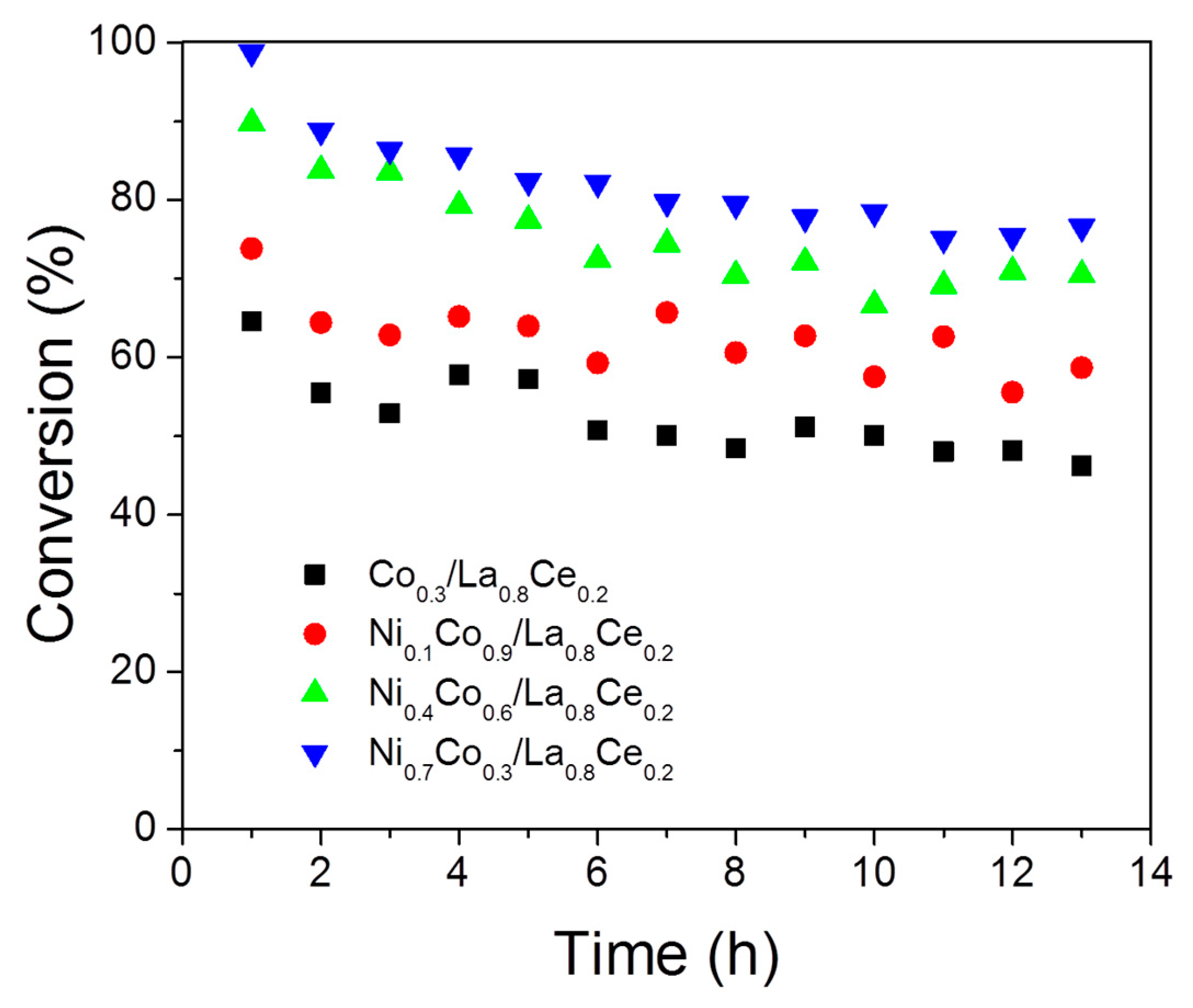
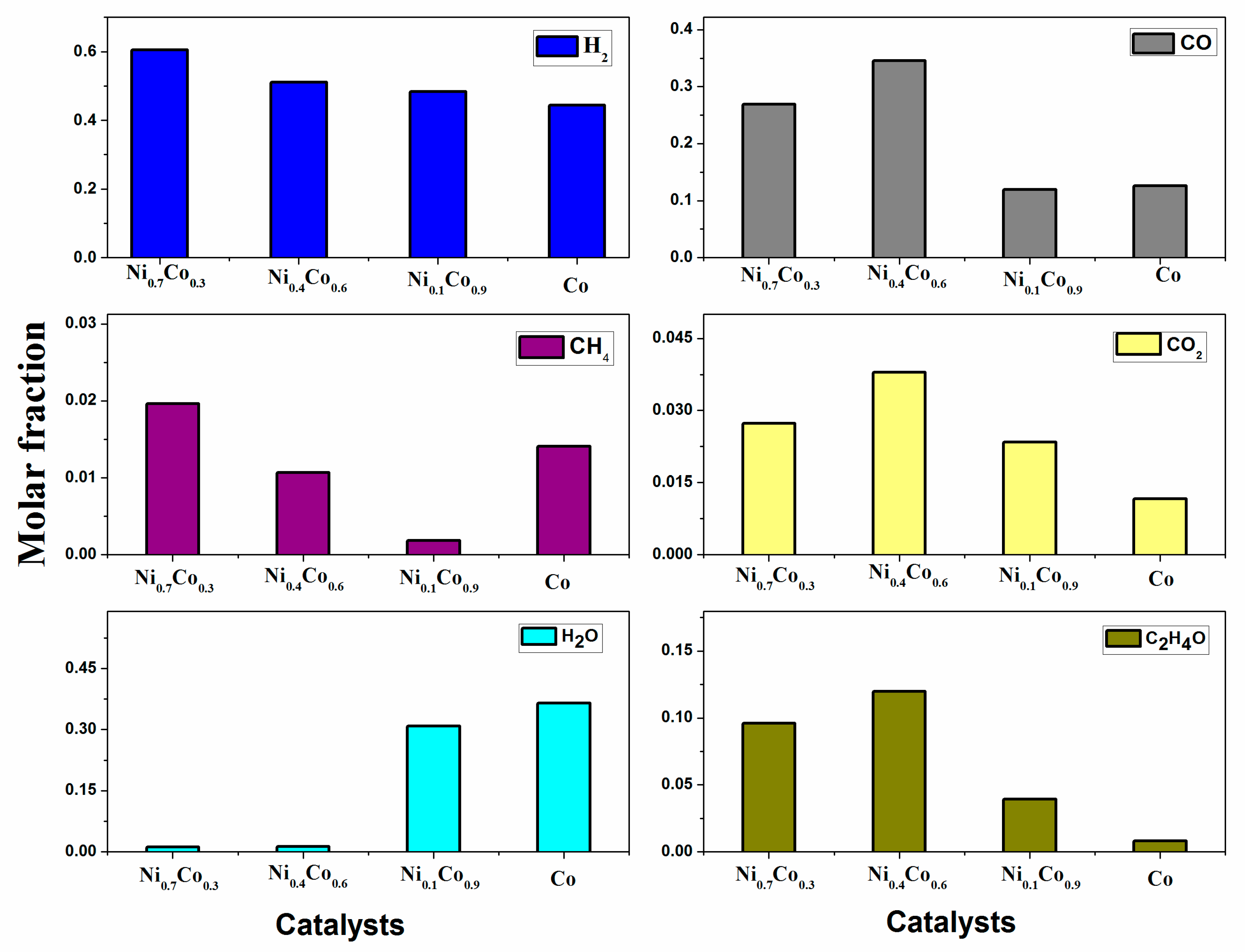
3.3. Ethanol Decomposition Catalytic Tests
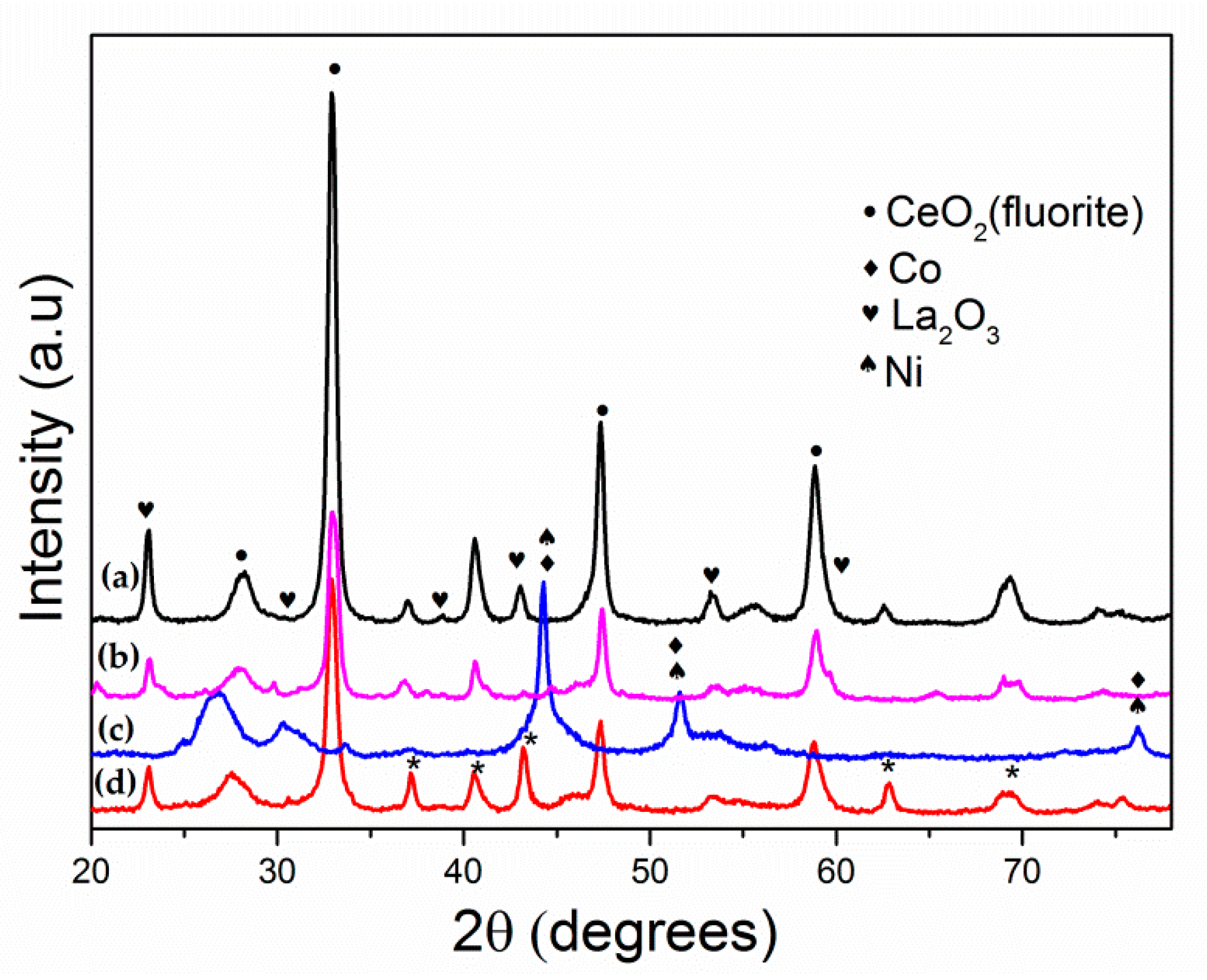
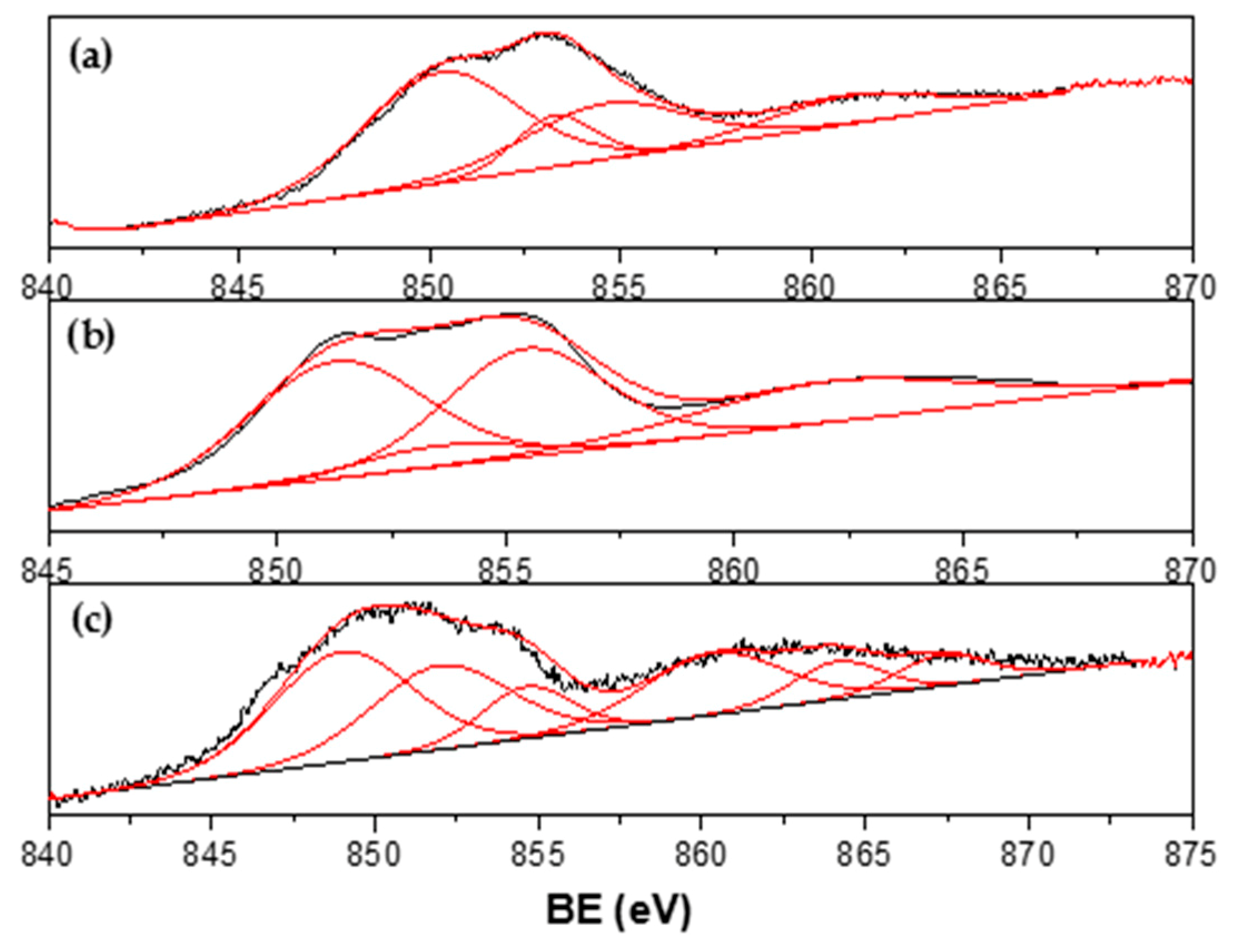
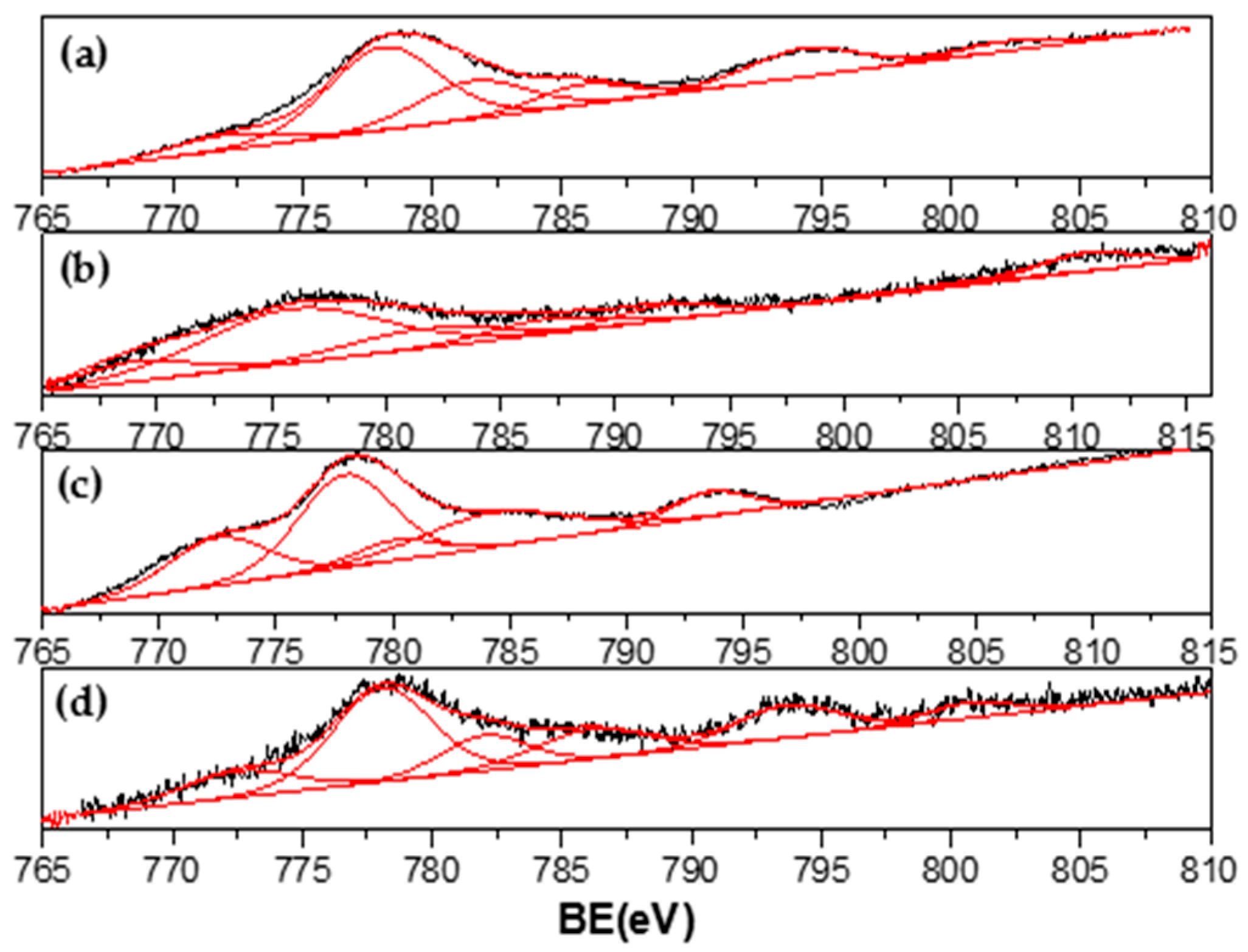
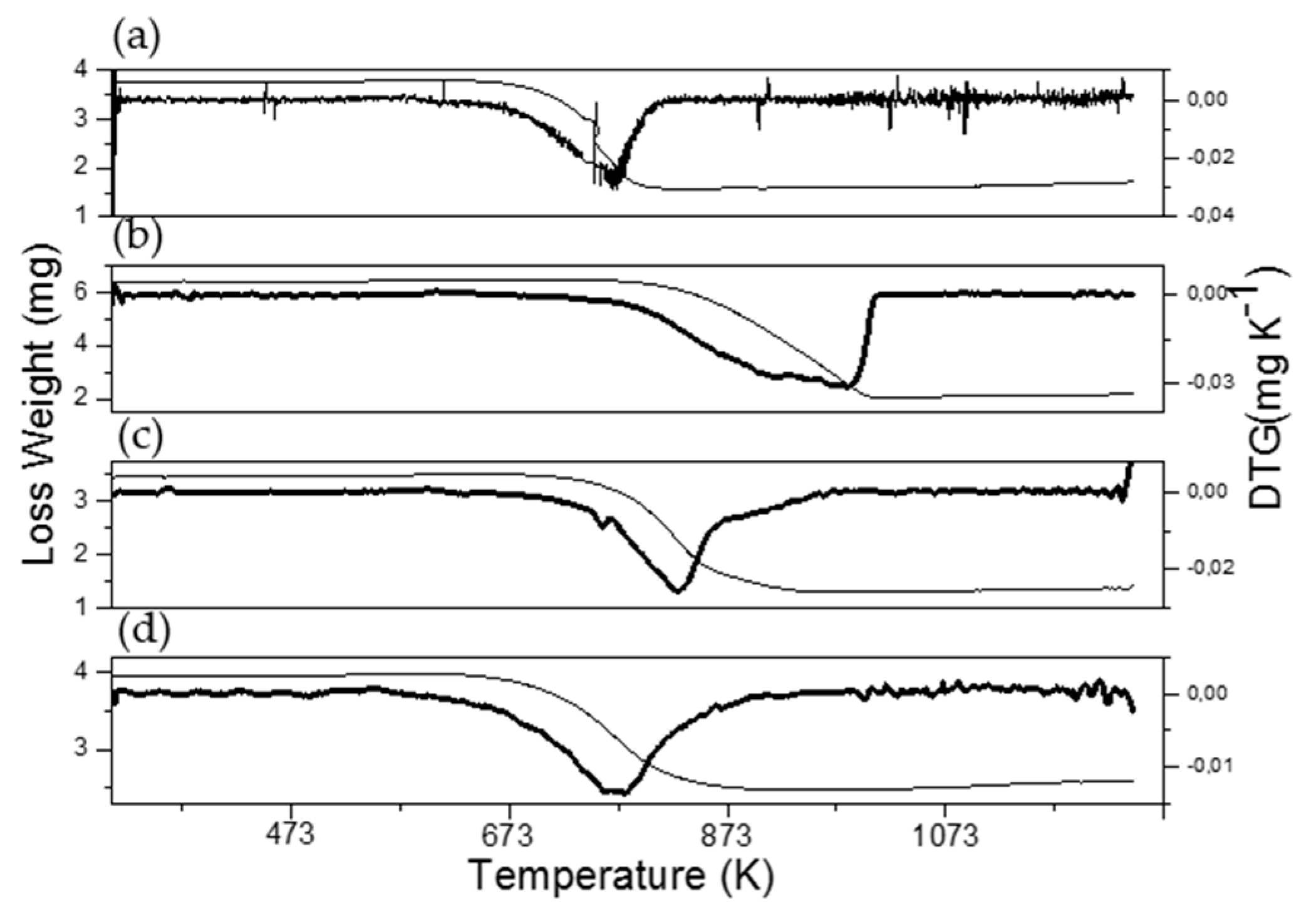
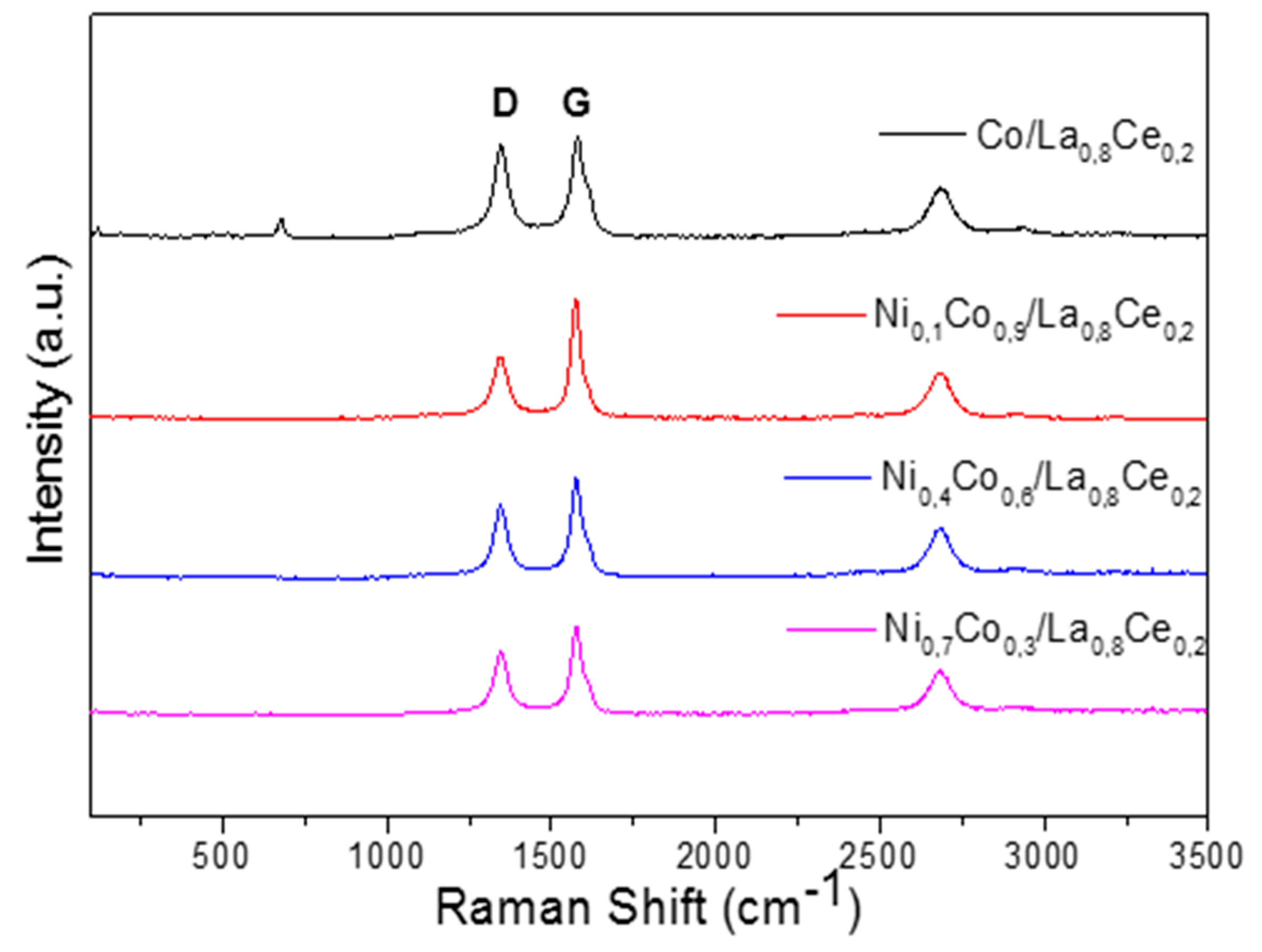
3.4. Characterization of Spent Catalysts
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ishizaki, H.; Hasumi, K. Ethanol Production from Biomass; Elsevier: Ámsterdam, The Netherlands, 2013; ISBN 9780124046092. [Google Scholar]
- Mattos, L.V.; Jacobs, G.; Davis, B.H.; Noronha, F.B. Production of hydrogen from ethanol: Review of reaction mechanism and catalyst deactivation. Chem. Rev. 2012, 112, 4094–4123. [Google Scholar] [CrossRef]
- De Lima, S.M.; da Silva, A.M.; da Costa, L.O.O.; Graham, U.M.; Jacobs, G.; Davis, B.H.; Mattos, L.V.; Noronha, F.B. Study of catalyst deactivation and reaction mechanism of steam reforming, partial oxidation, and oxidative steam reforming of ethanol over Co/CeO2 catalyst. J. Catal. 2009, 268, 268–281. [Google Scholar] [CrossRef]
- Campos, C.H.; Pecchi, G.; Fierro, J.L.G.; Osorio-Vargas, P. Enhanced bimetallic Rh-Ni supported catalysts on alumina doped with mixed lanthanum-cerium oxides for ethanol steam reforming. Mol. Catal. 2019, 469, 87–97. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R. Sulfur-passivated nickel catalysts for carbon-free steam reforming of methane. J. Catal. 1984, 85, 31–43. [Google Scholar] [CrossRef]
- He, S.; An, Z.; Wei, M.; Evans, D.G.; Duan, X. Layered double hydroxide-based catalysts: Nanostructure design and catalytic performance. Chem. Commun. 2013, 49, 5912–5920. [Google Scholar] [CrossRef] [PubMed]
- Manukyan, K.V.; Cross, A.J.; Yeghishyan, A.V.; Rouvimov, S.; Miller, J.J.; Mukasyan, A.S.; Wolf, E.E. Highly stable Ni-Al2O3 catalyst prepared from a Ni-Al layered double hydroxide for ethanol decomposition toward hydrogen. Appl. Catal. A Gen. 2015, 508, 37–44. [Google Scholar] [CrossRef]
- Ashok, A.; Kumar, A.; Ponraj, J.; Mansour, S.A.; Tarlochan, F. Effect of Ni incorporation in cobalt oxide lattice on carbon formation during ethanol decomposition reaction. Appl. Catal. B Environ. 2019, 254, 300–311. [Google Scholar] [CrossRef]
- Gallego, J.; Sierra, G.; Mondragon, F.; Barrault, J.; Batiot-Dupeyrat, C. Synthesis of MWCNTs and hydrogen from ethanol catalytic decomposition over a Ni/La2O3 catalyst produced by the reduction of LaNiO3. Appl. Catal. A Gen. 2011, 397, 73–81. [Google Scholar] [CrossRef]
- Morales, R.; Campos, C.H.; Fierro, J.L.G.; Fraga, M.A.; Pecchi, G. Stable reduced Ni catalysts for xylose hydrogenation in aqueous medium. Catal. Today 2018, 310, 59–67. [Google Scholar] [CrossRef]
- Kumar, A.; Miller, J.T.; Mukasyan, A.S.; Wolf, E.E. In situ XAS and FTIR studies of a multi-component Ni/Fe/Cu catalyst for hydrogen production from ethanol. Appl. Catal. A Gen. 2013, 467, 593–603. [Google Scholar] [CrossRef]
- De Lima, S.M.; Silva, A.M.; Graham, U.M.; Jacobs, G.; Davis, B.H.; Mattos, L.V.; Noronha, F.B. Ethanol decomposition and steam reforming of ethanol over CeZrO2 and Pt/CeZrO2 catalyst: Reaction mechanism and deactivation. Appl. Catal. A Gen. 2009, 352, 95–113. [Google Scholar] [CrossRef]
- Dancini-Pontes, I.; Fernandes-Machado, N.R.C.; De Souza, M.; Pontes, R.M. Insights into ethanol decomposition over Pt: A DFT energy decomposition analysis for the reaction mechanism leading to C2H6 and CH4. Appl. Catal. A Gen. 2015, 491, 86–93. [Google Scholar] [CrossRef]
- De Souza, G.; Balzaretti, N.M.; Marcílio, N.R.; Perez-Lopez, O.W. Decomposition of ethanol over Ni-Al catalysts: Effect of copper addition. Procedia Eng. 2012, 42, 335–345. [Google Scholar] [CrossRef]
- Diao, J.; Wang, H.; Li, W.; Wang, G.; Ren, Z.; Bai, J. Effect of C-supported Co catalyst on the ethanol decomposition to produce hydrogen and multi-walled carbon nanotubes. Phys. E Low-Dimensional Syst. Nanostruct. 2010, 42, 2280–2284. [Google Scholar] [CrossRef]
- Chen, R.; Xie, Y.; Zhou, Y.; Wang, J.; Wang, H. Production of hydrogen-rich gas and multi-walled carbon nanotubes from ethanol decomposition over molybdenum modified Ni/MgO catalysts. J. Energy Chem. 2014, 23, 244–250. [Google Scholar] [CrossRef]
- Wang, G.; Wang, H.; Tang, Z.; Li, W.; Bai, J. Simultaneous production of hydrogen and multi-walled carbon nanotubes by ethanol decomposition over Ni/Al2O3 catalysts. Appl. Catal. B Environ. 2009, 88, 142–151. [Google Scholar] [CrossRef]
- Li, W.; Wang, H.; Ren, Z.; Wang, G.; Bai, J. Co-production of hydrogen and multi-wall carbon nanotubes from ethanol decomposition over Fe/Al2O3 catalysts. Appl. Catal. B Environ. 2008, 84, 433–439. [Google Scholar] [CrossRef]
- Mezalira, D.Z.; Probst, L.D.; Pronier, S.; Batonneau, Y.; Batiot-Dupeyrat, C. Decomposition of ethanol over Ni/Al2O3 catalysts to produce hydrogen and carbon nanostructured materials. J. Mol. Catal. A Chem. 2011, 340, 15–23. [Google Scholar] [CrossRef]
- Laosiripojana, N.; Sutthisripok, W.; Assabumrungrat, S. Reactivity of high surface area CeO2 synthesized by surfactant-assisted method to ethanol decomposition with and without steam. Chem. Eng. J. 2007, 127, 31–38. [Google Scholar] [CrossRef]
- Manukyan, K.V.; Yeghishyan, A.V.; Danghyan, V.; Rouvimov, S.; Mukasyan, A.S.; Wolf, E.E. Structural transformations of highly porous nickel catalysts during ethanol conversion towards hydrogen. Int. J. Hydrogen Energy 2018, 43, 13225–13236. [Google Scholar] [CrossRef]
- Osorio-Vargas, P.; Flores-González, N.A.; Navarro, R.M.; Fierro, J.L.G.; Campos, C.H.; Reyes, P. Improved stability of Ni/Al2O3 catalysts by effect of promoters (La2O3, CeO2) for ethanol steam-reforming reaction. Catal. Today 2015, 259, 27–38. [Google Scholar] [CrossRef]
- Pechini, M.P. Method of Pre Parng Lead and Alkalne Earth Titanates and Nobates and Coat. U.S. Patent 3,330,697, 11 July 1967. [Google Scholar]
- Marinho, A.L.A.; Rabelo-Neto, R.C.; Noronha, F.B.; Mattos, L.V. Steam reforming of ethanol over Ni-based catalysts obtained from LaNiO3 and LaNiO3/CeSiO2 perovskite-type oxides for the production of hydrogen. Appl. Catal. A Gen. 2016, 520, 53–64. [Google Scholar] [CrossRef]
- Franchini, C.A.; Aranzaez, W.; Duarte de Farias, A.M.; Pecchi, G.; Fraga, M.A. Ce-substituted LaNiO3 mixed oxides as catalyst precursors for glycerol steam reforming. Appl. Catal. B Environ. 2014, 147, 193–202. [Google Scholar] [CrossRef]
- Yang, E.H.; Kim, N.Y.; Noh, Y.S.; Lim, S.S.; Jung, J.S.; Lee, J.S.; Hong, G.H.; Moon, D.J. Steam CO2 reforming of methane over La1-xCexNiO3 perovskite catalysts. Int. J. Hydrogen Energy 2015, 40, 11831–11839. [Google Scholar] [CrossRef]
- De Lima, S.M.; da Silva, A.M.; da Costa, L.O.O.; Assaf, J.M.; Mattos, L.V.; Sarkari, R.; Venugopal, A.; Noronha, F.B. Hydrogen production through oxidative steam reforming of ethanol over Ni-based catalysts derived from La1−xCexNiO3 perovskite-type oxides. Appl. Catal. B Environ. 2012, 121–122, 1–9. [Google Scholar] [CrossRef]
- Ma, F.; Chu, W.; Huang, L.; Yu, X.; Wu, Y. Steam reforming of ethanol over Zn-Doped LaCoO3 perovskite nanocatalysts. Chin. J. Catal. 2011, 32, 970–977. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, C.; Chen, S.; Liu, Y. Co-Ni bimetal catalyst supported on perovskite-type oxide for steam reforming of ethanol to produce hydrogen. Int. J. Hydrogen Energy 2014, 39, 5644–5652. [Google Scholar] [CrossRef]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquerol, J.; Siemieniewska, T. Reporting physisorption data for gas/solid systems with Special Reference to the Determination of Surface Area and Porosity (Recommendations 1984). Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Tang, Z.; Cao, H.; Tao, Y.; Heeres, H.J.; Pescarmona, P.P. Transfer hydrogenation from glycerol over a Ni-Co/CeO2 catalyst: A highly efficient and sustainable route to produce lactic acid. Appl. Catal. B Environ. 2020, 263, 118273. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Lau, L.W.M.; Gerson, A.; Smart, R.S.C. X-ray photoelectron spectroscopic chemical state Quantification of mixed nickel metal, oxide and hydroxide systems. Surf. Interface Anal. 2009, 41, 324–332. [Google Scholar] [CrossRef]
- Grosvenor, A.P.; Biesinger, M.C.; Smart, R.S.C.; McIntyre, N.S. New interpretations of XPS spectra of nickel metal and oxides. Surf. Sci. 2006, 600, 1771–1779. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Chuang, T.J.; Brundle, C.R.; Rice, D.W. Interpretation of the x-ray photoemission spectra of cobalt oxides and cobalt oxide surfaces. Surf. Sci. 1976, 59, 413–429. [Google Scholar] [CrossRef]
- Huck-Iriart, C.; Soler, L.; Casanovas, A.; Marini, C.; Prat, J.; Llorca, J.; Escudero, C. Unraveling the Chemical State of Cobalt in Co-Based Catalysts during Ethanol Steam Reforming: An in Situ Study by Near Ambient Pressure XPS and XANES. ACS Catal. 2018, 8, 9625–9636. [Google Scholar] [CrossRef]
- Zhai, Y.; Chu, M.; Shang, N.; Wang, C.; Wang, H.; Gao, Y. Bimetal Co8Ni2 catalyst supported on chitin-derived N-containing carbon for upgrade of biofuels. Appl. Surf. Sci. 2019, 144681. [Google Scholar] [CrossRef]
- Maslakov, K.I.; Teterin, Y.A.; Popel, A.J.; Teterin, A.Y.; Ivanov, K.E.; Kalmykov, S.N.; Petrov, V.G.; Petrov, P.K.; Farnan, I. XPS study of ion irradiated and unirradiated CeO2 bulk and thin film samples. Appl. Surf. Sci. 2018, 448, 154–162. [Google Scholar] [CrossRef]
- Bêche, E.; Charvin, P.; Perarnau, D.; Abanades, S.; Flamant, G. Ce 3d XPS investigation of cerium oxides and mixed cerium oxide (CexTiyOz). Surf. Interface Anal. 2008, 40, 264–267. [Google Scholar] [CrossRef]
- Paparazzo, E. XPS studies of damage induced by X-ray irradiation on CeO2 surfaces. Surf. Sci. 1990, 234, 1–6. [Google Scholar] [CrossRef]
- Fleisch, T.H.; Hicks, R.F.; Bell, A.T. An XPS study of metal-support interactions on Pd SiO2 and Pd La2O3. J. Catal. 1984, 87, 398–413. [Google Scholar] [CrossRef]
- Araujo, J.C.S.; Zanchet, D.; Rinaldi, R.; Schuchardt, U.; Hori, C.E.; Fierro, J.L.G.; Bueno, J.M.C. The effects of La2O3 on the structural properties of La2O3-Al2O3 prepared by the sol-gel method and on the catalytic performance of Pt/La2O3-Al2O3 towards steam reforming and partial oxidation of methane. Appl. Catal. B Environ. 2008, 84, 552–562. [Google Scholar] [CrossRef]
- De Lima, S.M.; da Cruz, I.O.; Jacobs, G.; Davis, B.H.; Mattos, L.V.; Noronha, F.B. Steam reforming, partial oxidation, and oxidative steam reforming of ethanol over Pt/CeZrO2 catalyst. J. Catal. 2008, 257, 356–368. [Google Scholar] [CrossRef]
- Mattos, L.V.; Noronha, F.B. Hydrogen production for fuel cell applications by ethanol partial oxidation on Pt/CeO2 catalysts: The effect of the reaction conditions and reaction mechanism. J. Catal. 2005, 233, 453–463. [Google Scholar] [CrossRef]
- Erdőhelyi, A.; Raskó, J.; Kecskés, T.; Tóth, M.; Dömök, M.; Baán, K. Hydrogen formation in ethanol reforming on supported noble metal catalysts. Catal. Today 2006, 116, 367–376. [Google Scholar] [CrossRef]
- Wang, H.; Dong, X.; Zhao, T.; Yu, H.; Li, M. Dry reforming of methane over bimetallic Ni-Co catalyst prepared from La(CoxNi1-x)0.5Fe0.5O3 perovskite precursor: Catalytic activity and coking resistance. Appl. Catal. B Environ. 2019, 245, 302–313. [Google Scholar] [CrossRef]
- Neagu, D.; Oh, T.S.; Miller, D.N.; Ménard, H.; Bukhari, S.M.; Gamble, S.R.; Gorte, R.J.; Vohs, J.M.; Irvine, J.T.S. Nano-socketed nickel particles with enhanced coking resistance grown in situ by redox exsolution. Nat. Commun. 2015, 6, 8120. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Jia, L.; Zheng, H.; Chi, B.; Pu, J.; Li, J. LaMnO3-based perovskite with in-situ exsolved Ni nanoparticles: A highly active, performance stable and coking resistant catalyst for CO2 dry reforming of CH4. Appl. Catal. A Gen. 2018, 564, 199–207. [Google Scholar] [CrossRef]
- Campos, C.H.; Osorio-Vargas, P.; Flores-González, N.; Fierro, J.L.G.; Reyes, P. Effect of Ni Loading on Lanthanide (La and Ce) Promoted γ-Al2O3 Catalysts Applied to Ethanol Steam Reforming. Catal. Lett. 2016, 146, 433–441. [Google Scholar] [CrossRef]
- Iglesias-Juez, A.; Beale, A.M.; Maaijen, K.; Weng, T.C.; Glatzel, P.; Weckhuysen, B.M. A combined in situ time-resolved UV-Vis, Raman and high-energy resolution X-ray absorption spectroscopy study on the deactivation behavior of Pt and PtSn propane dehydrogenation catalysts under industrial reaction conditions. J. Catal. 2010, 276, 268–279. [Google Scholar] [CrossRef]
- Mutz, B.; Sprenger, P.; Wang, W.; Wang, D.; Kleist, W.; Grunwaldt, J.D. Operando Raman spectroscopy on CO2 methanation over alumina-supported Ni, Ni3Fe and NiRh0.1 catalysts: Role of carbon formation as possible deactivation pathway. Appl. Catal. A Gen. 2018, 556, 160–171. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Surface Area BET (m2 g−1) | Pore Volume (cm3 g−1) | Pore Size (nm) | Dispersion (%) | Particle Size a (nm) | Particle Size b (nm) | Acidity (mol NH3 gcat−1) |
|---|---|---|---|---|---|---|---|
| Ni0.7Co0.3/La0.8Ce0.2 | 33 | 0.13 | 25 | 21 | 4.8 | 9.1 | 0.04 |
| Ni0.4Co0.6/La00.8Ce0.2 | 16 | 0.05 | 28 | 10 | 10 | 5.9 | 0.47 |
| Ni0.1Co0.9/La0.8Ce0.2 | 14 | 0.04 | 31 | 6 | 17 | 12.4 | 0.23 |
| Co/La0.8Ce0.2 | 23 | 0.06 | 45 | 3 | 33 | 16.8 | 0.35 |
| Catalysts | (−ra)0 (mol g−1 s−1) | TOF (s−1) | CWP | Ethanol Conversion (%) | Hydrogen Selectivity (%) |
|---|---|---|---|---|---|
| Ni0.7Co0.3/La0.8Ce0.2 | 6.2 × 10−4 | 0.06 | 6.3 × 10−3 | 98.4 | 75.0 |
| Ni0.4Co0.6/La00.8Ce0.2 | 5.6 × 10−4 | 0.11 | 2.8 × 10−3 | 90.0 | 64.5 |
| Ni0.1Co0.9/La0.8Ce0.2 | 4.7 × 10−4 | 0.15 | 2.8 × 10−3 | 74.6 | 56.3 |
| Co/La0.8Ce0.2 | 4.1 × 10−4 | 0.27 | 1.5 × 10−3 | 65.4 | 52.6 |
| Catalyst | mgcoke/gcat | Tpeak DTG | ID/IG |
|---|---|---|---|
| Co/La0.8Ce0.2 | 530.5 | 773 | 1.00 |
| Ni0.1Co0.9/La0.8Ce0.2 | 659.5 | 980 | 0.98 |
| Ni0.4Co0.6/La0.8Ce0.2 | 592.1 | 830 | 0.99 |
| Ni0.7Co0.3/La0.8Ce0.2 | 342.6 | 780 | 0.99 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vergara, H.R.; Brijaldo, M.H.; Martinez, J.J.; Rojas, H.A.; Pedraza, J.; Passos, F.B.; Pereira da Costa, L.; Gonzalez-Vera, D.; Osorio-Vargas, P. Effect of Metal Content on Ethanol Decomposition over Ni-Co Catalysts Supported on La-Ce Oxides. Materials 2020, 13, 759. https://doi.org/10.3390/ma13030759
Vergara HR, Brijaldo MH, Martinez JJ, Rojas HA, Pedraza J, Passos FB, Pereira da Costa L, Gonzalez-Vera D, Osorio-Vargas P. Effect of Metal Content on Ethanol Decomposition over Ni-Co Catalysts Supported on La-Ce Oxides. Materials. 2020; 13(3):759. https://doi.org/10.3390/ma13030759
Chicago/Turabian StyleVergara, Harold R., Maria H. Brijaldo, José J. Martinez, Hugo A. Rojas, José Pedraza, Fabio B. Passos, Luiz Pereira da Costa, Daniela Gonzalez-Vera, and Paula Osorio-Vargas. 2020. "Effect of Metal Content on Ethanol Decomposition over Ni-Co Catalysts Supported on La-Ce Oxides" Materials 13, no. 3: 759. https://doi.org/10.3390/ma13030759
APA StyleVergara, H. R., Brijaldo, M. H., Martinez, J. J., Rojas, H. A., Pedraza, J., Passos, F. B., Pereira da Costa, L., Gonzalez-Vera, D., & Osorio-Vargas, P. (2020). Effect of Metal Content on Ethanol Decomposition over Ni-Co Catalysts Supported on La-Ce Oxides. Materials, 13(3), 759. https://doi.org/10.3390/ma13030759