Preparation of Mesoporous Mn–Ce–Ti–O Aerogels by a One-Pot Sol–Gel Method for Selective Catalytic Reduction of NO with NH3

Abstract
1. Introduction
2. Materials and Methods
2.1. Catalyst Preparation
2.2. Catalyst Characterization
2.3. Catalytic Activity Measurement
3. Results and Discussion
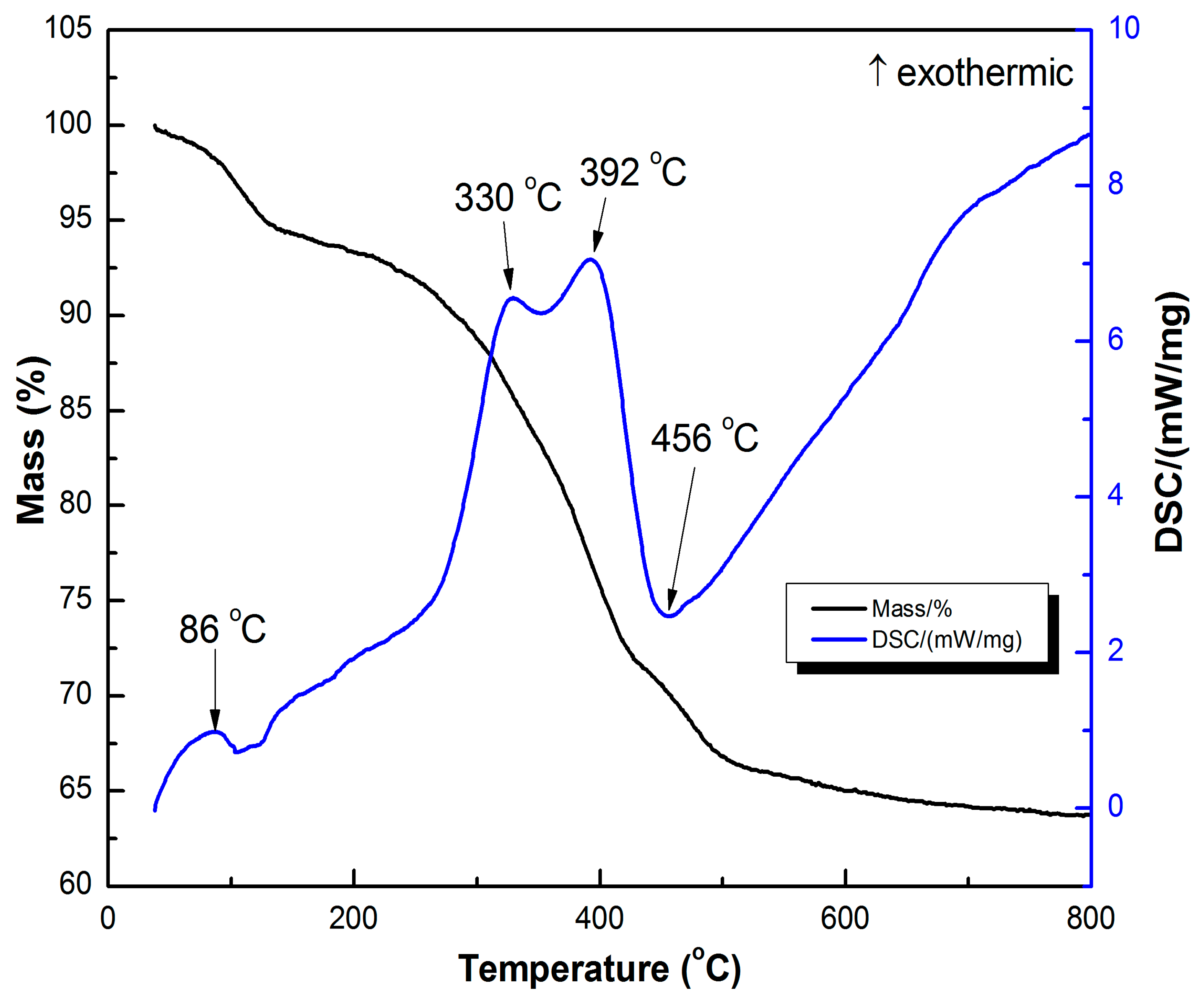
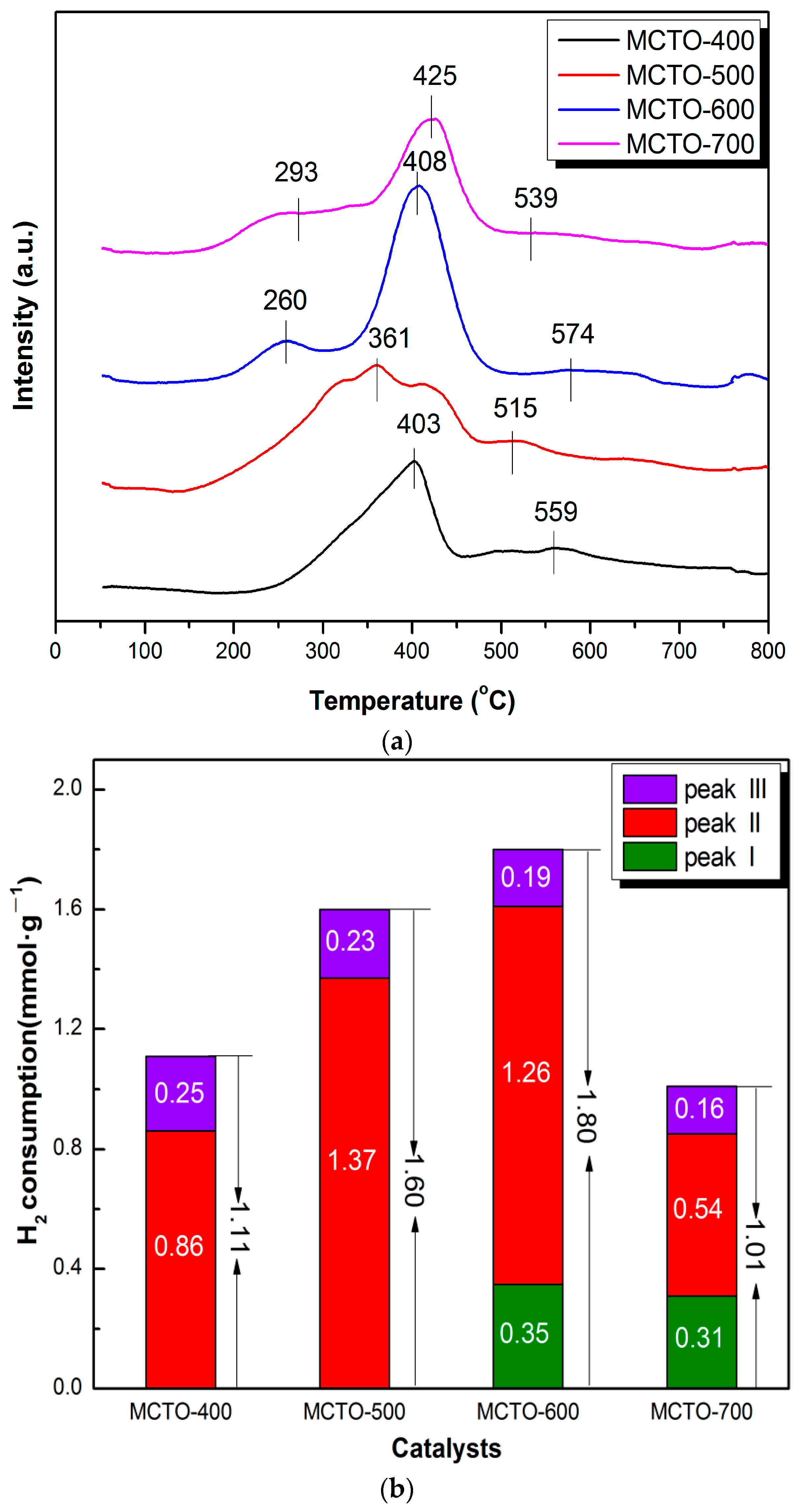
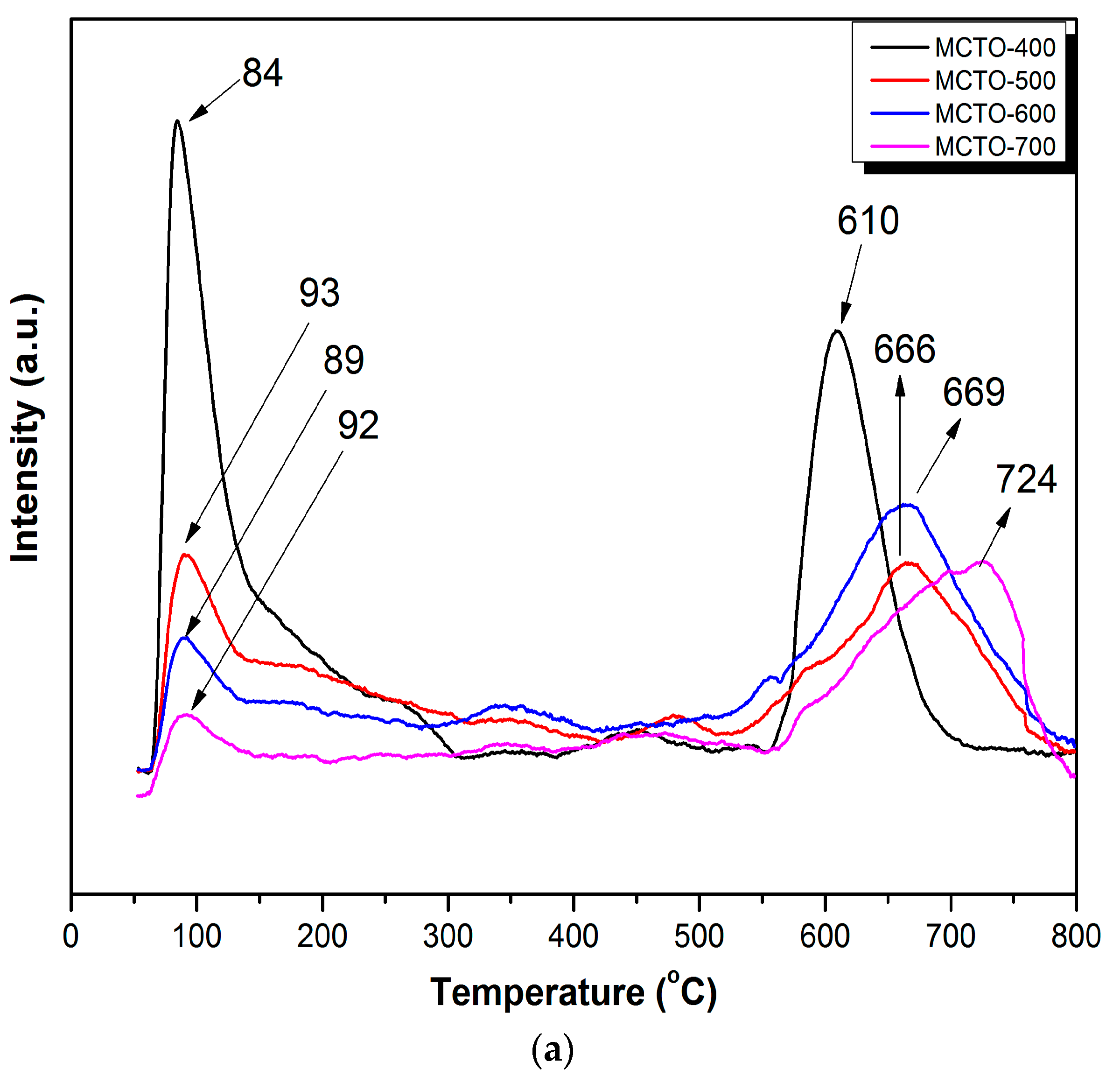
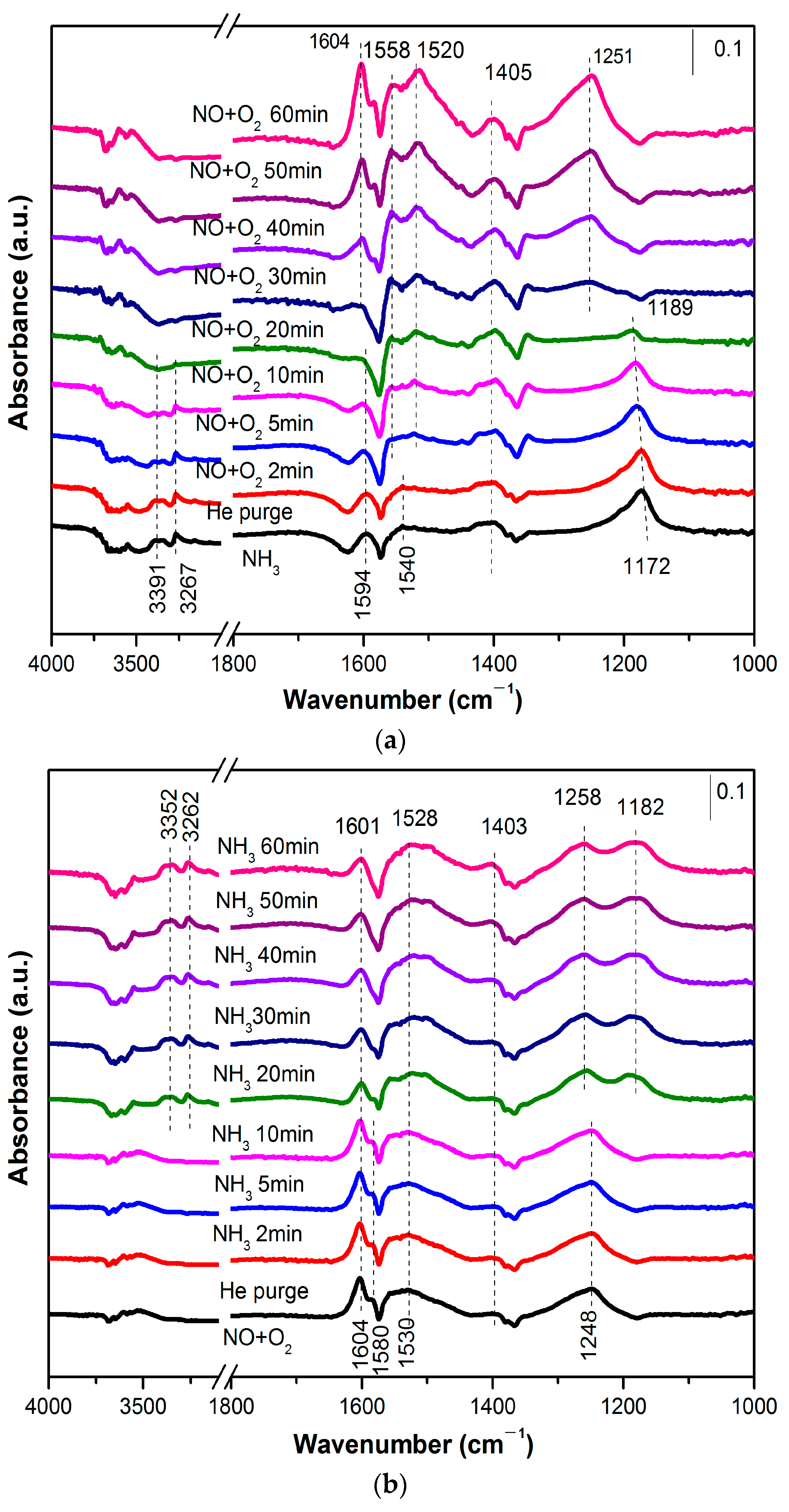
3.1. Catalyst Characterization
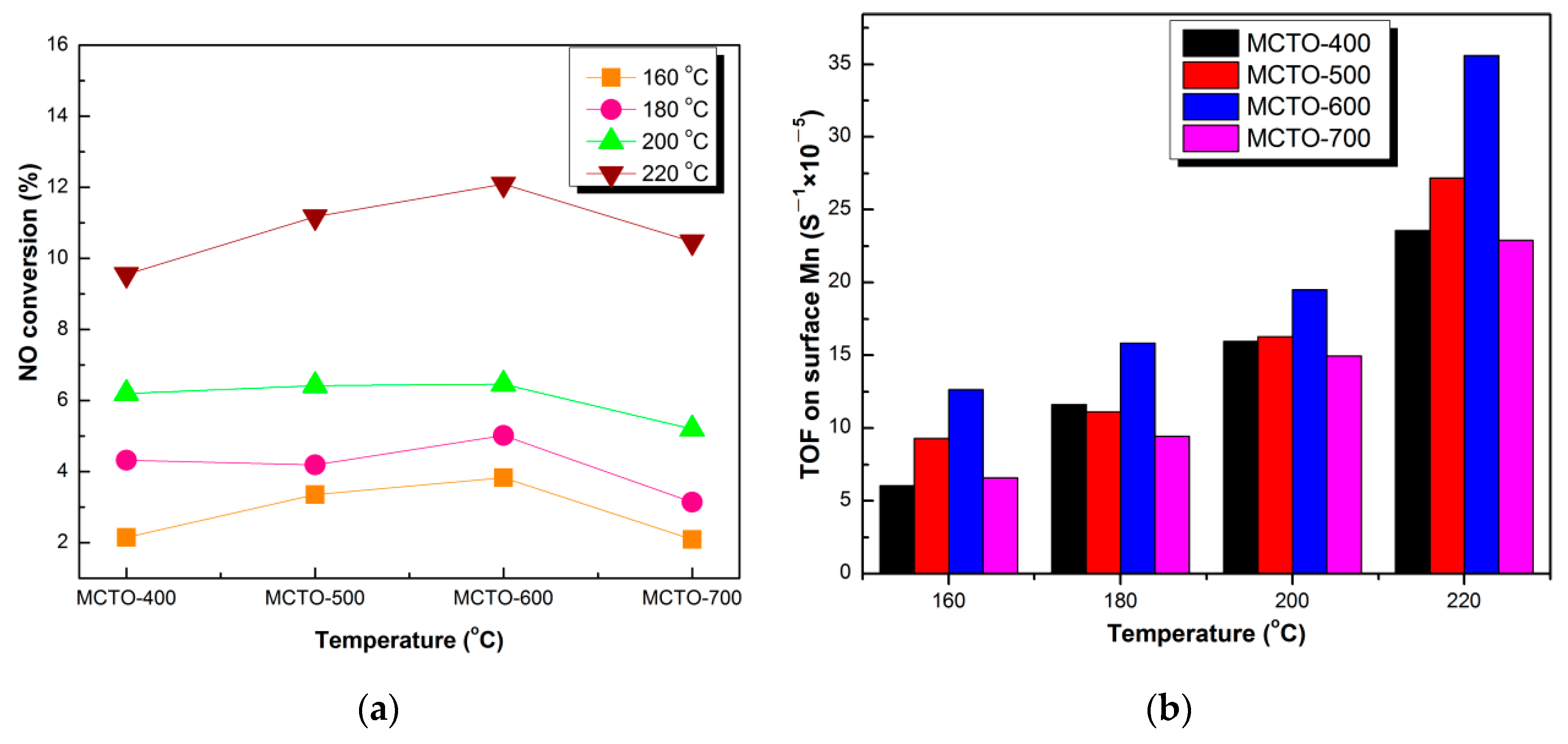
3.2. Catalytic Performance
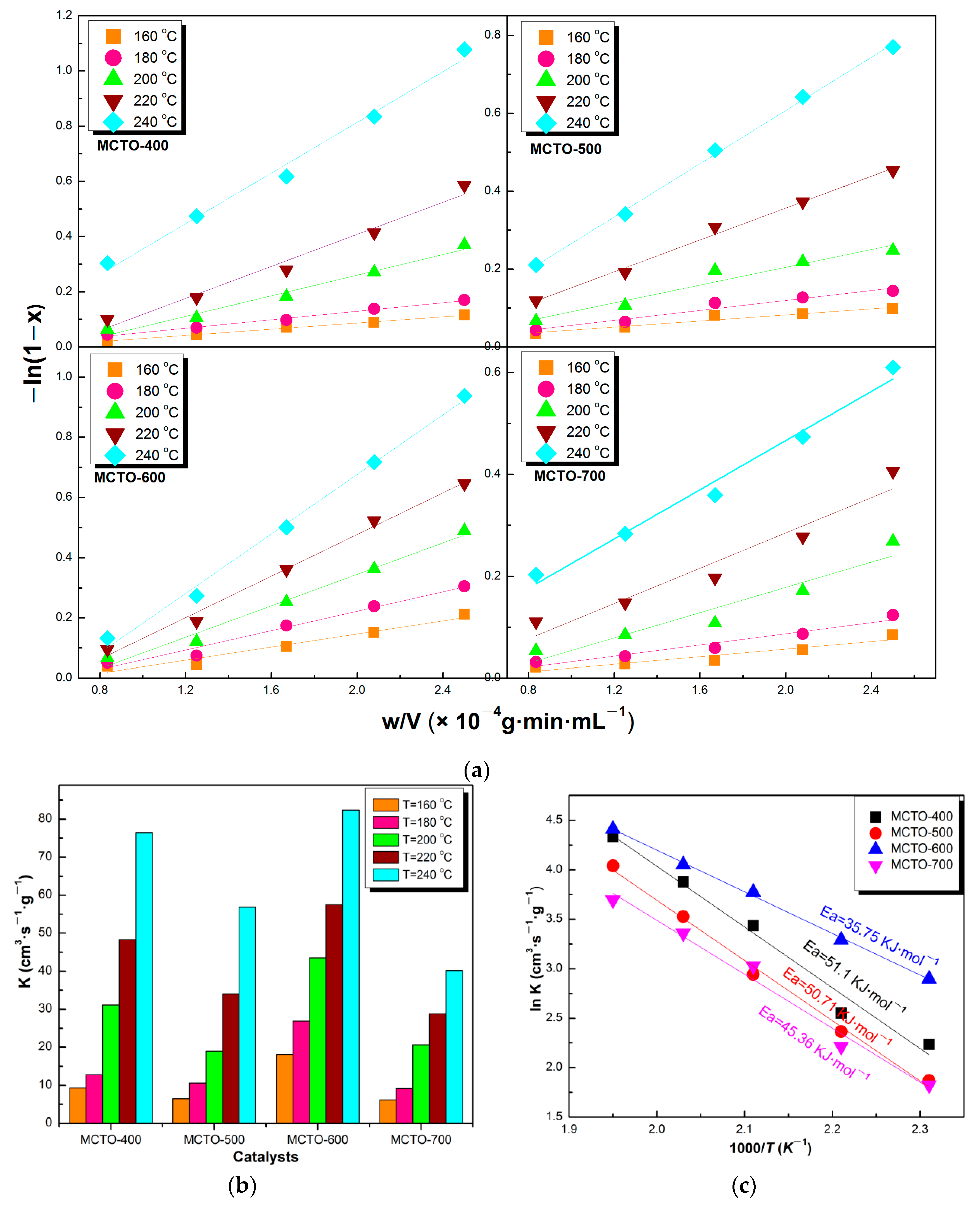
3.3. Kinetics Parameters
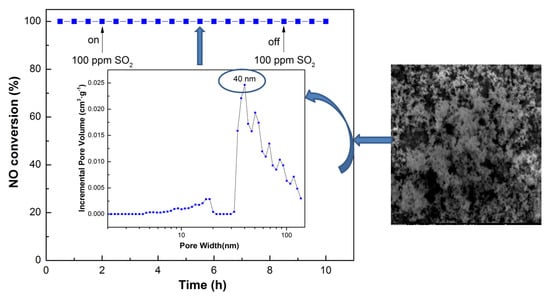
3.4. Influence of SO2 and H2O and Stability Test
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, L.; Zhang, D.; Zhang, J.; Cai, S.; Fang, C.; Huang, L.; Li, H.; Gao, R.; Shi, L. Design of meso-TiO2@MnOx–CeOx/CNTs with a core-shell structure as DeNOx catalysts: Promotion of activity, stability and SO2-tolerance. Nanoscale 2013, 5, 9821–9829. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, X.; Zhao, Q.; Ke, J.; Xiao, H.; Lv, X.; Liu, S.; Tadé, M.; Wang, S. Mechanistic investigation of the enhanced NH3-SCR on cobalt-decorated Ce–Ti mixed oxide: In situ FTIR analysis for structure-activity correlation. Appl. Catal. B Environ. 2017, 200, 297–308. [Google Scholar] [CrossRef]
- Xie, A.; Tang, Y.; Huang, X.; Jin, X.; Gu, P.; Luo, S.; Yao, C.; Li, X. Three-dimensional nanoflower MnCrOx/Sepiolite catalyst with increased SO2 resistance for NH3–SCR at low temperature. Chem. Eng. J. 2019, 370, 897–905. [Google Scholar] [CrossRef]
- Shao, J.; Lin, F.; Li, Y.; Tang, H.; Wang, Z.; Liu, P.; Chen, G. Co-precipitation synthesized MnOx–CeO2 mixed oxides for NO oxidation and enhanced resistance to low concentration of SO2 by metal addition. Catalysts 2019, 9, 519. [Google Scholar] [CrossRef]
- Huang, H.; Long, R.; Yang, R. Kinetics of selective catalytic reduction of NO with NH3 on Fe–ZSM-5 catalyst. Appl. Catal. A Gen. 2002, 235, 241–251. [Google Scholar] [CrossRef]
- Yang, S.; Li, J.; Wang, C.; Chen, J.; Ma, L.; Chang, H.; Chen, L.; Peng, Y.; Yan, N. Fe–Ti spinel for the selective catalytic reduction of NO with NH3: Mechanism and structure-activity relationship. Appl. Catal. B Environ. 2012, 117–118, 73–80. [Google Scholar] [CrossRef]
- Wei, B.; Yang, W.; Wang, J.; Tan, H.; Zhou, S.; Wang, F.; Ma, J. Study on reduction mechanism of Fe2O3 by NH3 under SNCR condition. Fuel 2019, 255, 115814. [Google Scholar] [CrossRef]
- Xi, H.; Zhou, S.; Zhou, J. New experimental results of NO removal from simulated marine engine exhaust gases by Na2S2O8/urea solutions. Chem. Eng. J. 2019, 362, 12–20. [Google Scholar] [CrossRef]
- Song, L.; Ma, K.; Tian, W.; Ji, J.; Liu, C.; Tang, S.; Jiang, W.; Yue, H.; Liang, B. An environmentally friendly FeTiSOx catalyst with a broad operation-temperature window for the NH3-SCR of NOx. AIChE J. 2019, 65, e16684. [Google Scholar] [CrossRef]
- Michalow-Mauke, K.; Lu, Y.; Kowalski, K.; Graule, T.; Nachtegaal, M.; Kröcher, O.; Ferri, D. Flame-Made WO3/CeOx–TiO2 Catalysts for Selective Catalytic Reduction of NOx by NH3. ACS. Catal. 2015, 5, 5657–5672. [Google Scholar] [CrossRef]
- Mu, J.; Li, X.; Sun, W.; Fan, S.; Wang, X.; Wang, L.; Qin, M.; Gan, G.; Yin, Z.; Zhang, D. Enhancement of Low-temperature catalytic activity over a highly dispersed Fe–Mn/Ti catalyst for selective catalytic reduction of NOx with NH3. Ind. Eng. Chem. Res. 2018, 57, 10159–10169. [Google Scholar] [CrossRef]
- Chen, X.; Wang, P.; Fang, P.; Ren, T.; Liu, Y.; Cen, C.; Wang, H.; Wu, Z. Tuning the property of Mn–Ce composite oxides by titanate nanotubes to improve the activity, selectivity and SO2/H2O tolerance in middle temperature NH3–SCR reaction. Fuel Process. Technol. 2017, 167, 221–228. [Google Scholar] [CrossRef]
- Gao, F.; Tang, X.; Yi, H.; Li, J.; Zhao, S.; Wang, J.; Chu, C.; Li, C. Promotional mechanisms of activity and SO2 tolerance of Co-or Ni-doped MnOx–CeO2 catalysts for SCR of NOx with NH3 at low temperature. Chem. Eng. J. 2017, 317, 20–31. [Google Scholar] [CrossRef]
- Leng, X.; Zhang, Z.; Li, Y.; Zhang, T.; Ma, S.; Yuan, F.; Niu, X.; Zhu, Y. Excellent low temperature NH3–SCR activity over MnaCe0.3TiOx (a = 0.1–0.3) oxides: Influence of Mn addition. Fuel Process. Technol. 2018, 181, 33–43. [Google Scholar] [CrossRef]
- Chen, L.; Yuan, F.; Li, Z.; Niu, X.; Zhu, Y. Synergistic effect between the redox property and acidity on enhancing the low temperature NH3–SCR activity for NOx removal over the Co0.2CexMn0.8-xTi10 (x = 0–0.40) oxides catalysts. Chem. Eng. J. 2018, 354, 393–406. [Google Scholar] [CrossRef]
- Ma, M.; Ma, X.; Cui, S.; Liu, T.; Tian, Y.; Wang, Y. Low Temperature NH3–SCR over Mn–Ce Oxides Supported on MCM-41 from Diatomite. Materials 2019, 12, 3654. [Google Scholar] [CrossRef]
- Meng, D.; Zhan, W.; Guo, Y.; Guo, Y.; Wang, L.; Lu, G. A highly effective catalyst of Sm–MnOx for the NH3–SCR of NOx at low temperature: Promotional role of Sm and its catalytic performance. ACS. Catal. 2015, 5, 5973–5983. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, R.; Shi, X.; Liu, X.; Qin, H.; Liu, Y.; Duan, C.; Guo, D.; Pan, W. The superior performance of CoMnOx catalyst with ball-flowerlike structure for low-temperature selective catalytic reduction of NOx by NH3. Chem. Eng. J. 2020, 381, 122753. [Google Scholar] [CrossRef]
- Liu, F.; Shan, W.; Lian, Z.; Xie, L.; Yang, W.; He, H. Novel MnWOx catalyst with remarkable performance for low temperature NH3-SCR of NOx. Catal. Sci. Technol. 2013, 3, 2699–2707. [Google Scholar] [CrossRef]
- Yang, S.; Wang, C.; Li, J.; Yan, N.; Ma, L.; Chang, H. Low temperature selective catalytic reduction of NO with NH3 over Mn–Fe spinel: Performance, mechanism and kinetic study. Appl. Catal. B Environ. 2011, 110, 71–80. [Google Scholar] [CrossRef]
- Wang, F.; Xie, Z.; Liang, J.; Fang, B.; Piao, Y.; Hao, M.; Wang, Z. Tourmaline-modified FeMnTiOx catalysts for improved low temperature NH3–SCR performance. Environ. Sci. Technol. 2019, 53, 6989–6996. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Yang, J.; Zhang, T.; Jiang, L.; Long, H.; Guo, F.; Kong, M. Role of cerium in improving NO reduction with NH3 over Mn–Ce/ASC catalyst in low-temperature flue gas. Chem. Eng. Res. Des. 2018, 133, 1–10. [Google Scholar] [CrossRef]
- Li, Q.; Li, X.; Li, W.; Zhong, L.; Zhang, C.; Fang, Q.; Chen, G. Effect of preferential exposure of anatase TiO2{001} facets on the performance of Mn–Ce/TiO2 catalysts for low-temperature selective catalytic reduction of NOx with NH3. Chem. Eng. J. 2019, 369, 26–34. [Google Scholar] [CrossRef]
- Jin, R.; Liu, Y.; Wang, Y.; Cen, W.; Wu, Z.; Wang, H.; Weng, X. The role of cerium in the improved SO2 tolerance for NO reduction with NH3 over Mn–Ce/TiO2 catalyst at low temperature. Appl. Catal. B Environ. 2014, 148–149, 582–588. [Google Scholar] [CrossRef]
- Han, L.; Cai, S.; Gao, M.; Hasegawa, J.Y.; Wang, P.; Zhang, J.; Shi, L.; Zhang, D. Selective Catalytic Reduction of NOx with NH3 by using novel catalysts: State of the art and future prospects. Chem. Rev. 2019, 119, 10916–10976. [Google Scholar] [CrossRef]
- Ma, D.; Yang, L.; Huang, B.; Wang, L.; Wang, X.; Sheng, Z.; Dong, F. MnOx–CeO2@TiO2 core-shell composites for low temperature SCR of NOx. New J. Chem. 2019, 43, 15161–15168. [Google Scholar] [CrossRef]
- Chang, H.; Chen, X.; Li, J.; Ma, L.; Wang, C.; Liu, C.; Schwank, J.W.; Hao, J. Improvement of activity and SO2 tolerance of Sn-Modified MnOx–CeO2 Catalysts for NH3–SCR at Low Temperatures. Environ. Sci. Technol. 2013, 47, 5294–5301. [Google Scholar] [CrossRef]
- Yu, J.; Guo, F.; Wang, Y.; Zhu, J.; Liu, Y.; Su, F.; Gao, S.; Xu, G. Sulfur poisoning resistant mesoporous Mn-base catalyst for low-temperature SCR of NO with NH3. Appl. Catal. B Environ. 2010, 95, 160–168. [Google Scholar] [CrossRef]
- Soh, B.W.; Nam, I.S. Effect of support morphology on the sulfur tolerance of V2O5/Al2O3 catalyst for the reduction of NO by NH3. Ind. Eng. Chem. Res. 2003, 42, 2975–2986. [Google Scholar] [CrossRef]
- Guo, K.; Fao, G.; Gu, D.; Yu, S.; Ma, K.; Liu, A.; Tan, W.; Wang, J.; Du, X.; Zhou, W.; et al. Pore size expansion accelerates ammonium bisulfate decomposition for improved sulfur resistance in low-temperature NH3–SCR. ACS Appl. Mater. Interfaces. 2019, 11, 4900–4907. [Google Scholar] [CrossRef]
- Ma, K.; Guo, K.; Li, L.; Zou, W.; Tang, C.; Dong, L. Cavity size dependent SO2 resistance for NH3–SCR of hollow structured CeO2–TiO2 catalysts. Catal. Commun. 2019, 128, 105719. [Google Scholar] [CrossRef]
- Lee, S.M.; Park, K.H.; Hong, S.C. MnOx/CeO2–TiO2 mixed oxide catalysts for the selective catalytic reduction of NO with NH3 at low temperature. Chem. Eng. J. 2012, 195–196, 323–331. [Google Scholar] [CrossRef]
- Sheng, Z.; Hu, Y.; Xue, J.; Wang, X.; Liao, W. SO2 poisoning and regeneration of Mn–Ce/TiO2 catalyst for low temperature NOx reduction with NH3. J. Rare Earths 2012, 30, 676–682. [Google Scholar] [CrossRef]
- Peng, Y.; Li, K.; Li, J. Identification of the active sites on CeO2–WO3 catalysts for SCR of NOx with NH3: An in-situ IR and Raman spectroscopy study. Appl. Catal. B Environ. 2013, 140–141, 483–492. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, L.; Shi, L.; Fang, C.; Li, H.; Gao, R.; Huang, L.; Zhang, J. In situ supported MnOx–CeOx on carbon nanotubes for the low-temperature selective catalytic reduction of NO with NH3. Nanoscale 2013, 5, 1127–1136. [Google Scholar] [CrossRef]
- Han, L.; Gao, M.; Hasegawa, J.; Li, S.; Shen, Y.; Li, H.; Shi, L.; Zhang, D. SO2–tolerant selective catalytic reduction of NOx over Meso–TiO2@Fe2O3@Al2O3 Metal-Based Monolith catalysts. Environ. Sci. Technol. 2019, 53, 6462–6473. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, Z.; Du, Y.; Wu, X.; She, H.; Jiang, G. Atomic-scale insights into the nature of active sites in Fe2O3-supported submonolayer WO3 catalysts for selective catalytic reduction of NO with NH3. Chem. Eng. J. 2020, 381, 122668. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, Q.; Ran, G.; Kong, M.; Ren, S.; Yang, J.; Li, J. V2O5–modified Mn–Ce/AC catalyst with high SO2 tolerance for low temperature NH3-SCR of NO. Chem. Eng. J. 2019, 370, 810–821. [Google Scholar] [CrossRef]
- Xiao, X.; Sheng, Z.; Yang, L.; Dong, F. Low-temperature selective catalytic reduction of NOx with NH3 over a manganese and cerium oxide/graphene composite prepared by a hydrothermal method. Catal. Sci. Technol. 2016, 6, 1507–1514. [Google Scholar] [CrossRef]
- Ye, B.; Lee, M.; Jeong, B.; Kim, J.; Lee, D.H.; Baik, J.M.; Kim, H.D. Partially reduced graphene oxide as a support of Mn–Ce/TiO2 catalyst for selective catalytic reduction of NOx with NH3. Catal. Today 2019, 328, 300–306. [Google Scholar] [CrossRef]
- Liu, F.; He, H.; Ding, Y.; Zhang, C. Effect of manganese substitution on the structure and activity of iron titanate catalyst for the selective catalytic reduction of NO with NH3. Appl. Catal. B Environ. 2009, 93, 194–204. [Google Scholar] [CrossRef]
- Koebel, M.; Elsener, M.; Madia, G. Reaction pathways in the selective catalytic reduction process with NO and NO2 at low temperatures. Ind. Eng. Chem. Res. 2001, 40, 52–59. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.; Li, X.; Tan, P.; Zhou, A.; Fang, Q.; Chen, G. Ho-modified Mn–Ce/TiO2 for low-temperature SCR of NOx with NH3: Evaluation and characterization. Chin. J. Catal. 2018, 39, 1653–1663. [Google Scholar] [CrossRef]
- Zhang, L.; Shi, L.; Huang, L.; Zhang, J.; Gao, R.; Zhang, D. Rational design of high-performance DeNOx catalysts bsed on MnxCo3–xO4 nanocages derived from Metal-Organic Frameworks. ACS Catal. 2014, 4, 1753–1763. [Google Scholar] [CrossRef]
- Li, C.; Tang, X.; Yi, H.; Wang, L.; Cui, X.; Chu, C.; Li, J.; Zhang, R.; Yu, Q. Rational design of template-free MnOx–CeO2 hollow nanotube as de-NOx catalyst at low temperature. Appl. Surf. Sci. 2018, 428, 924–932. [Google Scholar] [CrossRef]
- Marbán, G.; Solís, T.V.; Fuertes, A.B. Mechanism of low-temperature selective catalytic reduction of NO with NH3 over carbon-supported Mn3O4: Role of surface NH3 species: SCR mechanism. J. Catal. 2004, 226, 138–155. [Google Scholar] [CrossRef]
- Jiang, C.; Zhang, X.; Jia, D.; Song, L. Performance of Hβ zeolites modified by Ce3+ on synthesis of ethyl anthrquinone. J. China. Univ. Petrol. Nat. Sci. 2015, 39, 151–156. [Google Scholar]
- Liu, Z.; Zhang, S.; Li, J.; Ma, L. Promoting effect of MoO3 on the NOx reduction by NH3 over CeO2/TiO2 catalyst studied with in situ DRIFTS. Appl. Catal. B Environ. 2014, 144, 90–95. [Google Scholar] [CrossRef]
- Ding, S.; Liu, F.; Shi, X.; He, H. Promotional effect of Nb additive on the activity and hydrothermal stability for the selective catalytic reduction of NOx with NH3 over CeZrOx catalyst. Appl. Catal. B Environ. 2016, 180, 766–774. [Google Scholar] [CrossRef]
- Liu, L.; Su, S.; Xu, K.; Li, H.; Qing, M.; Hu, S.; Wang, Y.; Xiang, J. Insights into the highly efficient Co modified MnSm/Ti catalyst for selective catalytic reduction of NOx with NH3 at low temperature. Fuel 2019, 255, 115798. [Google Scholar] [CrossRef]
- Chen, L.; Li, J.; Ge, M.; Ma, L.; Chang, H. Mechanism of Selective Catalytic Reduction of NOx with NH3 over CeO2-WO3 Catalysts. Chin. J. Catal. 2011, 32, 836–841. [Google Scholar] [CrossRef]
- Marban, G.; Fuertes, A.B. Kinetics of the low-temperature selective catalytic reduction of NO with NH3 over activated carbon fiber composite-supported iron oxides. Catal. Lett. 2002, 84, 13–19. [Google Scholar] [CrossRef]
- Madia, G.; Koebel, M.; Elsener, M.; Wokaun, A. Side Reactions in the Selective Catalytic Reduction of NOx with Various NO2 Fractions. Ind. Eng. Chem. Res. 2002, 41, 4008–4015. [Google Scholar] [CrossRef]
- Grossale, A.; Nova, I.; Tronconi, E.; Chatterjee, D.; Weibel, M. The chemistry of the NO/NO2–NH3 “fast” SCR reaction over Fe-ZSM5 investigated by transient reaction analysis. J. Catal. 2008, 256, 312–322. [Google Scholar] [CrossRef]
- Wang, F.; Shen, B.; Zhu, S.; Wang, Z. Promotion of Fe and Co doped Mn-Ce/TiO2 catalysts for low temperature NH3-SCR with SO2 tolerance. Fuel 2019, 249, 54–60. [Google Scholar] [CrossRef]
- Ha, L.; Gao, M.; Feng, C.; Shi, L.; Zhang, D. Fe2O3-CeO2@Al2O3 nanoarrays on Al–Mesh as SO2–tolerant monolith catalysts for NOx reduction by NH3. Environ. Sci. Technol. 2019, 53, 5946–5956. [Google Scholar]
- Liu, F.; He, H.; Lian, Z.; Shan, W.; Xie, L.; Asakura, K.; Wang, W.; Deng, H. Highly dispersed iron vanadate catalyst supported on TiO2 for the selective catalytic reduction of NOx with NH3. J. Catal. 2013, 307, 340–351. [Google Scholar] [CrossRef]
- Yao, X.; Zhao, R.; Chen, L.; Du, J.; Tao, C.; Yang, F.; Dong, L. Selective catalytic reduction of NOx by NH3 over CeO2 supported on TiO2: Comparison of anatase, brookite, and rutile. Appl. Catal. B Environ. 2017, 208, 82–93. [Google Scholar] [CrossRef]
- Yu, S.; Lu, Y.; Cao, Y.; Wang, J.; Sun, B.; Gao, F.; Tang, C.; Dong, L. Composite catalytic systems: A strategy for developing the low temperature NH3–SCR catalysts with satisfactory SO2 and H2O tolerance. Catal. Today 2019, 327, 235–245. [Google Scholar] [CrossRef]
- Bordeje, E.G.; Pinilla, J.L.; Lazaro, M.J.; Moliner, R. NH3–SCR of NO at low temperatures over sulphated vanadia on carbon coated monoliths: Effect of H2O and SO2 traces in the gas feed. Appl. Catal. B Environ. 2006, 66, 281–287. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | DTiO2 (nm) | DCeO2 (nm) | DMn2O3 (nm) | DMn3O4 (nm) |
|---|---|---|---|---|
| MCTO-0 | 7.8 | - | - | - |
| MCTO-400 | 9.4 | - | 6.8 | 15.3 |
| MCTO-500 | 11.5 | - | 12.5 | 20.8 |
| MCTO-600 | 16.2 | 8.0 | 13.0 | 21.5 |
| MCTO-700 | 21.5 | 13.6 | 17.8 | 38.8 |
| Catalyst | Surface Area (m2·g−1) | Mean Pore Diameter (nm) | Pore Volume (cm3·g−1) |
|---|---|---|---|
| MCTO-400 | 127.1 | 14 | 0.44 |
| MCTO-500 | 71.6 | 22 | 0.38 |
| MCTO-600 | 35.8 | 32 | 0.29 |
| MCTO-700 | 18.9 | 20 | 0.095 |
| Sample | Atomic Composition (%) | Oα/(Oα + Oβ) (%) | Ce3+/Ce (%) | Mn4+/Mn (%) | Mn3+/Mn (%) | Mn2+/Mn (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mn | Ce | Ti | C | O | ||||||
| MCTO-400 | 5.4 | 1.8 | 16.4 | 13.3 | 63.2 | 57.2 | 17.5 | 25.9 | 31.3 | 42.8 |
| MCTO-500 | 5.5 | 1.3 | 23.1 | 6.6 | 63.5 | 34.9 | 17.9 | 27.9 | 30.6 | 41.5 |
| MCTO-600 | 4.6 | 1.7 | 25.3 | 5.1 | 63.2 | 41.3 | 24.0 | 29.3 | 29.9 | 40.8 |
| MCTO-700 | 4.9 | 2.4 | 24.9 | 2.3 | 65.6 | 29.0 | 13.8 | 24.3 | 30.5 | 45.3 |
| Catalysts | Average Pore Size | Preparation Methods | Reaction Conditions | NO Conversion Before and After Introducing SO2 | Refs |
|---|---|---|---|---|---|
| MnOx–CeO2@TiO2 | 13.3 nm | three-step method | T = 180 °C, [NO] = [NH3] = 500 ppm, [O2] = 5%, [SO2] = 200 ppm, GHSV = 24,000 h−1 | decline from 100% to 70% | [26] |
| Mn–Ce/TiO2 | 9.0 nm | co-precipitation method | T = 120 °C, [NO] = [NH3] = 600 ppm, [O2] = 3%, [SO2] = 700 ppm, GHSV = 40,000 h−1 | decline from 92.5% to 34.6% | [33] |
| Mn–Ce/TiO2 | 6.7 nm | sol–gel method | T = 150 °C, [NO] = [NH3] = 800 ppm, [O2] = 3%, [SO2] = 100 ppm, GHSV = 40,000 h−1 | decline from 100% to 60% | [24] |
| MnOx–CeO2/TiO2 | 13.7 nm | one-step hydrothermal method | T = 180 °C, [NO] = [NH3] = 500 ppm, [O2] = 5%, [SO2] = 200 ppm, GHSV = 24,000 h−1 | decline from 100% to 47% | [26] |
| MnOx/CeO2–TiO2 | 17.3 nm | wet impregnation | T = 180 °C, [NO] = 200 ppm, [NH3] = 220 ppm, [O2] = 8%, [SO2] = 100 ppm, GHSV = 60,000 h−1 | decline from 84% to 62% | [32] |
| MnCe/TNTs | 13.6 nm | hydrothermal method | T = 150 °C, [NO] = 720 ppm, [NH3] = 800 ppm, [O2] = 3%, [SO2] = 100 ppm, GHSV = 100,000 h−1 | decline from 92% to 84% | [12] |
| Mn–Ce–Ti–O composite aerogels | 32.5 nm | one-pot sol–gel method | T = 200 °C, [NO] = [NH3] = 500 ppm, [O2] = 5%, [SO2] = 100 ppm, GHSV = 14,400 h−1 | maintaining at 100% | in this work |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Y.; Jin, S.; Zhang, R.; Li, W.; Wang, J.; Yang, S.; Wang, H.; Yang, M.; Liu, Y.; Qiao, W.; et al. Preparation of Mesoporous Mn–Ce–Ti–O Aerogels by a One-Pot Sol–Gel Method for Selective Catalytic Reduction of NO with NH3. Materials 2020, 13, 475. https://doi.org/10.3390/ma13020475
Wei Y, Jin S, Zhang R, Li W, Wang J, Yang S, Wang H, Yang M, Liu Y, Qiao W, et al. Preparation of Mesoporous Mn–Ce–Ti–O Aerogels by a One-Pot Sol–Gel Method for Selective Catalytic Reduction of NO with NH3. Materials. 2020; 13(2):475. https://doi.org/10.3390/ma13020475
Chicago/Turabian StyleWei, Yabin, Shuangling Jin, Rui Zhang, Weifeng Li, Jiangcan Wang, Shuo Yang, He Wang, Minghe Yang, Yan Liu, Wenming Qiao, and et al. 2020. "Preparation of Mesoporous Mn–Ce–Ti–O Aerogels by a One-Pot Sol–Gel Method for Selective Catalytic Reduction of NO with NH3" Materials 13, no. 2: 475. https://doi.org/10.3390/ma13020475
APA StyleWei, Y., Jin, S., Zhang, R., Li, W., Wang, J., Yang, S., Wang, H., Yang, M., Liu, Y., Qiao, W., Ling, L., & Jin, M. (2020). Preparation of Mesoporous Mn–Ce–Ti–O Aerogels by a One-Pot Sol–Gel Method for Selective Catalytic Reduction of NO with NH3. Materials, 13(2), 475. https://doi.org/10.3390/ma13020475