Relationship between the Behavior of Hydrogen and Hydrogen Bubble Nucleation in Vanadium




Abstract
1. Introduction
2. Computational Details
3. Result and Discussion
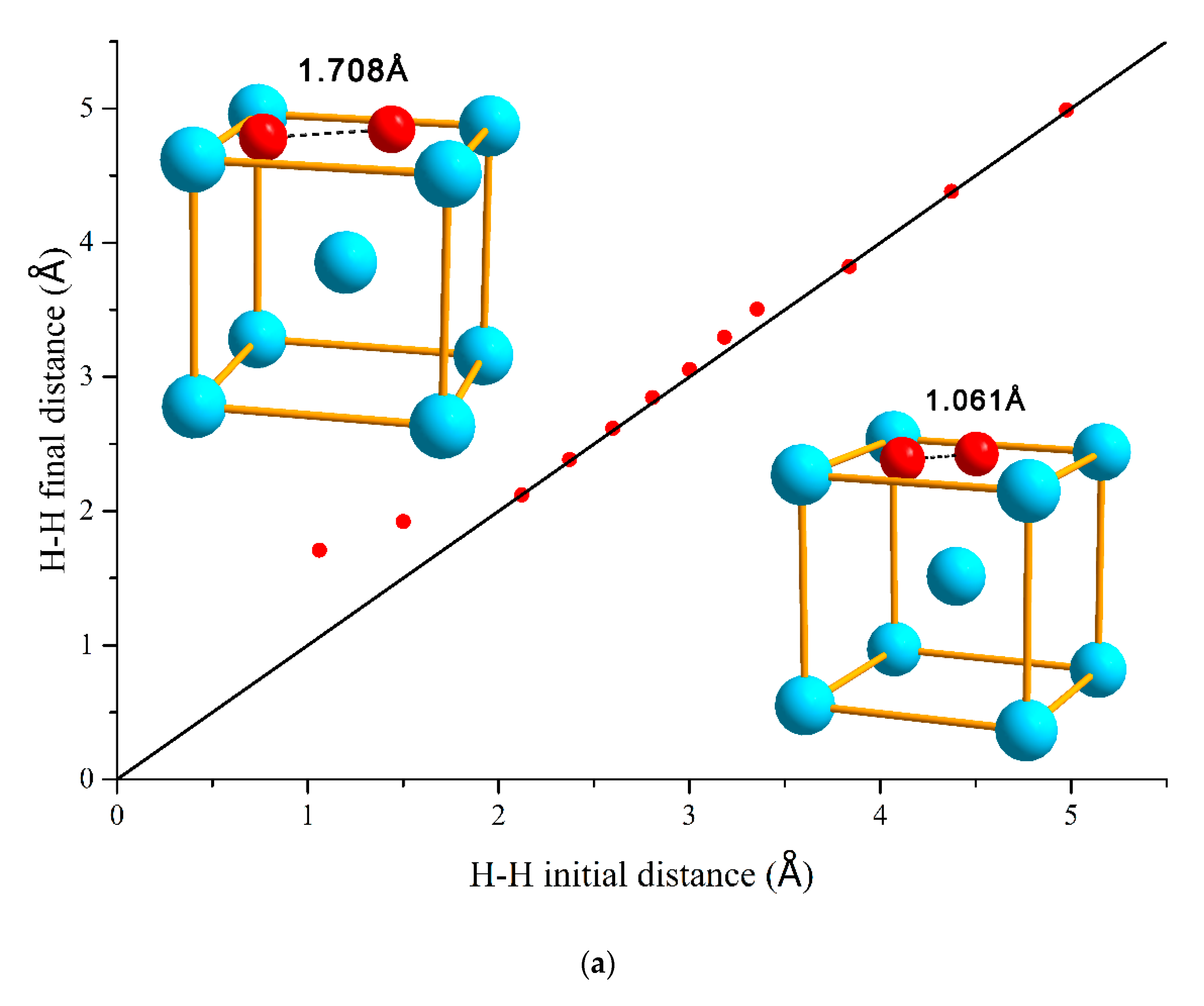
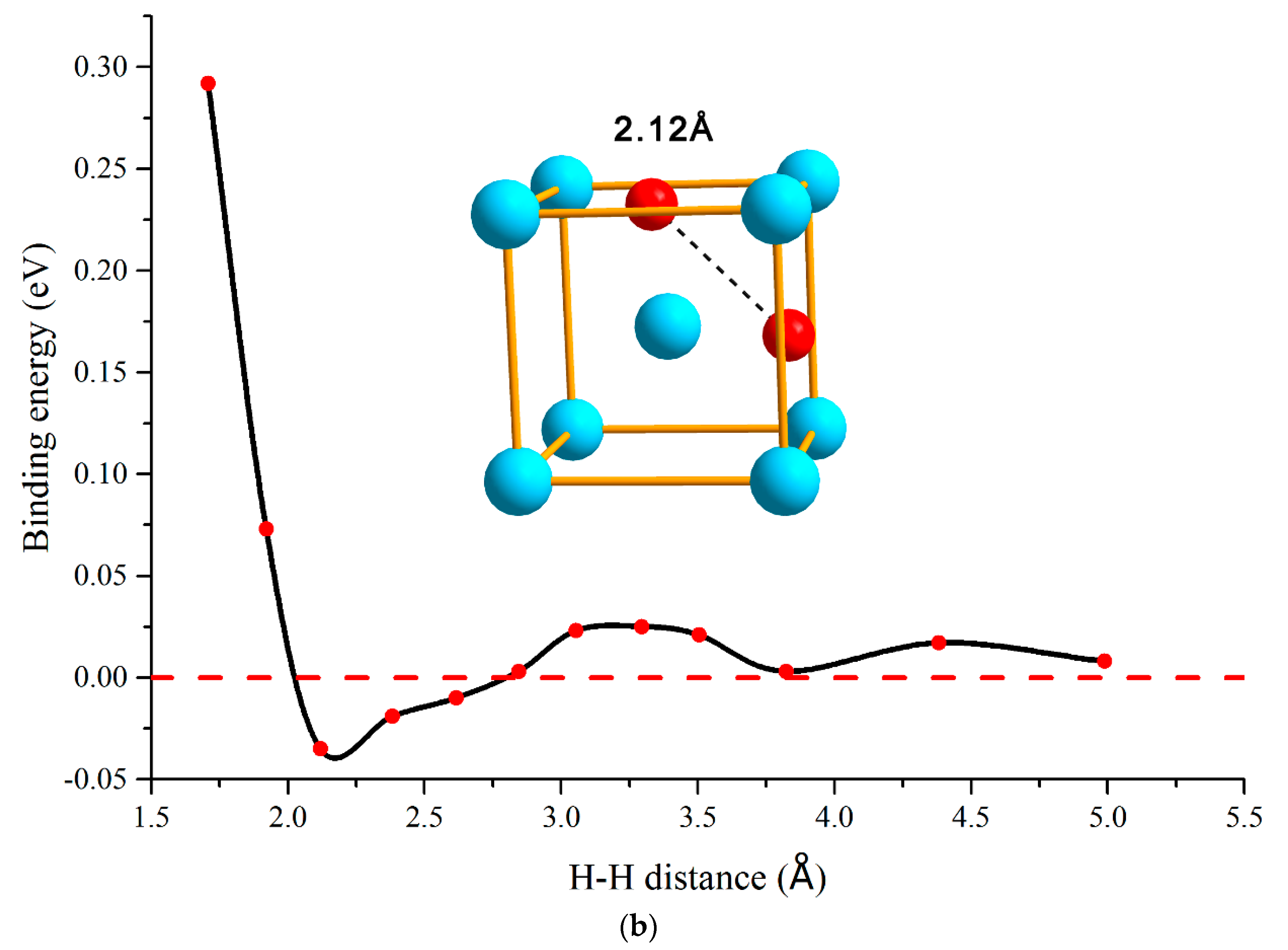
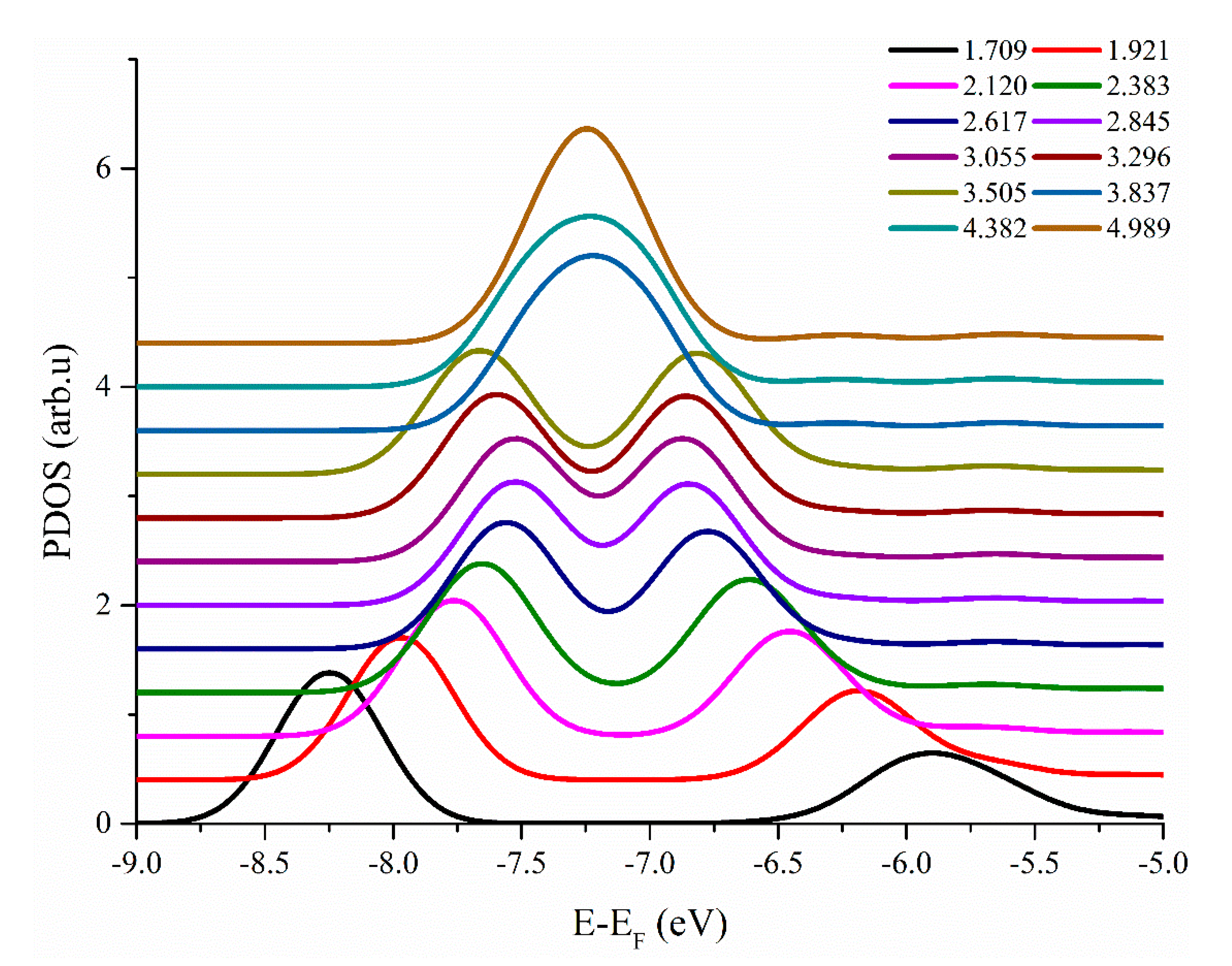
3.1. H–H Interaction
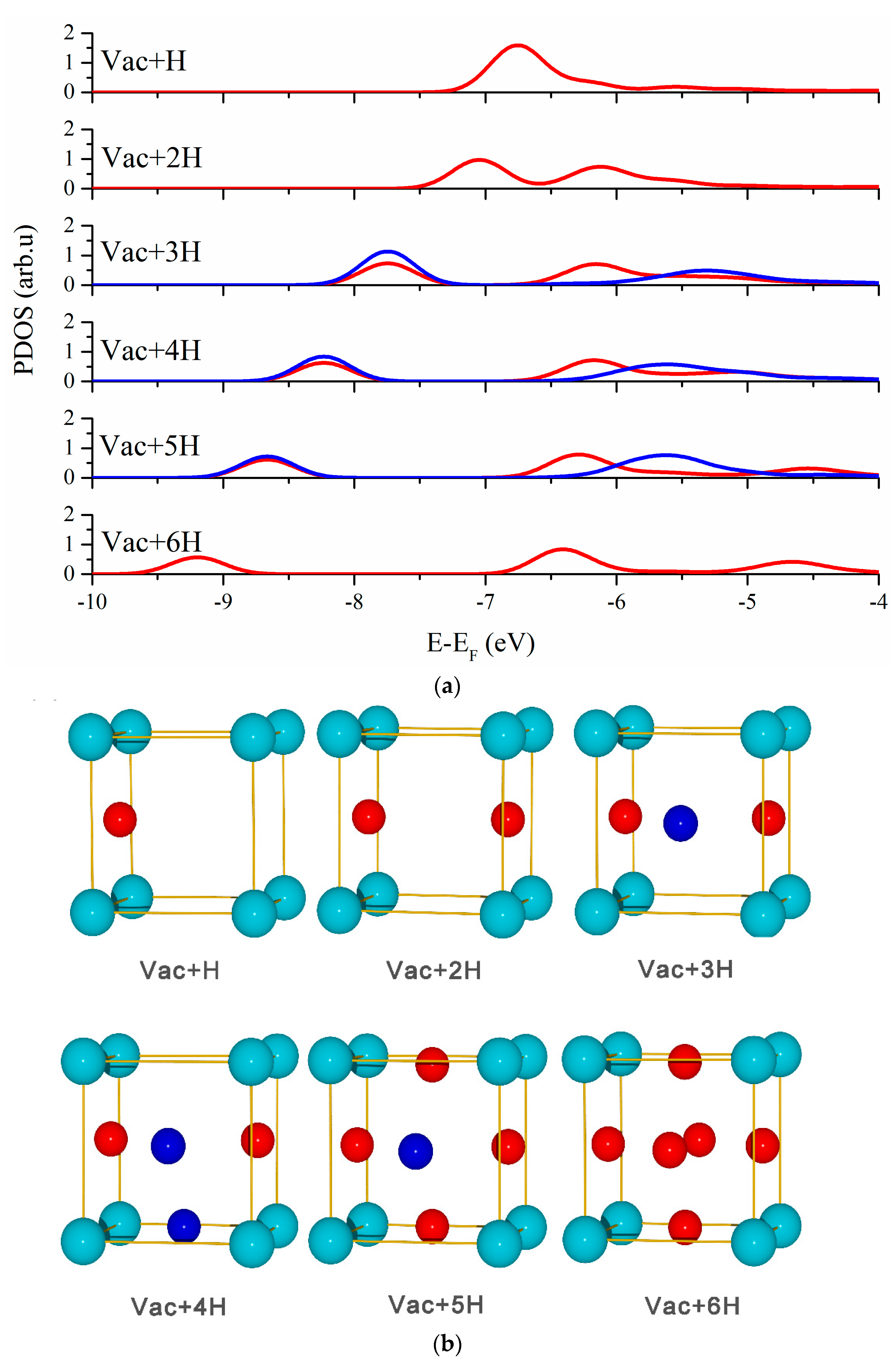
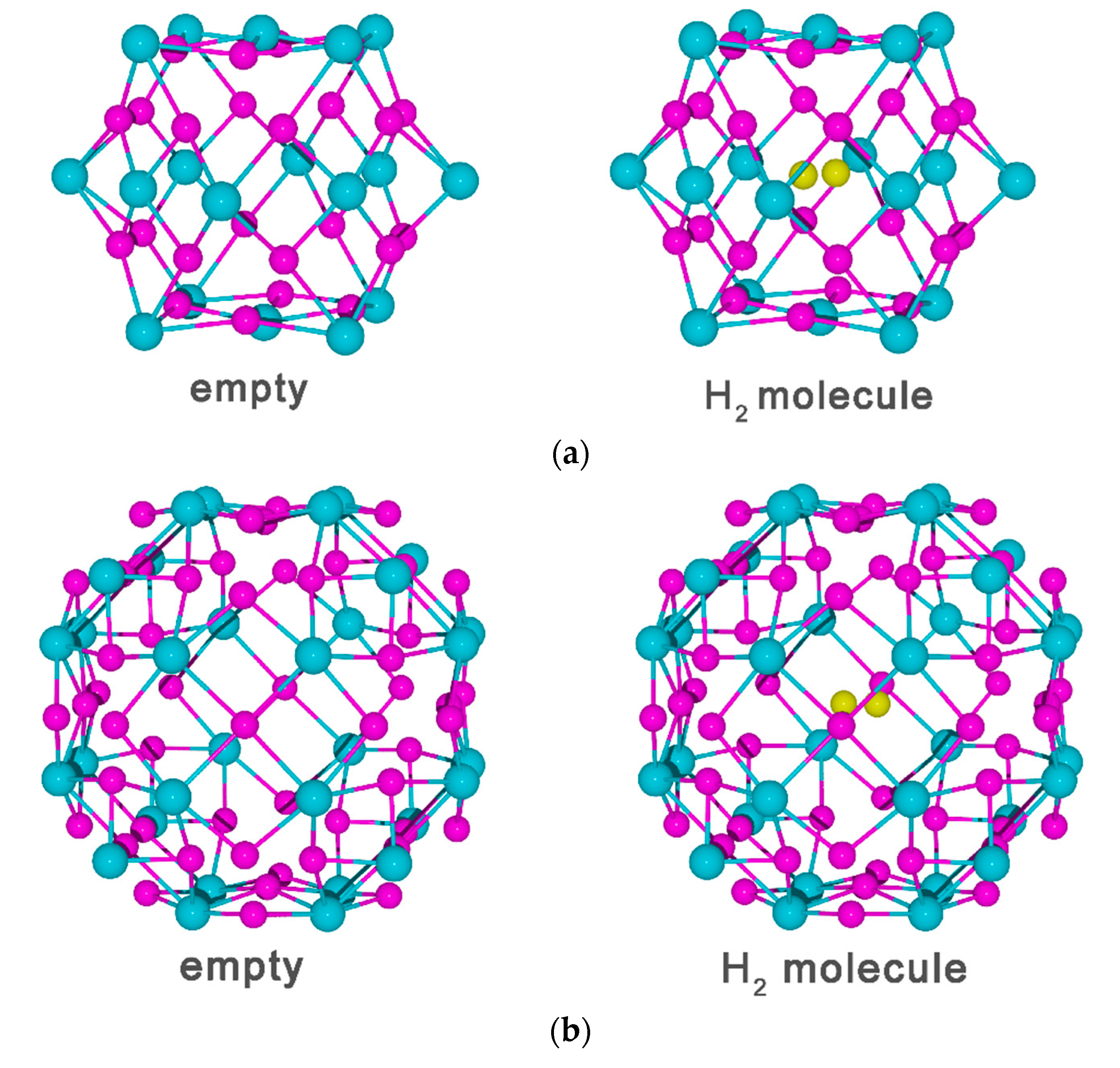
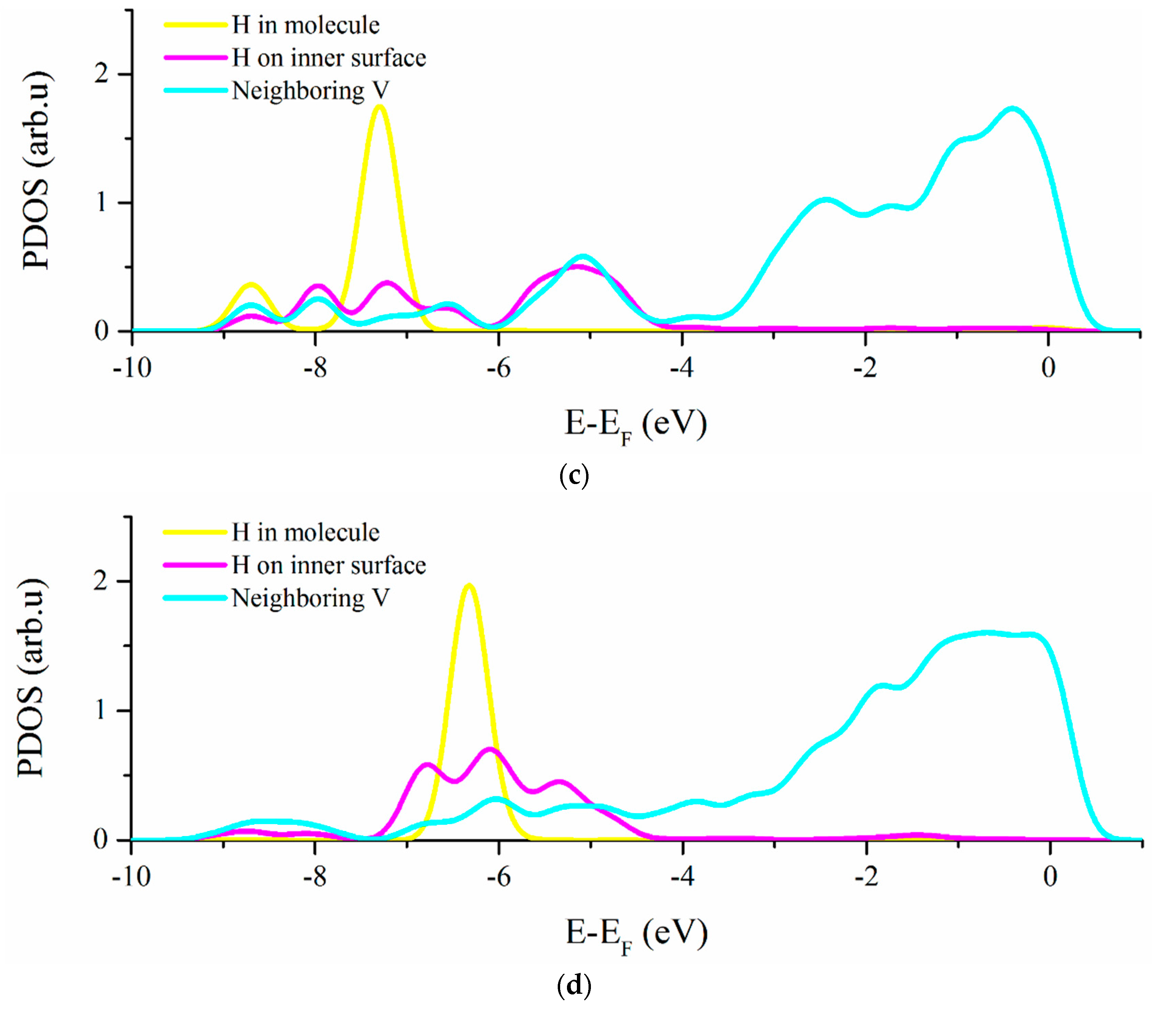
3.2. Vacancy-H Interaction
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Satou, M.; Abe, K.; Kayano, H. High-temperature deformation of modified V-Ti-Cr-Si type alloys. J. Nucl. Mater. 1991, 179, 757–761. [Google Scholar] [CrossRef]
- Rowcliffe, A.; Zinkle, S.; Hoelzer, D. Effect of strain rate on the tensile properties of unirradiated and irradiated V–4Cr–4Ti. J. Nucl. Mater. 2000, 283, 508–512. [Google Scholar] [CrossRef]
- Kurtz, R.J.; Hamilton, M.L. Biaxial thermal creep of V–4Cr–4Ti at 700 C and 800 C. J. Nucl. Mater. 2000, 283, 628–632. [Google Scholar] [CrossRef]
- Dyomina, E.; Fenici, P.; Kolotov, V.; Zucchetti, M. Low-activation characteristics of V-alloys and SiC composites. J. Nucl. Mater. 1998, 258, 1784–1790. [Google Scholar] [CrossRef]
- Jones, R.H.; Heinisch, H.L.; McCarthy, K. Low activation materials. J. Nucl. Mater. 1999, 271, 518–525. [Google Scholar] [CrossRef]
- Taylor, N.; Forty, C.; Petti, D.; McCarthy, K. The impact of materials selection on long-term activation in fusion power plants. J. Nucl. Mater. 2000, 283, 28–34. [Google Scholar] [CrossRef]
- Bloom, E.; Conn, R.; Davis, J.; Gold, R.; Little, R.; Schultz, K.; Smith, D.; Wiffen, F. Low activation materials for fusion applications. J. Nucl. Mater. 1984, 122, 17–26. [Google Scholar] [CrossRef]
- Barabash, V.; Peacock, A.; Fabritsiev, S.; Kalinin, G.; Zinkle, S.; Rowcliffe, A.; Rensman, J.W.; Tavassoli, A.A.; Marmy, P.; Karditsas, P.J.; et al. Materials challenges for ITER—Current status and future activities. J. Nucl. Mater. 2007, 367–370, 21–32. [Google Scholar] [CrossRef]
- Sun, Y.; Peng, Q.; Lu, G. Quantum mechanical modeling of hydrogen assisted cracking in aluminum. Phys. Rev. B 2013, 88, 104109. [Google Scholar] [CrossRef]
- Jia, Y.Z.; Liu, W.; Xu, B.; Qu, S.L.; Shi, L.Q.; Morgan, T.W. Subsurface deuterium bubble formation in W due to low-energy high flux deuterium plasma exposure. Nucl. Fusion 2017, 57, 034003. [Google Scholar] [CrossRef]
- Garner, F.A.; Simonen, E.P.; Oliver, B.M.; Greenwood, L.R.; Grossbeck, M.; Wolfer, W.; Scott, P. Retention of hydrogen in fcc metals irradiated at temperatures leading to high densities of bubbles or voids. J. Nucl. Mater. 2006, 356, 122–135. [Google Scholar] [CrossRef]
- Aoyagi, K.; Torres, E.; Suda, T.; Ohnuki, S. Effect of hydrogen accumulation on mechanical property and microstructure of V–Cr–Ti alloys. J. Nucl. Mater. 2000, 283, 876–879. [Google Scholar] [CrossRef]
- Mukouda, I.; Shimomura, Y.; Yamaki, D.; Nakazawa, T.; Aruga, T.; Jitsukawa, S. Microstructure in vanadium irradiated by simultaneous multi-ion beam of hydrogen, helium and nickel ions. J. Nucl. Mater. 2002, 307, 412–415. [Google Scholar] [CrossRef]
- Myers, S.; Richards, P.; Wampler, W.; Besenbacher, F. Ion-beam studies of hydrogen-metal interactions. J. Nucl. Mater. 1989, 165, 9–64. [Google Scholar] [CrossRef]
- Zibrov, M.; Ryabtsev, S.; Gasparyan, Y.; Pisarev, A. Experimental determination of the deuterium binding energy with vacancies in tungsten. J. Nucl. Mater. 2016, 477, 292–297. [Google Scholar] [CrossRef]
- Zhu, X.-L.; Zhang, Y.; Cheng, L.; Yuan, Y.; De Temmerman, G.; Wang, B.-Y.; Cao, X.-Z.; Lu, G.-H. Deuterium occupation of vacancy-type defects in argon-damaged tungsten exposed to high flux and low energy deuterium plasma. Nucl. Fusion 2016, 56, 036010. [Google Scholar] [CrossRef]
- Troev, T.; Markovski, A.; Petrova, M.; Peneva, S.; Yoshiie, T. Positron lifetime calculations of defects in vanadium containing hydrogen. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2006, 248, 297–304. [Google Scholar] [CrossRef]
- Xie, D.G.; Wang, Z.J.; Sun, J.; Li, J.; Ma, E.; Shan, Z.W. In situ study of the initiation of hydrogen bubbles at the aluminium metal/oxide interface. Nat. Mater. 2015, 14, 899–903. [Google Scholar] [CrossRef]
- Dudarev, S.L. Density Functional Theory Models for Radiation Damage. Annu. Rev. Mater. Res. 2013, 43, 35–61. [Google Scholar] [CrossRef]
- Fu, C.-C.; Torre, J.D.; Willaime, F.; Bocquet, J.-L.; Barbu, A. Multiscale modelling of defect kinetics in irradiated iron. Nat. Mater. 2004, 4, 68–74. [Google Scholar] [CrossRef]
- Domain, C.; Becquart, C.S.; Foct, J. Ab initiostudy of foreign interstitial atom (C, N) interactions with intrinsic point defects in α-Fe. Phys. Rev. B 2004, 69, 144112. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Zhang, Y.; Zhou, H.-B.; Lu, G.-H.; Liu, F.; Luo, G.N. Vacancy trapping mechanism for hydrogen bubble formation in metal. Phys. Rev. B 2009, 79, 172103. [Google Scholar] [CrossRef]
- Lu, G.; Kaxiras, E. Hydrogen embrittlement of aluminum: The crucial role of vacancies. Phys. Rev. Lett. 2005, 94, 155501. [Google Scholar] [CrossRef] [PubMed]
- Zepeda-Ruiz, L.A.; Rottler, J.; Wirth, B.D.; Car, R.; Srolovitz, D.J. Self-interstitial transport in vanadium. Acta Mater. 2005, 53, 1985–1994. [Google Scholar] [CrossRef]
- Heinola, K.; Ahlgren, T.; Nordlund, K.; Keinonen, J. Hydrogen interaction with point defects in tungsten. Phys. Rev. B 2010, 82, 094102. [Google Scholar] [CrossRef]
- Monasterio, P.R.; Lau, T.T.; Yip, S.; Van Vliet, K.J. Hydrogen-vacancy interactions in Fe-C alloys. Phys. Rev. Lett. 2009, 103, 085501. [Google Scholar] [CrossRef]
- Ueda, Y.; Shimada, T.; Nishikawa, M. Impacts of carbon impurities in hydrogen plasmas on tungsten blistering. Nucl. Fusion 2003, 44, 62. [Google Scholar] [CrossRef]
- Kong, X.-S.; You, Y.-W.; Fang, Q.F.; Liu, C.S.; Chen, J.-L.; Luo, G.N.; Pan, B.C.; Wang, Z. The role of impurity oxygen in hydrogen bubble nucleation in tungsten. J. Nucl. Mater. 2013, 433, 357–363. [Google Scholar] [CrossRef]
- Zhou, X.; Marchand, D.; McDowell, D.L.; Zhu, T.; Song, J. Chemomechanical Origin of Hydrogen Trapping at Grain Boundaries in fcc Metals. Phys. Rev. Lett. 2016, 116, 075502. [Google Scholar] [CrossRef]
- Valles, G.; Panizo-Laiz, M.; González, C.; Martin-Bragado, I.; González-Arrabal, R.; Gordillo, N.; Iglesias, R.; Guerrero, C.L.; Perlado, J.M.; Rivera, A. Influence of grain boundaries on the radiation-induced defects and hydrogen in nanostructured and coarse-grained tungsten. Acta Mater. 2017, 122, 277–286. [Google Scholar] [CrossRef]
- Hayashi, E.; Kurokawa, Y.; Fukai, Y. Hydrogen-induced enhancement of interdiffusion in Cu-Ni diffusion couples. Phys. Rev. Lett. 1998, 80, 5588. [Google Scholar] [CrossRef]
- Gui, L.-J.; Liu, Y.-L.; Wang, W.-T.; Jin, S.; Zhang, Y.; Lu, G.-H.; Yao, J.-E. First-principles investigation on vacancy trapping behaviors of hydrogen in vanadium. J. Nucl. Mater. 2013, 442, S688–S693. [Google Scholar] [CrossRef]
- Ouyang, C.; Lee, Y.-S. Hydrogen-induced interactions in vanadium from first-principles calculations. Phys. Rev. B 2011, 83, 045111. [Google Scholar] [CrossRef]
- Zhang, P.; Zhao, J.; Wen, B. Trapping of multiple hydrogen atoms in a vanadium monovacancy: A first-principles study. J. Nucl. Mater. 2012, 429, 216–220. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initiomolecular dynamics for open-shell transition metals. Phys. Rev. B 1993, 48, 13115–13118. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Han, S.; Zepeda-Ruiz, L.A.; Ackland, G.J.; Car, R.; Srolovitz, D.J. Self-interstitials in V and Mo. Phys. Rev. B 2002, 66, 220101. [Google Scholar] [CrossRef]
- Van de Walle, C.G.; Neugebauer, J. First-principles calculations for defects and impurities: Applications to III-nitrides. J. Appl. Phys. 2004, 95, 3851–3879. [Google Scholar] [CrossRef]
- Ohsawa, K.; Eguchi, K.; Watanabe, H.; Yamaguchi, M.; Yagi, M. Configuration and binding energy of multiple hydrogen atoms trapped in monovacancy in bcc transition metals. Phys. Rev. B 2012, 85, 094102. [Google Scholar] [CrossRef]
- Hou, J.; Kong, X.; Sun, J.; You, Y.; Wu, X.; Liu, C.; Song, J. Hydrogen bubble nucleation by self-clustering: Density Functional Theory and statistical models studies using tungsten as a model system. Nucl. Fusion 2018, 58, 096021. [Google Scholar] [CrossRef]
- Takagi, I.; Matsubara, N.; Akiyoshi, M.; Moritani, K.; Sasaki, T.; Moriyama, H. Deuterium trapping near vanadium surface bombarded with hydrogen ions. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2005, 232, 327–332. [Google Scholar] [CrossRef]
- Freysoldt, C.; Grabowski, B.; Hickel, T.; Neugebauer, J.; Kresse, G.; Janotti, A.; Van de Walle, C.G. First-principles calculations for point defects in solids. Rev. Mod. Phys. 2014, 86, 253–305. [Google Scholar] [CrossRef]
- Hayward, E.; Fu, C.-C. Interplay between hydrogen and vacancies in α-Fe. Phys. Rev. B 2013, 87, 174103. [Google Scholar] [CrossRef]
- Arbuzov, V.; Vykhodets, V.; Raspopova, G. The trapping of hydrogen ions in vanadium and titanium. J. Nucl. Mater. 1996, 233, 442–446. [Google Scholar] [CrossRef]
- Geng, W.T.; Wan, L.; Du, J.-P.; Ishii, A.; Ishikawa, N.; Kimizuka, H.; Ogata, S. Hydrogen bubble nucleation in α-iron. Scr. Mater. 2017, 134, 105–109. [Google Scholar] [CrossRef]
- Ferrin, P.; Kandoi, S.; Nilekar, A.U.; Mavrikakis, M. Hydrogen adsorption, absorption and diffusion on and in transition metal surfaces: A DFT study. Surf. Sci. 2012, 606, 679–689. [Google Scholar] [CrossRef]
- Dag, S.; Ozturk, Y.; Ciraci, S.; Yildirim, T. Adsorption and dissociation of hydrogen molecules on bare and functionalized carbon nanotubes. Phys. Rev. B 2005, 72, 155404. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Configuration | Ref. [34] (eV) | |
|---|---|---|
| −0.34 | −0.31 | |
| −0.43 | −0.40 | |
| −0.27 | −0.25 | |
| −0.28 | −0.25 | |
| −0.24 | −0.24 | |
| −0.16 | −0.12 | |
| 0.65 | 0.69 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, Z.; Wang, S.; Lu, C.; Peng, Q. Relationship between the Behavior of Hydrogen and Hydrogen Bubble Nucleation in Vanadium. Materials 2020, 13, 322. https://doi.org/10.3390/ma13020322
Su Z, Wang S, Lu C, Peng Q. Relationship between the Behavior of Hydrogen and Hydrogen Bubble Nucleation in Vanadium. Materials. 2020; 13(2):322. https://doi.org/10.3390/ma13020322
Chicago/Turabian StyleSu, Zhengxiong, Sheng Wang, Chenyang Lu, and Qing Peng. 2020. "Relationship between the Behavior of Hydrogen and Hydrogen Bubble Nucleation in Vanadium" Materials 13, no. 2: 322. https://doi.org/10.3390/ma13020322
APA StyleSu, Z., Wang, S., Lu, C., & Peng, Q. (2020). Relationship between the Behavior of Hydrogen and Hydrogen Bubble Nucleation in Vanadium. Materials, 13(2), 322. https://doi.org/10.3390/ma13020322